8. conferencia urp eje snc hipotalamo hipofiso testicular
65
-
Upload
anchi-hsu-xd -
Category
Health & Medicine
-
view
1.606 -
download
3
Transcript of 8. conferencia urp eje snc hipotalamo hipofiso testicular



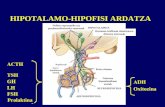
Sistema Nervioso Central
Los neurotransmisores del SNC actúan sobre el hipotálamo para regular la síntesis y secreción de la hormona liberadora de gonadotropinas (GnRH)
Estimulan:NorepinefrinaGABAAcetil Colina
Inhiben:SerotoninaDopaminaPéptidos opioides (endorfinas, encefalinas)

Hipotálamo
Sintetiza y secreta la GnRH
GnRH es un decapéptido
GnRH es transportada a la adenohipófisis por vía sanguínea porta-hipofisiaria
La secreción de GnRH es pulsátil y estimula la síntesis y secreción de (LH) y (FSH)
ESTIMULAN GnRHNorepinefrinaGABA
INHIBEN GnRHMelatoninaSerotoninaDopaminaPéptidos opioides

HIPOTALAMO CENTRO CICLICO: inactivado en la
vida fetalNúcleo supraquiasmáticoArea preóptica medial anterior
CENTRO TONICO: activado en la pubertadNúcleo arcuato
Núcleo ventro medial

HORMONAS HIPOTALAMICAS Péptidos de pequeño
tamaño con actividad fisiológica exclusiva en concentraciones elevadas observables en el sistema porta hipofisiario.
Las hormonas hipotalámicas se liberan de manera intermitente y las células blanco de la hipófisis anterior responden mejor a la administración intermitente de éstas hormonas que a una exposición continua.

HORMONAS HIPOTALAMICAS ESTIMULADORAS
Hormona liberadora de corticotropina (CRH)
Hormona liberadora de la hormona de crecimiento (GHRH)
Hormona liberadora de gonadotropina (GnRH ó LHRH)
Hormona liberadora de tirotropina (TSHRH)
Factores liberadores de prolactina (PRL)

Hormona liberadora de corticotropina (CRH)
La CRH es activadora de la secreción hipofisiaria de ACTH. Está constituida por 41 aminoácidos y su secreción procede
de neuronas hipotalámicas del núcleo paraventricular, núcleo supraóptico, núcleo arcuato y sistema límbico.
Actúa fijándose a receptores específicos de las células corticotropicas y solo estimula la liberación hormonal en presencia de calcio.
La concentración de AMPc aumenta paralelamente al efecto biológico.
El efecto estimulante de la CRH sobre la producción del AMPc es disminuido por los glucocorticoides.

Hormona liberadora de la hormona de crecimiento (GHRH)
La GHRH pertenece a una familia de moléculas entre las que se encuentran la secretina, el glucagón, el péptido intestinal vasoactivo (VIP) y el péptido gástrico inhibidor (GIP).
El núcleo arcuato del hipotálamo es el lugar principal de producción de GHRH.
Después de fijarse a la membrana de las células hipofisarias , estimula la secreción de GH por un mecanismo dependiente de calcio y activa a la adenilciclasa, con la acumulación del AMPc.
También activa el ciclo del fosfatidilinositol y ejercería una acción directa dentro de la célula mediante fosforilación de una enzima ligada al gránulo secretorio.
La GHRH aumenta también , la formación de nueva GH estimulando la trascripción del RNAm específico para GH, los efectos de la GHRH son bloqueados por la somatostatina y potenciados por los glucocorticoides.

Hormona liberadora de gonadotropina (GnRH ó LHRH)
La GnRH actúa sobre los receptores hipofisarios para estimular la producción y liberación de FSH y LH.
Se secreta principalmente por neuronas del área preóptica y está constituida por tan solo 10 aminoácidos.
La acción de la GnRH sobre la hipófisis se inicia con la fijación a receptores específicos de la superficie celular, y el proceso de liberación se activa mediante la movilización del calcio intracelular.
Los agonistas adrenérgicos facilitan la liberación de GnRH, mientras que los opiáceos endógenos la inhiben, los estrógenos aumentan la cantidad de receptores de GnRH y los andrógenos la reducen.

Hormona liberadora de tirotropina (TSHRH)
Es un tripéptido, se produce en el área hipotalámica anterior. La TSHRH estimula la secreción de TSH mediante el incremento del
calcio citoplasmático libre, probablemente el fosfatidilinositol y los fosfolípidos de membrana participan en la secreción de TSH mediada por la TSHRH y también estimula la liberación de prolactina.
Los efectos estimulantes de la TSHRH se inician con la fijación del péptido a los receptores específicos en la membrana plasmática de la célula hipofisaria.
La acción de la TSHRH se ejerce sobre la membrana y no depende de la internalización, aunque esta última tiene lugar.
Actúa mediante una hidrólisis calcio dependiente del fosfatidilinositol, con fosforilación de proteínquinasas para la activación posreceptor.
La TSHRH, estimula la formación de RNAm que codifica a la prolactina.

Factores liberadores de prolactina (PRL)
Los factores liberadores de PRL son neurotransmisores (serotonina, acetilcolina), sustancias opiáceas y estrógenos.
Otros factores estimulantes de la liberación de PRL en la especie humana son la TSHRH, péptido intestinal vasoactivo(VIP), la sustancia P, colecistoquinina, neurotensina, GHRH, GnRH, oxitocina, vasopresina y galanina.

HORMONAS HIPOTALAMICAS INHIBIDORAS
Factores inhibidores de PRL (dopamina)
Hormona inhibidora de GH (GHRIH o somatostatina)

HIPOFISIS
Forma esférica: 1.3 cm de longitud.
Peso: 600-700 mg
Adenohipófisis: Bolsa de Rathke
Neurohipófisis: Invaginación diencefálica


REGULACION E IRRIGACION
Regulación hipotalámica:Conexión vascularConexión nerviosa
Irrigación:Vena porta-hipofisiariaArteria hipofisiaria superiorFlujo retrógrado hipófiso-hipotalámico


Adenohipofisis Las hormonas que intervienen en la
regulación de la función testicular son:las gonadotropinas (LH y FSH) la prolactina
La síntesis y secreción de LH y FSH son estimuladas por la GnRH y reguladas por retroalimentación negativa por las hormonas testiculares (testosterona e inhibina)


ADENOHIPOFISIS La adenohipófisis deriva de la bolsa de Rathke y se localiza en la
silla turca.
Mediante distintas técnicas se lograron aislar diferente grupos celulares:
1. Células corticotrofas. Producen ACTH se sitúan en el centro.2. Células gonadotrofas. Sintetizan y segregan gonadotropinas,
activinas e inhibinas y se localizan en la pars distalis.3. Células lactotrofas: sintetizan y segregan PRL, se localizan en
la pars distalis. 4. Células melanotróficas: sintetizan y segregan hormonas
melanotropas (MSH) se localizan en la zona intermedia5. Células somatotrofas: sintetiza y segrega GH y se localiza en
las alas laterales.6. Células tirotrofas: sintetizan y segregan TSH su localización es
anteromedial.7. Células cromófobas: no secretoras, agranulares o nulas (Null
cells). No sintetizan hormonas.

Glándula Hormona Acción principal Mecanismo que controla su secreción
Tipo de molécula
Hormona de crecimiento (somatotropina)
Estimula el crecimiento del hueso, inhibe la oxidación de la glucosa, promueve la degradación de ácidos grasos
Hormona (s) hipotalámica (s)
Proteína
Prolactina Estimula la producción de leche
Hormona (s) hipotalámica (s)
Proteína
Hormona estimuladora de tiroides (TSH)
Estimula la glándula tiroides
Tiroxina en sangre; hormona (s) hipotalámica (s)
Glucoproteína
Hormona adrenocorticotrófica (ACTH)
Estimula la corteza suprarrenal
Cortisona en la sangre; hormona (s) hipotalámica (s)
Polipéptido (39 aminoácidos)
Hormona foliculoestimulante (FSH)*
Estimula al folículo ovárico, espermatogénesis
Estrógeno en la sangre; hormona (s) hipotalámica (s)
Glucoproteína
Hipófisis, lóbulo anterior
Hormona luteinizante (LH)
Estimula la ovulación y la formación del cuerpo lúteo en las hembras y las células intersticiales en el macho
Progesterona o testosterona en la sangre; hormona(s) hipotalámica (s)
Glucoproteína

HORMONAS ADENOHIPOFISIARIAS Células Acidófilas Células Basófilas Células Cromófobas

CELULAS ACIDOFILAS
Células secretoras de prolactina Células secretoras de GH

CELULAS ACIDOFILAS: PROLACTINA Tiene 198 aa PM: 22,500 (tres isoformas) Normal: 6-25 ng/ml (170-540 mU/l) Control inhibitorio tónico: Dopamina,
GABA PRF: VIP, PTH, 5-HT, TRH, Péptidos
opioides. PRLDa Pit 1: Factor de transcripción para la
expresión del gen de Prl.

PROLACTINA Hormona proteica
producida por las células eosinófilas de la adenohipófisis.
Potencia la acción de la LH en las células de Leydig aumentando la producción de testosterona (Up Regulation).
Su exceso (>25 ng/ml) produce inhibición de la respuesta de LH en Células de Leydig (Down Regulation)
ESTIMULAN PRLSerotoninaPTHVIPTRH
INHIBEN PRLDAGABA

CELULAS BASOFILAS
Células secretoras de LH Células secretoras de FSH Células secretoras de TSH Células productoras de pro-opio-
melanocortina.

LH Actúa sobre las células de Leydig del
testículo favoreciendo la producción y secreción de testosterona.
Estimula la síntesis de la proteína reguladora esteroidogénica aguda (biosíntesis de hormonas esteroideas)

FSH Actúa sobre las células de Sertoli,
favoreciendo la síntesis y secreción de:inhibina, proteína ligadora de andrógenos,activador de plasminógeno, transferrinalactato fluido testicular.
Estimulante del crecimiento de los tubulos seminiferos.

REGULACIONLa testosterona por retroalimentación
negativa inhibe a nivel hipofisiario la secreción de LH y a nivel de hipotálamo la de GnRH.
La inhibina inhibe la FSH a nivel hipofisiario.


TESTICULOS
Son dos en los cuales se producen los espermatozoides y la testosterona
Están situados fuera de la cavidad abdominal pélvica en una bolsa llamada el escroto.
FSH, favorece la produccion de espermatozoides
LH, las células de Leydig secretan los andrógenos.

TESTICULOS
Células intersticiales Células de Leydig Macrófagos Vasos sanguíneos y vasos linfáticos
Túbulos seminíferosCélulas mioidesCélulas de Sertoli Células germinales.

Células de Leydig Su función es la producción de
andrógenos bajo el estimulo de la LH. La T tiene función paracrina sobre las
células mioides, de Sertoli y células germinales,
La T tiene función endocrina sobre diferentes órganos (Masa muscular, eritropoyesis, etc).
Favorece la espermatogénesis, la movilidad de los espermatozoides, y la erección.

TESTOSTERONA Se equilibra
rápidamente entre sangre y órganos.
Valor en suero=saliva.
98% unido a proteína (SHBG, Albúmina)
95% producido en testículo
FavoreceEspermatogenesisEspermiaciónMovilidad de SPZ
Protege el metabolismo de esteroides en hígado

Células mioides Con C. Sertoli secretan componentes
de la matríz extracelular. Rodean a los túbulos seminíferos
(movimiento del fluído y de los SPZ ) Tienen receptores para testosterona Dos funciones:
Facilitar la contracción de los túbulos (Oxitocina, serotonina y prostaglandinas)
Función paracrina ante un estimulo androgénico actuando sobre las células de Sertoli.

Células de Sertoli Forman parte del epitelio seminífero Tiene receptores para FSH y
testosterona Es célula blanco para:
FSHTriyodotironinaInsulinaIGF 1 Testosterona.

Produce diversos compuestos como:InhibinaProteína ligadora de andrógenos(ABP)TransferrinaActivador de plasminógeno Lactato.
Las células de Sertoli se comunican entre si a través de uniones oclusivas, que son la base de la barrera hematotesticular.

HIPOGONADISMO (1) El hipogonadismo en el varón se define como la incapacidad
del testículo para realizar sus funciones y de acuerdo a la edad del sujeto, producir testosterona (T), espermatozoides o ambos.
La testosterona es una hormona que afecta a muchos tejidos del organismo.
Algunos de sus efectos, como la diferenciación sexual son tiempo -dependientes y no reversibles.
Otros, como sus acciones sobre la aparición y mantenimiento de los caracteres sexuales secundarios y proporciones corporales del varón, son parcialmente reversibles.
Otras acciones como su acción sobre el hueso, músculo, eritropoyesis, próstata, energía o función sexual son reversibles

HIPOGONADISMO (2) Clásicamente el hipogonadismo se divide en primario y
secundario, según la función testicular se afecte primariamente o de forma secundaria a una alteración hipotálamo-hipofisaria.
También se puede clasificar, de una forma más clínica, según el momento biológico en que se produce la insuficiencia testicular:
1) prenatal, durante la diferenciación sexual; 2) puberal, cuando el eje hipotálamo-hipófiso-testicular
debe madurar y producir el desarrollo puberal 3) edad adulta cuando el testículo asume la producción
definitiva de T y espermatozoides.

HIPOGONADISMO (3) Dependiendo de la edad e intensidad de la
alteración de la secreción de testosterona y producción espermática, la presentación clínica del hipogonadismo varía.
El diagnóstico se puede hacer en el periodo neonatal por genitales ambiguos, micropene, criptorquidia o hipospadias; en el adolescente, por pubertad retrasada; en el adulto, por problemas sexuales, regresión de caracteres sexuales o infertilidad.

HIPOGONADISMO (4) La incidencia del hipogonadismo es muy
variable según su etiología, pero es una enfermedad relativamente frecuente: el síndrome de Klinefelter oscila entre 1:400 y 1:1.000 recién nacidos vivos. El retraso constitucional del crecimiento y de la pubertad supone el 2,5 % de la población. Se han visto niveles de testosterona bajos en el 20 % de hombres con fractura de cadera, por lo que la frecuencia de formas adquiridas de hipogonadismo podría ser incluso más alta.

TABLA 1. Clasificación de hipogonadismos en el varón 1. Retraso constitucional del crecimiento y
pubertad (variante de la normalidad) 2. Hipogonadismo hipogonadotrópico
(lesión hipotálamo-hipofisaria) 3. Hipogonadismo hipergonadotrópico
(lesión testicular)

TABLA 2. – Causas de hipogonadismo hipogonadotrópico (1)1. CAUSAS ORGÁNICAS: LESIONES PRENATALES- Lesiones embriológicas del SNC: displasia septoóptica, anencefalia,
holoprosencefalia- Síndromes polimalformativos LESIONES ADQUIRIDAS
- Tumores intraselares o extraselares: craneofaringioma, astrocitoma, germinoma, glioma
- Irradiación craneal
- Hidrocefalia
- Enfermedades infiltrativas: histiocitosis, sarcoidosis, hemocromatosis
- Infecciones, alteraciones vasculares, traumatismos craneales, hipofisitis, síndrome de silla turca vacía

TABLA 2. – Causas de hipogonadismo hipogonadotrópico (2)
2. CAUSAS IDIOPÁTICAS: - Síndrome de Maestre de San Juan o de Kallmann - Alteraciones en el gen DAX-1 - Hipogonadismo hipogonadotrópico de origen en el adulto - Formas parciales de deficiencia de GnRH: eunuco fértil,
deficiencia aislada de FSH, retraso constitucional del crecimiento y pubertad
- Deficiencia genética en el receptor de GnRH - Deficiencia de FSH por alteración en el gen de subunidad
beta - Deficiencia de LH por alteración en el gen de subunidad
beta - Deficiencias congénitas de múltiples hormonas
hipofisarias

TABLA 2. – Causas de hipogonadismo hipogonadotrópico (3)3. CAUSAS FUNCIONALES: - Ayuno - Ejercicio físico - Enfermedades graves: sepsis, coma hepático,
estrés severo, etc. - Enfermedades crónicas: celiaquía, enfermedad
inflamatoria intestinal, hepatopatías, hemocromatosis, betatalasemia, síndrome de Cushing, obesidad, hipotiroidismo
- Medicamentos: andrógenos, estrógenos, progestagénos, productores de hiperprolactinemia.

HIPOGONADISMO HIPOGONADOTROFICO-LESIONES PRENATALES
Síndromes polimalformativos poco comunes. Síndrome de Prader-Willi: su prevalencia es de 1/10.000-1/50.000 recién
nacidos vivos. Está caracterizado por obesidad, talla baja, manos y pies pequeños, retraso mental, hipotonía en periodo neonatal, micropene y criptorquidia. Aunque puede ser esporádico, en el 95 % de los casos se encuentra una base genética, y en el 75% se encuentra una deleción de la región q11-13 del brazo largo del cromosoma15 de origen paterno, pudiendo encontrarse otras deleciones, duplicaciones y traslocaciones en este cromosoma. En el resto, mediante análisis molecular,se puede demostrar dos copias de la región q11-13 de origen materno (disomía materna uniparental) o diversas alteraciones en el ADN (10).
Síndrome de Laurence-Bardet-Moon-Biedl: se caracteriza por obesidad,
talla baja, polidactilia, retinitis pigmentosa, deficiencia mental e hipoplasia
genital por hipogonadismo-hipogonadotrópico. .

TABLA 3 – Síndromes poco frecuentes que cursan con hipogonadismoSíndrome y Características Lowe : Síndrome de Fanconi, cataratas, glaucoma, hipotonía, retraso mental Leopard : Léntigo generalizado, sordera neurosensorial, talla baja Rud: Ictiosis, retraso mental, retinitis pigmentosa Charge: Coloboma, cardiopatía, retraso de crecimiento y desarrollo Martsolf :Talla baja, retraso mental, raza judía Rothmun-Thompson : Alopecia, contracturas, poiquilodermia, talla baja, anemia,
cataratas Borjeson :Arcos supraorbitarios prominentes, ptosis, hipotonía, retraso mental Juberg: Retraso mental, fallo de crecimiento, ceguera y microngenitalismo Pollit : Tricodistrofia, ictiosis, talla baja, xeroderma-pigmentoso Lynch :Talla baja, retraso mental, anosmia, ictiosis Louis Bar: Telangiectasias, ataxia, inmunodeficiencia Johnson : Alopecia, anosmia, sordera, anomalías de pabellones auriculares Biemond : Coloboma, obesidad, retraso mental, polidactilia. Kraus-Ruppert :Microcefalia, retraso mental, sindactilia Richard-Rundle :Cetoacidosis, retraso mental, cifosis progresiva, y ataxia troncal Boucher-Neuhauser : Distrofia coriorretiniana, ataxia cerebelosa
.

Causas idiopáticas (1) Síndrome de Maestre de San Juan o de Kallman En 1856 el español Maestre de San Juan describió
la relación anatómica entre ausencia de nervios olfatorios y el hipogenitalismo. En 1944, Kallmann describió tres familias con esta enfermedad y dio su nombre al síndrome. Se caracteriza por la ausencia de neuronas GnRH asociado a anosmia. Es enfermedad genética probablemente autosómica dominante con varios genes autosómicos implicados. En algunos enfermos es debido a alteraciones del gen KAL localizado en Xp22.3,

Causas idiopáticas (2)
Alteraciones en el gen DAX-1. Localizado en Xp21.3-21.2, posiblemente se trata de un gen represor de la transcripción. La pérdida de su función da lugar a distintas alteraciones: hipogonadismo hipotalámico y en ocasiones
hipofisario, asociado a distintos grados de hipoplasia adrenal congénita

Causas idiopáticas (3) Hipogonadismo hipogonadotrópico de
inicio en el adulto.
Se trata de una deficiencia aislada de GnRH que se inicia en el adulto. La alteración neuroendocrina que conlleva a la ausencia de secreción de GnRH es permanente y de causa desconocida. Produce una regresión pospuberal de la libido y fertilidad en un varón que había desarrollado una pubertad normal

Causas idiopáticas (4) Formas parciales de deficiencia de GnRH. 1) Síndrome de eunuco fértil: Hay una deficiencia en la
secreción de LH, probablemente por una insuficiente secreción de GnRH.
2) Deficiencia aislada de FSH: Es una entidad rara en la que se ve alterada la espermatogénesis. A veces se asocia a criptorquidia, hipospadias, sordera, onfalocele, displasia olfatogenital o talla baja.
3) Retraso constitucional del crecimiento y pubertad: En la población general la incidencia de RCCP supónese 1-2,5 %. Algunos autores sugieren que un RCCP pese a que progrese a un desarrollo sexual normal podría considerarse la forma más benigna del amplio espectro clínico de la deficiencia de GnRH

Causas idiopáticas (5) Deficiencia de gonadotropinas por alteración
genética en el receptor de GnRH.
No se han encontrado hasta la fecha alteraciones genéticas en humanos del gen de GnRH, localizado en 8p21-8p11.2. Sin embargo, desde la clonación en 1994 del receptor de GnRH, localizado en cromosoma 4, se han descrito varios grupos familiares de con hipogonadismo hipogonadotrópico debido a diferentes alteraciones en este gen.

Causas idiopáticas (6) Deficiencia de FSH debida a alteraciones del gen de la subunidad
beta.
Se presenta como un paciente con hipogonadismo con valores indetectables de FSH, que no aumentan tras la administración de GnRH exógeno, valores bajos de inhibina y síntomas clínicos de falta de producción de andrógenos, probablemente porque la FSH desempeñe un papel más importante del que se piensa en la síntesis de andrógenos testiculares. Se debe a alteraciones genéticas (deleciones) del gen que codifica la subunidad beta de la FSH, con lo que se produce una proteína truncada sin acción biológica.
Deficiencia de LH debida a alteraciones en el gen de la subunidad beta.
Se ha comunicado un paciente con clínica de pubertad retrasada. Sus niveles de LH inmunorreactiva son elevados y bajos los de LH bioactiva. La LH mutante tiene una sustitución en Gln54 por Arg y una disminución en la actividad de ligar al receptor

Causas idiopáticas (7) Deficiencias congénitas de múltiples hormonas
hipofisarias.
Aunque la mayoría de casos de panhipopituitarismo idiopático congénito son esporádicas, se han descrito casos familiares con posible herencia autosómica recesiva (tipo I) y una forma ligada al cromosoma X (tipo II) . Hasta ahora se sabe que las mutaciones en el gen PROP 1 y en el gen LHX3 causan défícit de LH y FSH, además de defecto de GH, TSH y prolactina. Anomalías en el gen GH 1 cursan con micropene

TABLA 4. – Causas de hipogonadismo hipergonadotrópico (1)
ALTERACIONES GENÉTICAS EN EL RECEPTOR DE GONADOTROPINAS:
- Hipofunción del receptor de LH,
- Hipofunción del receptor de FSH,
- Síndrome de glicoproteínas deficientes en carbohidratos (varón que después de alcanzar una pubertad normal, experimenta una regresión del volumen testicular)
ALTERACIONES CROMOSÓMICAS:
- Síndrome de Klinefelter (47,XXY, 93%)
- Varón XX
- Síndrome de Down
-Sindrome de Noonan(46,XY, genitales externos masculinos y estigmas de síndrome de Turner. El volumen testicular está disminuido, la función de las células de Leydig alterada y es frecuente la criptorquidia y la esterilidad)
- Síndrome de cromosoma X frágil. Además de un fenotipo característico,
existe un macroorquidismo con ligera disminución de la espermatogénesis,
aunque las células de Sertoli y Leydig son normales. Se produce por una alteración en el cromosoma X (Xq,27fra).

TABLA 4. – Causas de hipogonadismo hipergonadotrópico (2) ALTERACIONES GENÉTICAS QUE CONLLEVAN
ALTERACIONES EN LA DIFERENCIACIÓN SEXUAL:
Cualquier estado intersexual en el varón genético se puede considerar una situación de hipogonadismo y, por ende, cualquier alteración en los mecanismos normales del desarrollo de las gónadas, genitales internos y externos indiferenciados hacia estructuras masculinas son causas de hipogonadismo. Cada vez se conocen más genes implicados en la determinación de sexo, como son: FTZ-FI, SRY, SOX 9, SF1,WT1, DMRT1-DMRT2, DAX1 AHCH. Se han descrito diferentes alteraciones en la diferenciación sexual, asociadas o no a hipoplasia suprarrenal y alteraciones somáticas

TABLA 4. – Causas de hipogonadismo hipergonadotrópico (3) DEFECTOS GENÉTICOS EN LA BIOSÍNTESIS DE ESTEROIDES
SEXUALES SUPRARRENALES Y TESTICULARES SÍNDROME DE RESISTENCIA TOTAL O PARCIAL A LOS
ANDRÓGENOS ANORQUIA CONGÉNITA O SÍNDROME DE TESTES
EVANESCENTES
Se define como la ausencia de testículos en un varón sin otras anomalías. Al acompañarse de un pene de configuración normal y ausencia de estructuras mullerianas, indica que ha existido una función testicular normal hasta la semana 13 de gestación. Si el pene es de tamaño normal, indica que incluso posteriormente ha habido una adecuada androgenización. Podría tratarse de atrofia testicular prenatal por compromiso vascular.
CRIPTORQUIDIA

TABLA 4. – Causas de hipogonadismo hipergonadotrópico (4)
ENFERMEDADES CRÓNICAS: ANEMIA FALCIFORME (por microinfartos testiculares), DISTROFIA MIOTÓNICA (degeneración de los túbulos seminíferos en el 75 % de los pacientes), FIBROSIS QUÍSTICA (Muchos pacientes con fibrosis quística tienen ausencia de conductos deferentes), INSUFICIENCIA RENAL (niveles altos sobre todo de LH, ya que su aclaramiento está muy disminuido; se puede asociar a hipeprolactinemia), LESIÓN MEDULAR (Ocasionalmente se encuentran niveles elevados de LH. La mayoría de los pacientes con lesión medular crónica presentan alteraciones en la eyaculación y en la calidad de semen, quizás por alteración en la termorregulación del testículo), ENFERMEDAD TIROIDEA AUTOINMUNE (niveles elevados de gonadotropinas y ginecomastia, aunque la función testicular es generalmente normal. Se ha sugerido una asociación entre la enfermedad tiroidea autoinmune y alteraciones autoinmunes de la espermatogénesis), DIABETES MELLITUS (niveles de T bajos durante la cetosis), NEOPLASIAS(Leucemias y Hodgkin pueden presentar disfunción testicular antes de iniciar el tratamiento, sobre todo si hay pérdida de peso.

TABLA 4. – Causas de hipogonadismo hipergonadotrópico (5) OTRAS ENFERMEDADES CRÓNICAS
En general, cualquier enfermedad crónica, como artritis reumatoide, enfermedad respiratoria crónica, enfermedad cardiovascular, etc., puede producir, por mecanismos no bien conocidos, niveles bajos de T y elevados de FSH y LH.
INFECCIONES:
- HIV (aparece un fallo testicular primario y más tarde un hipogonadismo hipogonadotrópico en los estados de pérdida de peso y enfermedad grave. Puede haber una lesión directamente testicular por el HIV, gérmenes oportunistas y tumores malignos).
- PAROTIDITIS (60 % de los pacientes puberales con orquitis por el virus de la parotiditis resulta infértil, niveles bajos de testosterona y elevados de gonadotropinas).
- LEPRA (Mycobacterium leprae afecta a las células de Leydig o a los túbulos seminíferos)

TABLA 4. – Causas de hipogonadismo hipergonadotrópico (6) IRRADIACIÓN TESTICULAR
Cuando el testículo recibe 15 rads disminuye la espermiogénesis; con 50 rads puede aparecer azoospermia reversible; con 400 rads la infertilidad es irreversible, y con 800 rads se afectan también las células de Leydig.
TRAUMATISMO Y TORSIÓN TESTICULAR
La torsión unilateral puede afectar al otro testículo, probablemente por un mecanismo autounmune.
MEDICAMENTOS (ver el cuadro siguiente…)

TABLA 5. – Medicamentos que causan hipogonadismo en el varón (1) MEDICAMENTO : Mecanismo de acción
ANDRÓGENOS, ANABOLIZANTES ESTEROIDEOS, PROGESTÁGENOS, GLUCOCORTICOIDES :Supresión de gonadotropinas + < SHBG
ESTRÓGENOS, HORMONAS TIROIDEAS, ANTIANDRÓGENOS Y ESPIRONOLACTONA : Supresión de gonadotropinas + > SHBG
INHIBIDORES DE 5-REDUCTASA (Finasteride en HPB): Modulación del receptor esteroideo
AGONISTAS Y ANTAGONISTAS DE GNRH: Supresión de gonadotropinas
BLOQUEANTES DE ESTEROIDOGÉNESIS(aminoglutetimida, ketoconazol): Lesión testicular primaria

TABLA 5. – Medicamentos que causan hipogonadismo en el varón (2)
MEDICAMENTO : Mecanismo de acción
BARBITÚRICOS, BENZODIAZEPINAS: Supresión de gonadotropinas + Hiperprolactinemia
ANTICONVULSIVANTES: PRIMIDONA, CARBAMACEPINA: >SHBG
FENITOÍNA :Supresión gonadotropinas + hiperprolactinemia + > SHBG
VALPROATO: Lesión testicular primaria
FENOTIAZINAS:Mecanismo neuroendocrino + hiperprolactinemia + supresión de gonadotropinas

TABLA 5. – Medicamentos que causan hipogonadismo en el varón (3)
MEDICAMENTO : Mecanismo de acción BUTIROFENONAS (haloperidol): Mecanismo neuroendocrino
ANTIDEPRESIVOS TRICÍCLICOS (Imiporamina y amitriptilina) E INHIBIDORES DE MONOAMINOOXIDASA(Moclobemida, Toloxantona): Hiperprolactinemia + mecanismo neuroendocrino
BLOQUEANTES DE CANALES DE CALCIO, INHIBIDORES DE ACE, BETA- BLOQUEANTES, METILDOPA, RESERPINA, GUANETIDINA, CLONIDINA, HIDRALAZINA, SIMPATICO-MIMÉTICOS, DIGOXINA, NITRATOS: Mecanismo neuroendocrino (+ algunos producen hiperprolactinemia)

TABLA 5. – Medicamentos que causan hipogonadismo en el varón (4)MEDICAMENTO : Mecanismo de acción CITOTÓXICOS:(ALQUILANTES Y ANTIMETABOLITOS): Lesión
testicular primaria
ANTIMICROBIANOS (SULFAPIRIDINA ,SULFASALAZINA, NITROFURANTOÍNA, METRONIDAZOL GRISEOFULVINA, RIFAMPICINA, DAPSONA): Lesión testicular primaria
HIPOLIPEMIANTES (INHIBIDORES DE LA HMG-CoA REDUCTASA, CLOFIBRATO, GEMFIBROZIL): Lesión testicular primaria
ANTIHISTAMÍNICOS: Mecanismo neuroendocrino

TABLA 5. – Medicamentos que causan hipogonadismo en el varón (5) ALCOHOL: Lesión testicular primaria + supresión de
gonadotropinas
MARIHUANA: Lesión testicular primaria + supresión de gonadotropinas + hiperprolactinemia