
1 La secuencia de reacciones que sigue se emplea en la...
26
EJERCICIOS SOBRE COMPUESTOS AROMÁTICOS 1 La secuencia de reacciones que sigue se emplea en la síntesis del ftalilsulfatiazol (profármaco del sulfatiazol) que presenta en N 4 un grupo polar que hace que la absorción oral sea escasa o nula. Sin embargo, en el tramo final del intestino experimenta una activación metabólica por acción de microorganismos de la flora intestinal que hidrolizan el grupo carboxamido liberando el sulfatiazol, por lo que el ftalilsulfatiazol es utilizado en el tratamiento de las infecciones bacterianas a nivel intestinal. Propón las estructuras de los compuestos A, B, C, D, E. Anilina A B ClCO 2 Et C clorosulfonación D N S H 2 N O O O E NaOH S N H H N N S O O O HO O Ftalilsulfatiazol SOLUCIÓN A B ClCO 2 Et C clorosulfonación N S H 2 N NH 2 NHCO 2 Et NHCO 2 Et SO 2 Cl ClSO 3 H O O O D E NaOH NHCO 2 Et SO 2 NH N S NH 2 SO 2 NH N S S N H H N N S O O O HO O Ftalilsulfatiazol
Transcript of 1 La secuencia de reacciones que sigue se emplea en la...

EJERCICIOS SOBRE COMPUESTOS AROMÁTICOS 1 La secuencia de reacciones que sigue se emplea en la síntesis del ftalilsulfatiazol
(profármaco del sulfatiazol) que presenta en N4 un grupo polar que hace que la absorción oral sea escasa o nula. Sin embargo, en el tramo final del intestino experimenta una activación metabólica por acción de microorganismos de la flora intestinal que hidrolizan el grupo carboxamido liberando el sulfatiazol, por lo que el ftalilsulfatiazol es utilizado en el tratamiento de las infecciones bacterianas a nivel intestinal. Propón las estructuras de los compuestos A, B, C, D, E.
Anilina
A BClCO2Et
Cclorosulfonación
DN
SH2N
O
O
OE
NaOH SNH
HN
N
SO
O
OHO O
Ftalilsulfatiazol SOLUCIÓN
A B
ClCO2Et
C
clorosulfonación
NSH2NNH2
NHCO2EtNHCO2Et
SO2Cl
ClSO3H
O
O
O
D E
NaOH
NHCO2Et
SO2NHN
S
NH2
SO2NHN
S
SNH
HN
NS
O
O
OHO O
Ftalilsulfatiazol

2 La siguiente secuencia sintética forma parte de un programa encaminado a la síntesis de fenoles que inhiben la acción de las hormonas que intervienen en el proceso de la coagulación de la sangre. La primera reacción es una alquilación de Friedel-Crafts a partir de un alcohol. ¿Cuál es la estructura de A y C? ¿Y la de los reactivos B y D?
etapa 1A
BCH3CH3CH3
CH3OH
Cetapa 2
D+
CH3
CH3OH
(CH2)5COOH
F
Br
SOLUCIÓN
A
BCH3HOCH3
CH3OH
C
D
(CH2)5COOH
F
H3O+
(CH2)5COOH
FBr2 / AlBr3
3 La octopamina es una ariletanolamina simpatomimética, usada como estimulante cardíaco.
OH
CHCH2NH2HO
Octopamina A continuación se muestra una posible vía sintética de la octopamina, partiendo de fenol e
hidrocloruro de aminoacetonitrilo, mediante una reacción de sustitución electrofílica aromática en la primera etapa. Completa el esquema sintético.
OH
+ N C CH2NH2
. HCl
AlCl3 OctopaminaH2O
A
SOLUCIÓN
OH
+ N C CH2NH2.HClAlCl3
OH
C CH2NH2O
H2
Ni-Raney
H2O
B
A

OH
CHCH2NH2HO
Octopamina
4 La dopamina es un neurotransmisor excitador a nivel de sistema nervioso central. Tiene una estructura molecular muy sencilla, se trata de una fenetilamina, y dada su similitud estructural con las ariletanolaminas se comporta también como agonista adrenérgico. A nivel periférico, esta acción justifica el uso de la dopamina como cardiotónico por su capacidad de estimular los receptores β1 cardíacos. Su síntesis se inicia a partir del o-dimetoxibenceno, haciendo uso de una reacción
clorometilación de anillos bencénicos activados, con metanal y cloruro de hidrógeno. Escribe las estructuras de los compuestos intermedios hasta llegar a la dopamina.
HCl KCNCH3O
CH3O
+ CH2O H2
catalizadorA B
HBr HO
HO
CH2CH2NH2
Dopamina
C H2
catalizador
SOLUCIÓN
HCl KCNCH3O
CH3O
+ CH2O CH3O
CH3O
CH2Cl
A
HBr H2
catalizadorCH3O
CH3O
CH2CN CH3O
CH3O
CH2CH2NH2
B C
HBr HO
HO
CH2CH2NH2
Dopamina

5 La dobutamina es un análogo del neurotransmisor dopamina, capaz de estimular los receptores β1 cardíacos, lo que justifica el uso de la dobutamina como agente cardiotónico. Completa el esquema indicado a continuación que conduce a la obtención de la dobutamina.
HCHOBACH3O
CH3OHCl
(C9H11ClO2)
KCN
(C10H11NO2)
H2
Ni-RaC
(C10H15NO2)
HBrD
(C8H11NO2)
D + OCH3CH2CH2CO
CH3H2
Pd-CE
(C19H25NO3)
HIDobutamina
(C18H23NO3) SOLUCIÓN
KCN
A
CH3O
CH3O
CH2ClCH3O
CH3O
HCHO
HClB
CH3O
CH3O
CH2CN
H2
Ni-RaC
HBr
D
CH3O
CH3O
CH2CH2NH2 HO
HO
CH2CH2NH2
D
+ OCH3CH2CH2CO
CH3HO
HO
CH2CH2NH2
H2
Pd-C
HO
HO
CH2CH2N OCH3CH2CH2CCH3
E
HIHO
HO
CH2CH2N OCH3CH2CH2CHCH3H

Dobutamina
HO
HO
CH2CH2N OHCH2CH2CHCH3H
6 La observación de que las sulfamidas antibacterianas producían acidosis metabólica y
alcalinización de la orina condujo a la obtención de diversas familias de fármacos diuréticos, a una de las cuales pertenece la bumetanida, utilizado en clínica como antihipertensivo y para el tratamiento del edema asociado a insuficiencia cardíaca, enfermedad renal, hepática o pulmonar. Completa la siguiente secuencia sintética que lleva a la obtención de la bumetanida.
COOH
Cl ClSO3HA
HNO3B
H2SO4
D
PhONa
NH3C
EHCl
SnCOOH
OPhH2NO2S N
HCH3
Bumetanida
F
SOLUCIÓN
COOH
Cl ClSO3H
A
HNO3
B
H2SO4COOH
ClSO2Cl
COOH
ClSO2Cl
O2N
D
PhONaNH3
CCOOH
ClSO2NH2
O2N COOH
PhOSO2NH2
O2N COOH
PhOSO2NH2
H2NE
HCl
Sn
F
I CH3
COOH
OPhH2NO2S N
HCH3
Bumetanida

7 La furosemida es uno de los diuréticos más efectivos, desarrollado a partir de las sulfonamidas antibacterianas. Propón una posible vía de síntesis de la furosemida a partir del ácido 2,4-diclorobenzoico, furfurilamina y los reactivos que consideres necesarios.
OCH2
NHCOOH
SO2NH2
Cl
Furosemida
OCH2NH2 Furfurilamina
ClCOOH
ClÁcido 2,4-diclorobenzoico
SOLUCIÓN
ClCOOH
SO2NH2
Cl
ClCOOH
SO2ClCl
NH3
ClCOOH
Cl
ClSO3H
ClCOOH
SO2NH2
Cl
OCH2NH2
OCH2
NHCOOH
SO2NH2
Cl
8 La tolpropamina, utilizada como antialérgico, pertene a una de las familias clásicas de
antihistamínicos H1, las propilaminas. Completa su secuencia sintética.
A
HCHO
HN(CH3)2
B C D calor
BrMgTolpropamina
H2CH3
Pd-C
H3O+ ClCOCH3
AlCl3 SOLUCIÓN
ClCOCH3
AlCl3
CH3O
A
HCHO
HN(CH3)2
O
N(CH3)2
B
BrMg CH3
H2O

C
D
calor
Tolpropamina
H2
Pd-C
H3O+
HO
N(CH3)2N(CH3)2 N(CH3)2
CH3 CH3 CH3
9 El cloranfenicol (cloromicetina) fue un agente antibacteriano muy usado en la década de los cincuenta. En la actualidad, a pesar de presentar problemas tóxicos (puede producir anemia aplásica por su posible interacción con el ADN) que han restringido su uso, sigue siendo importante en el tratamiento de ciertas infecciones.
OH
NO2
HO
NHCOCl2
H
HCloranfenicol
Fue el primer producto farmacéutico de origen natural que tenía un grupo nitro y el primero que
se obtuvo por síntesis química total, aunque también era viable aplicar la ruta de fermentación. La primera ruta sintética industrial del cloranfenicol se iniciaba con la condensación del
benzaldehído y el β-nitroetanol. Tras la separación de los isómeros eritro y treo, se llevaba a cabo la reducción del grupo nitro en la mezcla racémica de los isómeros treo (R,R y S,S); seguidamente se resolvía la mezcla racémica con ácido (+)-tartárico, aislándose el isómero (R,R), se introducía el grupo nitro en posición aromática y por último se realizaba una dicloroacilación del grupo amino.
Escribe todas las reacciones descritas que conducen al antibacteriano cloranfenicol. SOLUCIÓN
CHO + HOCH2CH2NO2 C CHH
OH
NO2
CH2OH
1. Separación2. H2 / Pd
(eritro/treo 2:1)
OH
O2N
HO
NH2
H
H
C CHH
OH
NH2
CH2OH
Mezcla racémica delos isómeros treo
1. ac. (+)-tartárico2. Acetilación
3. Nitración (HNO3/H2SO4)4. Desacetilación

Cl2CHCOOCH3
O2N
OH
HO
NHCOCHCl2
H
H
10 Aplica el análisis retrosintético al antiséptico nitrofural y sintetízalo, en una sola etapa, partiendo de los reactivos adecuados.
O CHO2N N NH C NH2O
Nitrofural
SOLUCIÓN Una desconexión adecuada es la que se señala en la estructura.
O CHO2N N NH C NH2O
Síntesis en un solo paso. Formación de semicarbazona a partir del aldehído correspondiente y
de la semicarbazida. El mecanismo es el de adición-eliminación de un nucleófilo con nitrógeno sobre un grupo
carbonilo.
H2OO CO2N
OH + H2N
NC
NH2
H
O
O CO2N N
H
NH C NH2
O
Nitrofural5-Nitrofurfural Semicarbazida 11 La última etapa de una de las síntesis industriales del antiinflamatorio no esteroidal
ibuprofeno, utilizado terapéuticamente como antiinflamatorio vaginal, consiste en la carbonilación del alcohol con monóxido de carbono dando el ácido carboxílico.
CHCH3CH3
CH2 CH CH3OH
CO PdCl2 HCl
PPh3 CH3COCH2CH3
Ibuprofeno
Esta síntesis no es estereoespecífica y se obtiene una mezcla racémica de ambos
enantiómeros. Escribe la estructura de los dos enantiómeros del ibuprofeno. SOLUCIÓN La estructura del ibuprofeno sin estereoquímica es

CHCH3CH3
CH2 CH COOHCH3
*
Los dos enantiómeros de este fármaco tienen las siguientes estructuras moleculares:
CHCH3CH3
CH2 COOH
CH3HCHCH2HOOC
CH3 H
CH3
CH3
12 La sulfanilamida fue uno de los primeros quimioterápicos que se sintetizaron. Completa el
siguiente esquema de síntesis.
NCH3
O
H
A BClSO3H NH3 NaOH
Sulfanilamida
SOLUCIÓN El compuesto A es el cloruro de 4-acetamidobencenosulfonilo y el compuesto B es la 4-
acetamidobencenosulfonamida. El esquema de síntesis completo es:
NCH3
O
H A
ClSO3H NH3
NCH3
O
H
SO2Cl
NaOH
SulfanilamidaB
NCH3
O
H
SO2NH2
H2N
SO2NH2
13 La obtención del tuberculostático PAS (ácido p-aminosalicílico) es un ejemplo de orto-
carboxilación de un fenol dando el correspondiente ácido orto-hidroxibencenocarboxílico (Síntesis de Kolbe-Schmitt).
Escribe la reacción para obtener PAS (ácido 4-amino-2-hidroxibenzoico) a partir de m-
aminofenol. SOLUCIÓN

+ CO2
OH
COOHNH2 5-10 at
H2N
OHKHCO3
PAS 14 Escribe el mecanismo de la síntesis del tuberculostático isoniazida.
N
C
NH2
N
C
Isonicotinatode etilo
O OCH2CH3 O NHNH2
NH2
Isoniazida
SOLUCIÓN La formación de isoniazida a partir de isonicotinato de etilo e hidrazina es un proceso de
adición + eliminación similar a la formación de amidas a partir de ésteres por reacción con NH3.
La hidracina ataca nucleofílicamente al carbono del carboxilo y sale eliminado el etanol.
N
CNH2
O OCH2CH3
NH2
N
C OCH3CH2O
NNH2HH
N
CO NHNH2
+ CH3CH2OH
hidrazida del ácido isonicotínico
N
C OCH3CH2O
NHNH2H
15 Dibuja las estructuras de los productos intermedios representados por letras en la secuencia
de reacciones que integran la síntesis total de la mostaza nitrogenada antineoplásica clorambucilo.
HNO3 HCl, CH3OH(CH2)3CO2CH3
H2, Pd
H2SO4
BA C

O H2OD E
N
CH2CH2CH2COOH
CH2CH2ClClCH2CH2
Clorambucilo
POCl32
SOLUCIÓN
NO2
CH2CH2CH2COOH
A
B
NO2
CH2CH2CH2COOCH3
NH2
CH2CH2CH2COOCH3
C
N
CH2CH2CH2COOCH3
CH2CH2OHHOCH2CH2
D
N
CH2CH2CH2COOCH3
CH2CH2ClClCH2CH2
E
16 El (S)-naproxeno es un fármaco antiinflamatorio no esteroideo perteneciente al grupo de los ácidos α-arilpropiónicos. El proceso de síntesis desarrollado por los Laboratorios Zambon permite la obtención del (S)-naproxeno enantioméricamente puro mediante la utilización de tartrato de metilo como auxiliar quiral, evitándose así la resolución óptica.
CH3O
CCOOH
HH3C
(S)-Naproxeno
La estrategia sintética, partiendo de la 1-(6-metoxi-2-naftil)-propan-1-ona, consta de cinco
etapas. Escribe los reactivos necesarios para cada una de las cinco etapas:

CH3O CH3O
O O O
COOCH3CH3OOC1)
CH3O
O O
COOCH3CH3OOC
CH3O
O O
COOCH3CH3OOC
Br
BrRRS >> RRR
2)
CH3O
O O
COOHHOOC
Br
BrRRS
CH3O
O O
COOCH3CH3OOC
Br
BrRRS
3)
CH3O
O O
COOHHOOC4)
Br
Br
CH3O
CCOOH
HCH3
Br
CH3O
CCOOH
HCH3
(S)-Naproxeno
CH3O
CCOOH
HCH3
Br
5)
SOLUCIÓN 1. Utilización del auxiliar quiral (2R,3R)-tartrato de dimetilo: entra en la primera etapa y sale en la
cuarta etapa.
CH3O
CH3O
OO O
COOCH3CH3OOC
HCOOCH3
OH
COOCH3HO H+
2. Reacción de bromación: presenta una alta diastereoselectividad (RRS >> RRR).

CH3O
O O
COOCH3CH3OOC
CH3O
O O
COOCH3CH3OOC
Br
Br
Br2
3. Hidrólisis del diéster.
CH3O
CH3
O O
COOHHOOC
Br
Br
HCH3O
CH3
O O
COOCH3CH3OOC
Br
Br
H
H3O+
4. Transposición del arilo y conversión del cetal cíclico en ácido carboxílico.
CH3OCH3
O OCOOHHOOC
Br
BrH CH3O
CH3C
Br
H
OHO
H+
∆
CH3O
HCH3
Br
COOH
5. Reducción del bromo en la posición 5 del anillo aromático.
CH3O
HCH3
Br
COOH H2
Ni (Raney) CH3O
HCH3COOH
17 Los derivados de ácidos arilacéticos y arilpropiónicos constituyen dos de las familias más
representativas de fármacos antiinflamatorios no esteroideos. Dado el enorme interés terapéutico de estos fármacos, se han desarrollado numerosos métodos sintéticos para su obtención. A continuación se muestran dos vías alternativas para la síntesis del ibuprofeno (ácido 2-arilpropiónico antiinflamatorio).
Propón las estructuras de los compuestos indicados con letras.

1. ClCOCH(CH3)2 AlCl3
2. Zn-Hg/HClA
ClCH2COOEtAlCl3
B1. EtONa
2. ClCOOEtC
DE
1. H3O+
2. calor
COOH
Ibuprofeno
FClCOCH3
AlCl3KCN
H3O+G
HI/P
SOLUCIÓN
ClCH2COOEt
AlCl3
B
1. EtONa
2. ClCOOEt
1. ClCOCH(CH3)2 AlCl3
2. Zn-Hg/HCl
A
COOEt
C
EtOOC COOEt
D
E
HNa
ICH3
EtOOC COOEt1. H3O+
2. calor
COOH
Ibuprofeno
G
HI/P
A
ClCOCH3
AlCl3
O
F
KCNH3O+
HOCN
Ibuprofeno
COOH
18 Los derivados de ácidos arilalcanoicos (ibuprofeno, flubiprofeno, naproxeno, indometacina,
etc) constituyen el grupo más numeroso de AINES (antiinflamatorios no esteroideos), grupo en el que se encuentran descritos hasta la fecha los antinflamatorios más potentes. A continuación se muestra una posible vía de síntesis del antiinflamatorio flubiprofeno.
a) Completa la secuencia sintética que conduce a la obtención del flubiprofeno. b) Sugiere un mecanismo para la conversión de A en B.
A
B C1. HNa
2. CH3I
1. KOH2. HCl
3. calor
F CH2 CO
OEt EtO OEtO
EtONaFlubiprofeno
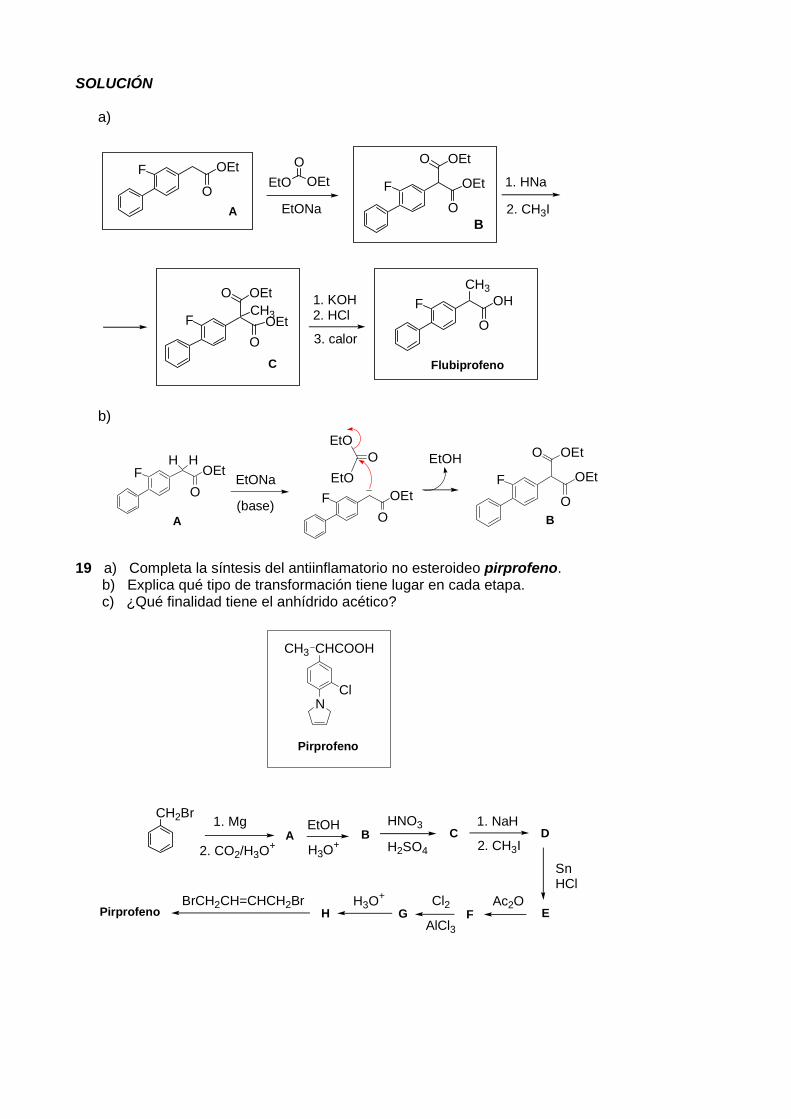
SOLUCIÓN a)
A
EtO OEtO
EtONa
FO
OEt
B
1. HNa
2. CH3I
FO
OEt
O OEt
Flubiprofeno
FO
OHCH3
C
1. KOH2. HCl
3. calorF
OOEt
O OEtCH3
b)
A
EtO
EtOO
EtONaFO
OEt
FO
OEt
EtOH
(base)
HH
B
FO
OEt
O OEt
19 a) Completa la síntesis del antiinflamatorio no esteroideo pirprofeno. b) Explica qué tipo de transformación tiene lugar en cada etapa. c) ¿Qué finalidad tiene el anhídrido acético?
CHCOOHCH3
NCl
Pirprofeno
CH2Br 1. Mg
2. CO2/H3O+A
EtOH
H3O+B
HNO3
H2SO4C
1. NaH
2. CH3ID
SnHCl
EAc2O
FCl2
AlCl3G
H3O+
HBrCH2CH=CHCH2Br
Pirprofeno

SOLUCIÓN a)
A C D
CH2COOH
B
CH2COOEt CH2COOEt
NO2
CHCOOEt
NO2
CH3
E
CHCOOEt
NH2
CH3
F
CHCOOEt
NHCOCH3
CH3
G
CHCOOEt
NHCOCH3
CH3
Cl
H
CHCOOH
NH2
CH3
Cl
b) ¬ Formación del magnesiano y carboxilación: compuesto A ¬ Esterificación: compuesto B ¬ Nitración: compuesto C ¬ Alquilación en el carbono α al carbonilo del grupo éster: compuesto D ¬ Reducción del grupo nitro: compuesto E ¬ Formación de la amida: compuesto F ¬ Cloración: compuesto G ¬ Hidrólisis de la amida: compuesto H ¬ Formación del pirprofeno a partir de H:
CHCOOHCH3
NCl
Pirprofeno
CHCOOH
NH2
CH3
Cl CH2CH=CHCH2BrBr
CHCOOH
HN
CH3
Cl
Br
− HBr
− HBr
c) El uso del anhídrido acético tiene como objetivo convertir el grupo amino, que es un grupo
fuertemente activante, en un grupo carboxamido, que es moderadamente activante; de esta forma, se puede conseguir en la cloración que el cloro entre exclusivamente en una de las dos posiciones orto al grupo carboxamido.
20 La benzocaína es un anestésico local de aplicación exclusivamente tópica debido a su baja
solubilidad en agua.
COOCH2CH3N
H
HBenzocaínap-aminobenzoato de etilo
a) Analiza las desconexiones sucesivas que deben aplicarse a la estructura molecular de la
benzocaína para su obtención.

b) Escribe una estrategia de síntesis de la benzocaína a partir de tolueno. SOLUCIÓN
a) Análisis retrosintético
COO - CH2CH3H2N
b) Estrategia sintética
CH3
HNO3
H2SO4CH3O2N
KMnO4COOHO2N
COOHH2NH2
Pd/C
EtOHH2SO4
COOCH2CH3H2N
Benzocaína
21 ¿Cómo se podría preparar el analgésico derivado del ácido salicílico, la etenzamida (2-
etoxibenzamida), a partir de la salicilamida (2-hidroxibenzamida)?
CONH2OCH2CH3
Etenzamida
CONH2OH
Salicilamida SOLUCIÓN Formación de un etilariléter, por reacción del fenol correspondiente con un agente alquilante, el
sulfato de dietilo.
CONH2OH
Salicilamida
+ CH3CH2 O SO
OO CH2CH3
NaOH
Sulfato de dietilo
CONH2OCH2CH3
Etenzamida El hidróxido sódico transforma el fenol en fenóxido de sodio, que es un buen nucleófilo.
CONH2OH
+ NaOH
CONH2O Na
+ H2O

La alquilación tiene lugar mediante un mecanismo SN2, con el sulfato de dietilo (Et2SO4) como sustrato y el anión fenóxido (PhO− Na+) como nucleófilo.
CONH2O
+ CH3CH2 O SO
OO CH2CH3
SN2
CONH2OCH2CH3
Etenzamida 22 La búsqueda de compuestos estructuralmente relacionados con la morfina condujo a la
obtención de la metadona, interesante por sus propiedades analgésicas, pero que conserva los efectos secundarios de la morfina, aunque con menor intensidad, por lo que se suele utilizar en el tratamiento de los síntomas del síndrome de abstinencia. A continuación se muestra una secuencia sintética que lleva a la obtención de metadona racémica.
a) Dibuja las estructuras de los compuestos indicados por letras.
b) La metadona presenta un centro estereogénico. Indica qué procedimiento permitirá disponer de (R)-metadona enantioméricamente pura.
CHO1.BrMgPh
ASOCl2
BKCN
C1. base
2.
DEH3O+
Metadonaracémica
2. H3O+
ClCH2CHCH3
N(CH3)2
BrMgEt
SOLUCIÓN a)
CH
A
OHPh CH
B
ClPh CH
C
CNPh C
D
CNPh CH2CHN(CH3)2
CH3
C
E
C
Ph CH2CHN(CH3)2
CH3
Et NH
C
Metadona racémica
C
Ph CH2CHN(CH3)2CH3
Et O
b) Para obtener (R)-metadona enantioméricamente pura podríamos llevar a cabo una resolución
de la mezcla racémica de (R) y (S)-metadona, utilizando ácido (-)-dibenzoiltartárico. La reacción de la mezcla racémica con el ácido (−)-dibenzoil-tartárico daría lugar a la formación

de las sales diastereoméricas correspondientes, cuya separación por cristalización y posterior hidrólisis conduciría a la obtención de la (R)-metadona enantioméricamente pura.
C
(R)-Metadona
CPh
Et O
NH CH3
CH3
CH3
23 El paracetamol es un analgésico antitérmico, que proviene del metabolismo tanto de la acetanilida como de la acetofenetidina, utilizados inicialmente con esta finalidad terapéutica.
Completa el esquema de síntesis del paracetamol, escribiendo sobre las flechas los reactivos necesarios para cada uno de los procesos químicos implicados.
OH OH
NH2
OH
NO2
Paracetamol
OH
NH C
O
CH3
SOLUCIÓN El procedimiento de obtención del paracetamol tal como se muestra en el esquema consiste
en tres etapas:
¬ La primera transformación es la nitración del fenol, según una reacción SEAr.
¬ En la siguiente etapa: El p-nitrofenol se reduce mediante una hidrogenación catalítica, transformándose en 4-aminofenol
OH
HNO3 H2
Ni-RaneyOH
NH2
OH
NO2
¬ Por último, el 4-aminofenol se acetila con anhídrido acético sobre el átomo de nitrógeno del
grupo amino, más nucleófilo que el oxígeno del hidroxilo.
Paracetamol
OH
NH2CH3 O CH3
O O
OH
NH CO
CH3
+ CH3COOH

24 La entacapona es un fármaco de reciente aparición, utilizado para el tratamiento de la
enfermedad de Parkinson asociado con la L-Dopa. La entacapona es resultado del diseño de inhibidores selectivos de la COMT (catecol O-metiltransferasa), enzima que metaboliza las catecolaminas. Esta inhibición enzimática reduce el metabolismo de la dopamina en el cerebro y potencia la acción de la L-Dopa.
La síntesis de la entacapona consta de las etapas siguientes:
CHO
OCH3
HO
NO2CHO
OCH3
HO CH3COOH
HNO3 HBr conc
∆
CHO
OHHO
NO2
NC N CH3
O
CH3CHO
OHHO
NO2+
N H
OHHO
NO2 N
O
CH3
CH3CN Entacapona
Propón mecanismos para: a) La rotura del metiléter. b) La condensación que tiene lugar en la última etapa de la síntesis. SOLUCIÓN a) Ruptura del éter en medio ácido. La protonación del grupo metoxilo favorece el desplazamiento
del fenol que es un buen grupo saliente y el ataque nucleofílico del bromuro sobre el metilo.
CHO
OHO
O2N
CH3
H Br
CHO
OHO
O2N
CH3 H
Br
CHO
OHO
O2N
H
b) Se trata de una condensación de tipo aldólico. La piperidina es una base suficientemente fuerte para arrancar el protón del metileno activo y
formar un anión nucleófilo que ataca posteriormente al carbonilo del benzaldehído. El alcohol que se forma pierde agua transformándose en la amida α,β-insaturada que es la entacapona.
NC N CH3
O
CH3
+ NH NC N CH3
O
CH3
+ NH
H

C
OHHO
O2N
O
HNC N CH3
O
CH3
OHHO
O2N N
O
CH3
CH3CN
O
- H2O
OHHO
O2N N
O
CH3
CH3CN
EntacaponaOH
HO
O2N N
O
CH3
CH3CN
OH
25 El haloperidol, cabeza de serie de los neurolépticos derivados de la butirofenona, es un
antipsicótico de uso muy generalizado, cuyo descubrimiento fue resultado de la manipulación estructural de la petidina, uno de los análogos estructurales más antiguos de la morfina.
NCH3
CO2EtN
O
CO2Et
Petidina (meperidina)(analgésico)
Análogo butirofenónicode la Petidina(analgésico y neuroléptico)
N
F
O
OH
ClHaloperidol(neuroléptico)
El haloperidol, como es una molécula relativamente compleja, se obtiene por la reacción de dos sintones, que a su vez se preparan por separado, a partir de compuestos más simples.
a) Aplica el análisis retrosintético a la molécula de haloperidol y propón la desconexión más adecuada.
b) Escribe una posible vía de síntesis de ambos sintones. SOLUCIÓN a) La desconexión está señalada sobre la estructura:
N
F
OOH
Cl
Los dos sintones que deben reaccionar para dar el haloperidol son:

F CO
CH2CH2CH2Cl
γ-cloro-p-fluoro-butirofenona
NHOH
Cl
+ Haloperidol
4-(4-clorofenil)-piperidin-4-ol b) El agente alquilante, la γ-cloro-p-fluorobutirofenona, se prepara por una acilación de
Friedel-Crafts sobre el anillo de fluorobenceno.
AlCl3F + ClCOCH2CH2CH2Cl F CO
CH2CH2CH2Cl
La piperidina disustituida en la posición 4 se obtiene mediante la siguiente secuencia de
reacciones químicas:
NH3Cl CCH2
CH3 + 2 O CH
HC CH2
CH3
CH2 NH
CH2HO
Cl
ClO
NHCH3 H3O+
Cl C CH2
CH3CH2 N CH2
OH H
H2O
Cl C CH2
CH2
CH2 N
CH2
H
Cl NHHBr H2O
Cl NH
HO
26 El antidepresivo melitraceno se prepara a partir de la 10H-antracen-9-ona. En la primera etapa de la secuencia sintética hay una doble alquilación en medio básico sobre el metileno activo de la posición 10 del antraceno. Como agente alquilante empleamos yoduro de metilo. El compuesto resultante de la alquilación anterior se trata con el magnesiano adecuado. Por último, la eliminación de agua en medio ácido conduce al fármaco.
O
+ I CH3NaOH
O
CH3 CH3

ClMg N CH3
CH3 HO
CH3 CH3
N CH3CH3
HCl
CH3 CH3
NCH3
CH3
Melitraceno Escribe el mecanismo detallado de la primera reacción de la síntesis. ¿A qué tipo de reacción
pertenece? SOLUCIÓN En medio básico se forma un anión, al perderse un hidrógeno ácido en el metileno del
antraceno. El anión resultante ataca nucleofílicamente al ioduro de metilo.
O
H H
OH
O
H
I CH3
O
H CH3
SN2
La segunda alquilación (metilación) se produce siguiendo un mecanismo idéntico, con el otro
hidrógeno ácido que queda sobre el carbono 10 del antraceno.
O
H CH3
OH− I CH3
O
CH3 CH3 27 La maprotilina es un prototipo de los antidepresivos tetracíclicos. En su síntesis se aprovecha
la reactividad del sistema diénico del antraceno frente a dienófilos en reacciones de Diels-Alder. Identifica los reactivos que corresponden a cada una de las etapas.
O
NaOCH3
O
CH2CH2CN
H2O
a b
O
CH2CH2COOH
c
CH2CH2COOH

CH2 CH2
CH2CH2COOHd
CH2CH2COCl
CH2CH2CONHCH3
f
CH2CH2CH2NHCH3
Maprotilina
e
SOLUCIÓN ¬ Reacción de adición de Michael e hidrólisis ácida del grupo nitrilo.
O
CH2 CH
NaCH3OO
CH2CH2CNH2SO4
H2O
abCN
¬ Reducción del grupo carbonilo y deshidratación del alcohol correspondiente.
O
CH2CH2COOHZn/NH3
CH2CH2COOHc
¬ Reacción de Diels-Alder entre el sistema diénico del antraceno y el eteno como dienófilo,
creando un nuevo puente en la molécula. Conversión del ácido carboxílico en cloruro de ácido.
CH2 CH2
CH2CH2COOHSOCl2
CH2CH2COCld
¬ El cloruro de acilo formado se transforma en la amida N-metilsustituida, que se reduce, por último, hasta la amina correspondiente.
CH3NH2CH2CH2CONHCH3
LiAlH4CH2CH2CH2NHCH3
Maprotilina
e f

28 Completa el esquema de síntesis del ácido acetrizoico, medio de contraste para rayos X,
escribiendo los reactivos que faltan.
COOH COOH
O2N
COOH
NH2
a b c
COOH
NH2
I
I
I COOH
NH
I
I
I
CCH3
O
Ácido Acetrizoico
d
Explica la causa de que se produzca directamente la trihalogenación del anillo aromático. SOLUCIÓN
a) Partimos del ácido benzoico, que se nitra con ácido nítrico y ácido sulfúrico. El grupo nitro se reduce a amina con hierro en medio ácido. Posteriormente, se introducen tres átomos de yodo en las posiciones 2, 4 y 6, mediante el cloruro de yodo como electrófilo. La última etapa de la síntesis consiste en la acetilación del grupo amina, empleando anhídrido acético.
COOH + HNO3 (conc)H2SO4 (conc)
COOH
aO2N
Fe / HClCOOH
NH2
reducción halogenaciónCOOH
NH2
I
I
IICl
b c
COOH
NH2
I
I
IAc2O
COOH
NH
I
I
I
CCH3
O
Ácido Acetrizoicod
b) El grupo amino es fuertemente activante, por lo que la sustitución electrofílica está favorecida. La entrada del primer átomo de yodo en el anillo aromático aumenta la reactividad y las sucesivas sustituciones electrofílicas aromaticas de los átomos de yodo están cinéticamente favorecidas.

29 La adipiodona se emplea como medio de contraste radiológico. Propón una síntesis para la
adipiodona, partiendo del ácido 3-amino-2,4,6-triiodobenzoico.
COOH
NH
I I
I
COOH
HN
II
I
O
O
Adipiodona
SOLUCIÓN Dos moléculas del ácido 3-amino-2,4,6-triodobenzoico reaccionan con el dicloruro de
hexanodioilo obteniéndose la adipiodona.
COOH
NH2
I
I
Iformaciónde la amida
+ Cl ClO
O
COOH
NH
I I
I
COOH
HN
II
I
O
O
Adipiodona


















