
CONTRIBUCIÓN AL ESTUDIO DE DIHIDROPIRIMIDINA...
76
CONTRIBUCIÓN AL ESTUDIO DE DIHIDROPIRIMIDINA DESHIDROGENASA (DHPD) DE LA RUTA CATALÍTICA DE PIRIMIDINAS EN ORYZA SATIVA L. Estudiantes Ángela Yazmín García Fonseca Wendy Johana Montero Ovalle UNIVERSIDAD DISTRITAL FRANCISCO JOSÉ DE CALDAS FACULTAD DE CIENCIAS Y EDUCACIÓN PROYECTO CURRICULAR DE LICENCIATURA EN QUÍMICA BOGOTÁ D.C. 2016
Transcript of CONTRIBUCIÓN AL ESTUDIO DE DIHIDROPIRIMIDINA...

CONTRIBUCIÓN AL ESTUDIO DE DIHIDROPIRIMIDINA DESHIDROGENASA (DHPD) DE LA RUTA CATALÍTICA DE PIRIMIDINAS EN ORYZA SATIVA L.
Estudiantes Ángela Yazmín García Fonseca Wendy Johana Montero Ovalle
UNIVERSIDAD DISTRITAL FRANCISCO JOSÉ DE CALDAS
FACULTAD DE CIENCIAS Y EDUCACIÓN PROYECTO CURRICULAR DE LICENCIATURA EN QUÍMICA
BOGOTÁ D.C. 2016

CONTRIBUCIÓN AL ESTUDIO DE DIHIDROPIRIMIDINA DESHIDROGENASA (DHPD) DE LA RUTA CATALÍTICA DE PIRIMIDINAS EN Oryza sativa L.
Ángela Yazmín García Fonseca
Wendy Montero Ovalle
Proyecto Trabajo de Grado para optar por el título de Licenciado en Química
Dirigido por:
Barbara H. Zimmermann, Ph.D
Universidad de los Andes
Codirigido por:
Berta Inés Delgado Fajardo
Universidad Distrital Francisco José de Caldas
UNIVERSIDAD DISTRITAL FRANCISCO JOSÉ DE CALDAS
FACULTAD DE CIENCIAS Y EDUCACIÓN
PROYECTO CURRICULAR DE LICENCIATURA EN QUÍMICA
BOGOTÁ D.C.
2016

Nota de aceptación:
______________________________
______________________________
______________________________
______________________________
______________________________
______________________________
_____________________________
Firma del presidente del jurado
_____________________________
Firma del jurado
_____________________________
Firma del jurado
_____________________________
Bogotá. D.C., __________ de_________ 2017

CONTENIDO
Contenido RESUMEN ...................................................................................................................................... 1
INTRODUCCIÓN ............................................................................................................................. 3
1. PROBLEMA DE INVESTIGACIÓN ............................................................................................ 5
1.1 Planteamiento del problema ........................................................................................ 5
1.2 Justificación ................................................................................................................... 6
2. OBJETIVOS ............................................................................................................................. 7
2.1 General .......................................................................................................................... 7
2.2 Específicos ..................................................................................................................... 7
3. MARCO TEÓRICO ................................................................................................................... 8
3.1 Antecedentes ................................................................................................................ 8
3.2 Oryza sativa L. ............................................................................................................. 11
3.3 Taxonomía y Anatomía de Oryza sativa L. .................................................................. 11
3.3.1 Órganos Vegetativos ........................................................................................... 12
3.3.2 Órganos Reproductivos ....................................................................................... 13
3.4 Características Genéticas del arroz ............................................................................. 14
3.5 Estrés Abiótico ............................................................................................................. 15
3.5.1 Salinidad .............................................................................................................. 16
3.5.2 Sequía .................................................................................................................. 17
3.6 Nucleótidos de pirimidina ........................................................................................... 18
3.7 Síntesis de Novo de pirimidinas .................................................................................. 19
3.8 Vía de reciclaje de pirimidinas .................................................................................... 21
3.9 Vía de degradación de pirimidinas .............................................................................. 21
3.9.1 Dihidropirimidina Deshidrogenasa ...................................................................... 23
3.9.2 Gluatamato Sintasa ............................................................................................. 24
3.10 Vectores de Clonación ................................................................................................. 28
3.10.1 Vector de Clonación pET .................................................................................... 29
3.10.2 Vector de Clonación pGEM-T EASY .................................................................... 29
4. METODOLOGÍA .................................................................................................................... 30
4.1 Descripción del área de estudio .................................................................................. 30
4.2 Procedimientos ........................................................................................................... 30

4.2.1 Búsqueda de las secuencias en las bases de datos ............................................. 30
4.2.2 Diseño de primers ............................................................................................... 30
4.2.3 Extracción de ARN ............................................................................................... 31
4.2.4 Digestión con DNAsa ........................................................................................... 31
4.2.5 Síntesis de ADNc .................................................................................................. 32
4.2.6 PCR ...................................................................................................................... 32
4.2.7 Preparación del vector para subclonación .......................................................... 33
4.2.8 Preparación del inserto para subclonación ......................................................... 35
4.2.9 Ligación y transformación del inserto con pET19b ............................................. 35
4.2.10 Mini preparación del Vector recombinante y restricción enzimática ................. 36
5. Congelados .......................................................................................................................... 37
6. RESULTADOS ....................................................................................................................... 38
6.1 Búsqueda de las secuencias en las bases de datos ..................................................... 38
6.2 Diseño de primers ....................................................................................................... 39
6.3 Extracción de ARN ....................................................................................................... 42
6.4 Síntesis de ADNc y PCR ................................................................................................ 44
6.5 Restricción Enzimática ................................................................................................. 46
6.6 Clonación en pET 19b .................................................................................................. 48
6.7 Análisis de secuencia ................................................................................................... 51
7. DISCUSIÓN ........................................................................................................................... 57
8. CONCLUSIONES ................................................................................................................... 61
9. ANEXOS ............................................................................................................................... 62
10. REFERENCIAS BIBLIOGRÁFICAS ....................................................................................... 66

1
RESUMEN
El arroz es un alimento esencial dentro de la canasta familiar lo que lo hace uno de los
cereales más consumidos, pese a esto, en los últimos años se han evidenciado
cambios climáticos severos como altas temperaturas y continuas precipitaciones que
afectan el crecimiento, reproducción y ciclos vitales de las plantas dado por la
disminución en la captación de nutrientes a través del suelo (Peñuelas, 2004), lo cual
conlleva a que estos factores abióticos representen una gran problemática, ya sea en
la estructura de la planta o en las diferentes rutas metabólicas ocasionando una
expresión muy alta o muy baja de ciertos genes regulados implicados en la tolerancia
frente al estrés abiótico como mecanismo de defensa en la planta de arroz (Soren,
2010). Una de las rutas presentes en la tolerancia frente a factores abióticos como
sequía y salinidad podría ser la ruta de pirimidinas, ya que, se ha reportado una sobre
expresión frente a este tipo de estrés en la primera enzima de degradación de
pirimidinas la Dihidropirimidina Deshidrogenasa (DHPD) (Liu et al. 2009). La proteína
se ha estudiado en hígado de cerdo demostrando que está conformada por 5 dominios
siendo uno de ellos una subunidad donde FMN y NADPH se acoplan (Dobritzsch, et
al. 2002), y que en plantas se desconoce, por lo que la DHPD tiene reportada tan solo
la mitad del tamaño en comparación a la DHPD de mamíferos, faltando la subunidad
en la que se ensambla NADPH y se da el transporte de electrones desde este al
sustrato (Zrenner et al., 2006). Esto sugiere que en plantas se requiere de una
interacción entre DHPD y la proteína que se encargue de la transferencia de
electrones para un óptimo funcionamiento en la vía de degradación de Uracilo o
Timina; sin embargo, esto no ha sido reportado para ninguna especie vegetal. Un
posible candidato del fragmento carente es la β-subunidad de Nicotinamida Adenina
Dinucleótido Fosfato-Glutamato Sintasa (NADPH-GltS) por su homología reportada en
bancos de secuencias con el segmento faltante de DHPD (Vanoni et al. 1999). A partir
de esto, el trabajo realizado tuvo como fin obtener los genes que posiblemente hacen
parte de la subunidad complementaria al dominio de la proteína DHPD de la ruta de
degradación de pirimidinas en Oryza sativa L. y así contribuir a futuros estudios con
esta enzima y su importancia en la tolerancia frente al estrés abiótico ocasionado por
salinidad y altas sequias que trae consigo los problemas climáticos que actualmente
se presentan. Para esto, se clonaron las dos isoformas de la β-subunidad de NADH-
GltS en el vector de expresión pET19b para futuros ensayos de complementación
entre las Isoformas de la β-subunidad de NADH-GltS y la DHPD. La clonación se

2
realizó mediante la extracción de ARN en plantas de arroz y posteriormente de obtuvo
ANDc, desde este se diseñaron primers que funcionen como templado para la
amplificación de las secuencias de las dos isoformas de la β-subunidad de NADH-
GltS. La β-subunidad de NADH-GltS 1 se clonó en vector de expresión pET-19b
utilizando tanto para la secuencia de NADH-GltS 1 como para el vector la enzima de
restricción XhoI. Los fragmentos fueron purificados con QIAquick Gel Extraction Kit de
QIGEN, para posterior ligación y transformación en células quimicompetentes DH5α.
En el caso de la β-subunidad de NADH-GltS 2 se realizó el mismo procedimiento pero
cambiando las enzimas de restricción, empleando XhoI y BamHI para la secuencia y el
vector pET-19b. Finalmente se obtuvo la clonación de la β-subunidad de NADH-GltS 1
corroborándose mediante PCR, restricción enzimática y secuenciación de los clones.
Sin embargo, la clonación de la β-subunidad de NADH-GltS 2 no se consiguió debido
a la similitud entre las isoformas planteando posibles estrategias a partir de los primers
para próximos estudios. Por último, se dejaron creciendo las bacterias transformadas
de la isoforma 1 en medio LB para realizar congelados de las mismas con la finalidad
de preservar las secuencias de interés.
Como conclusión de esta investigación se obtuvieron células transformadas con la β-
subunidad de NADH-GltS 1, estas contribuirán a estudios de interacción entre el
posible dominio transferente de electrones y la enzima DHPD correspondiente a la
primera ruta de degradación de pirimidinas en Oryza sativa L, proporcionando
herramientas para investigaciones de la ruta y la expresión de la enzima al ser
sometida a factores de estrés abiótico como sequía y salinidad.

3
INTRODUCCIÓN
La planta de arroz (Oryza sativa L.) es considerada como uno de los cultivos más
antiguos en el mundo y hace parte del alimento principal en 17 países
aproximadamente por su importancia como fuente primordial de suministro de energía
alimenticia. Este cereal proporciona diferentes tipos de aminoácidos como ácido
glutámico y aspártico, vitaminas como riboflavina, tiamina, fibra en el caso del arroz
integral entre otros compuesto esenciales para el ser humano (FAO, 2004).La planta
Oryza Sativa L. representa una fuente económica de gran valor gracias al aumento
significativo en su demanda y por ende en su producción, por ejemplo, en América
latina, la planta de arroz aumentó su rendimiento durante los años de 1975 y 2005 y ya
para 2008 se producen más de 25 millones de toneladas de arroz con cascara según
datos registrados por el Instituto Internacional de investigación sobre el Arroz
(Degiovanni, 2010). En Colombia específicamente el arroz es uno de los cultivos de
ciclos cortos de mayor trascendencia siendo el tercer alimento más importante
después del café y el maíz, constituyendo el 12% del área cultivada en Colombia y en
su producción el 10% de la actividad agrícola en este país (Martinez et al. 2004).
A partir de los datos reportados se demuestra la relevancia y el alcance que tiene el
cultivo de arroz en el mundo, no obstante, las plantas de este cereal son unas de las
más abatidas por las variaciones climáticas drásticas que perjudica de una u otra
manera su metabolismo y por consiguiente su desarrollo y cosecha. Los motivos
primordiales que alteran el ciclo de vida de este cereal son la sequía y la salinidad
(Singh et al. 2008) y como respuesta a esto, se genera una sobre expresión de ciertas
proteínas que están implicadas en la regulación de la expresión génica que originan
una cascada de señalización útil para aminorar el estrés ocasionado por los problemas
ambientales (Soren et al. 2010). Por ello, es de gran importancia realizar análisis
minuciosos que conlleven a soportar estas problemáticas que desfavorecen el
rendimiento de la agricultura. Para este caso, el trabajo se ha enfocado en la ruta de
degradación de pirimidinas, ya que, previos experimentos han sugerido que en la
tolerancia al estrés abiótico, la ruta de pirimidinas juega un papel importante, las
investigaciones apoyan la relevancia de la DHPD, primera enzima que actúa en la ruta
de degradación de pirimidinas en el crecimiento y desarrollo de las plantas, además se
ha reportado que su ausencia retarda la germinación y afecta la evolución en plantas
como Arabidopsis (Cornelius et al. 2011) Otro estudio revela, que la exposición a
salinidad y sequía perturba en gran medida la planta de arroz, y generan una sobre
expresión de la DHPD indicando que está relacionada con la tolerancia a estos

4
estreses abióticos (Liu et al. 2009). Sin embargo, la secuencia de la proteína de la
investigación, fue identificada como dihidroorato deshidrogenasa tipo 1 (DHOD)
(perteneciente a la ruta anabólica de pirimidinas), a pesar de esto, en el laboratorio de
la Dr. Zimmermann se desarrollaron técnicas moleculares con el fin de comparar y
comprender estas dos enzimas (DHPD y DHOD), concluyendo que en la publicación la
proteína que realmente se analizó fue la DHPD mencionada erróneamente como
DHOD.
Por lo tanto y a partir de los estudios anteriormente mencionados, la ruta de
degradación de pirimidinas en plantas de arroz se ha convertido en centro de interés
de este proyecto y más exactamente la primera enzima de la ruta DHPD. Se ha
demostrado que en cerdo la estructura de DHPD es bastante compleja conformada
por 5 dominios, los cuales tienen sitios de unión a cofactores como clusters [4Fe-4S] ,
Flavín Adenin Dinucleótido (FAD) y Flavín Mononucleótido (FMN), que favorecen el
transporte de electrones desde Nicotinamina Adenina Dinucleótido Fosfato (NADPH)
hasta el sustrato (Dobritzsch, et al. 2002). En toda especie vegetal es desconocido
aquel dominio encargado de la transferencia de electrones que permite su óptimo
funcionamiento, es decir, que la DHPD tiene reportada tan solo una parte del tamaño
de todo el complejo enzimático en comparación a la DHPD de mamíferos.(Zrenner et
al., 2006) El objetivo de la investigación, fue la obtención de los candidatos que
posiblemente cumplen esta función para futuros estudios de esta enzima y de la ruta
como tal, ya que se cree que esta vía puede llegar a contribuir en la solución efectiva
frente a los problemas ambientales que se observan actualmente siendo una vía
metabólica que favorezca la tolerancia al estrés ambiental beneficiando la cosecha y
conservación de este cereal fuente principal de consumo en Colombia y en el mundo.

5
1. PROBLEMA DE INVESTIGACIÓN
1.1 Planteamiento del problema
Los factores ambientales alteran de diferentes formas el ecosistema en el que se
desarrolla el reino vegetal. La salinidad y la sequía son unas de las razones
importantes que hoy en día debilita la siembra y progreso del cultivo de arroz a nivel
mundial (Fernández, 2010) y aunque no se tienen registros exactos del área afectada
por estos factores se sabe que deterioran en gran medida los suelos para agricultura.
El estrés salino ocasiona cambios fisiológicos y bioquímicos en el metabolismos de
plantas como Oryza sativa L. (A. Lamz et al. 2013), mientras que la sequía ocasiona
una pérdida total en el rendimiento de productividad, debido a que reduce el
crecimiento, desarrollo y floración (R. Serraj et al. 2009). Es por esto que actualmente
es de suma relevancia entender cómo actúa y se defiende el cultivo frente a este tipo
de estrés abiótico de tal manera que logra hacerse más tolerante a los cambios
climáticos. Uno de los ejes de estas investigaciones para sobrellevar la problemática
ambiental radica precisamente en aquellos cambios bioquímicos y genéticos que
emplea la planta para ser más resistente, como es el caso de la regulación genética, o
productos bioactivos que potencializan los diferentes mecanismos utilizados por la
planta (Reyes et al. 2008).
Para este proyecto se investigó la primera enzima de la ruta de degradación de
pirimidinas, es decir la DHPD, ya que se cree que es una de las vías que puede ser
alterada como mecanismo de defensa al estrés abiótico por sequía y salinidad
(Stasolla et al. 2003, Liu et al. 2009). La vía de Pirimidinas participa en muchos
procesos metabólicos de las plantas como lo es la síntesis de ácidos nucleicos,
síntesis de azucares como sacarosa, polisacáridos empleados en la construcción de la
pared celular de las plantas, síntesis de lípidos y fosfolípidos, demostrando que esta
ruta es de vital trascendencia en el desarrollo y crecimiento óptimo de las plantas
(Zrenner et al. 2009, Stasolla et al. 2003) y que por esto mismo puede ser perjudicada
por factores ambientales los cuales, como se ha descrito anteriormente desfavorecen
la productividad de la cosecha. Por tanto, en el actual trabajo se quiere conocer en
primer lugar el complejo enzimático de la DHPD, más precisamente el dominio que se
encarga de la transferencia de electrones para su óptimo funcionamiento, el cual se
desconoce en plantas, esto con el fin de contribuir en próximos estudios al
entendimiento de la estructura y función de este complejo proteico y por ende lograr
desarrollar en laboratorio experimentos con la proteína y su relación con el estrés
abiótico.

6
1.2 Justificación
El cereal de arroz es un cultivo que requiere de ciertos factores importantes si se
desea proveer una cosecha; uno de estos es un buen suministro de agua y calidad en
los suelos y a consecuencia de los diferentes problemas ambientales, continentes
como África y Asia actualmente no pueden suplir la demanda de este cultivo,
conociendo de ante mano que este último continente tiene el 58% de la población
mundial que consume arroz (SAG, 2003), Por otro lado, la FAO pronostica que en los
próximos 35 años se tendrá que cosechar hasta 3000 millones de toneladas de arroz
pero con menores recursos de agua, suelos y productos agroquímicos ocasionado por
los fenómenos meteorológicos drásticos dados por el cambio climático (Unidas, 2014).
Esta investigación se realizó por la necesidad de buscar soluciones que aminoren las
consecuencias y efectos que han traído los cambios climáticos tan drásticos a nivel
mundial para el cultivo de arroz, fuente primordial de alimento de más de un tercio de
la población en el mundo. La sequía y alta concentración de sales en los suelos
perturba cada fase de desarrollo de la cosecha de este cereal, específicamente la
etapa de la plántula, la cual es el momento donde más sensible esta al estrés por
salinidad (Reyes et al. 2008) y que a causa de esto trae grandes pérdidas tanto de una
fuente principal de alimento como gran perjuicio económico para aquellos países que
viven en gran medida de la producción de este cereal.
Este trabajo también se llevó a cabo porque se considera que el estudio de la probable
subunidad complementaria de la primera enzima de degradación DHPD, contribuirá en
al análisis conformacional y funcional de esta proteína. Las isoformas de la β-
subunidad de NADH-GltS son posibles candidatos de la subunidad carente, por lo
tanto, la obtención de clones de esta proteína permitirá caracterizar este segmento
mediante secuenciación y a su vez hacer posibles pruebas de interacción proteína-
proteína y de esta forma identificar si es aquel dominio que hoy se desconoce de este
complejo proteico en la especie vegetal.
Asimismo, esta investigación facilitará los futuros experimentos relacionados con el
papel que tiene la vía de pirimidinas y la enzima DHPD en los problemas de estrés
abiótico y así proveer soluciones frente a la tolerancia a la sequía y salinidad en Oryza
sativa L. de tal manera, que supla la demanda que en próximos años tendrá este
cereal según los reporte de la FAO.

7
2. OBJETIVOS
2.1 General
Obtener los diferentes genes de interés que posiblemente hacen parte de la subunidad
complementaria al dominio de la enzima DHPD de la ruta de degradación de
pirimidinas en Oryza sativa L.
2.2 Específicos
Clonar la β-subunidad de las isoformas de Glutamato Sintasa [NADH] en un
vector de expresión para futuros ensayos de interacción proteína-proteína con
DHPD.
Identificar la secuencia codificante para la β-subunidad de las isoformas de
Glutamato Sintasa [NADH] a partir de ADNc generado de ARN de plantas de
arroz.
Contribuir al estudio de la vía de degradación de pirimidinas en Oryza sativa L.
para posteriores investigaciones relacionadas con el estrés abiótico.

8
3. MARCO TEÓRICO
3.1 Antecedentes
Los cambios climáticos drásticos que se han dado en los últimos años han provocado
impactos severos en los cultivos y escenarios agricultores que hacen parte de un gran
porcentaje de consumo a nivel mundial. El aumento de temperatura y el cambio en las
precipitaciones tienen efectos directos sobre el rendimiento de los cultivos, ya sea
limitando el aporte de nutrientes, minerales y carbohidratos a estos o causando la
represión en el desarrollo vegetativo, generando así estreses abióticos en las plantas
(Nelson et al. 2009). Otros factores que influyen en el desarrollo adecuado de los
cultivos además de los mencionados anteriormente, incluyen la humedad, fuertes
vientos, la salinidad y la sequía, siendo estos dos últimos de los más alarmantes en la
actualidad (Hidalgo, 2005).
La salinidad ha sido considerada como uno de los inconvenientes más grandes
causantes de pérdidas de diversos cultivos, según la Organización mundial de la
salud, de 230 millones de hectáreas que se encuentran disponible para cultivos 45
millones son afectados por altas concentraciones de sal provocando la pérdida de 1.5
millones de hectáreas de cultivos (Bronwyn et al. 2007). Una alta concentración de
sales en el suelo genera un cambio en la homeostasis dado por la acumulación
excesiva de iones como lo es el Cl— y el Na+ conllevando a su vez a cambios
metabólicos donde se ven afectadas todas las reacciones de síntesis evidenciándose
en el bajo crecimiento de la planta. Las sales hacen parte del estrés por factores
ambientales que generan toxicidad en las plantas, ocasionado por el alto depósito de
Na+ en los tejidos, no obstante, la habilidad para responder a este sometimiento de
sales determina la capacidad de resistencia y tolerancia que una planta puede tener
frente a estas problemáticas (Morales et al. 2006).
La Sequía ha originado un 82% de las pérdidas de producción agrícola en el mundo
(FAO, 2015). Este factor de estrés es producido por un déficit del potencial hídrico en
la planta, lo cual afecta el alargamiento y el tamaño final de las hojas, la senescencia
foliar y la pérdida de follaje, todo esto se ve reflejado en la intercepción de la energía
solar provocando limitaciones en el proceso de fotosíntesis y por ende el crecimiento
óptimo vegetal (Ritchie et al. 2000).

9
Uno de los cultivos más importantes a nivel mundial al ser fuente primordial de
suministro de energía y principal alimento en 17 países es el de Oryza sativa L. (FAO,
2004), y su cosecha se está viendo limitada a causa de la sequía y salinidad y debido
a que estos inciden negativamente en la productividad de arroz, se ha visto la
necesidad de crear soluciones que aminoren estas problemáticas. En el 2005 García y
colaboradores emplearon el uso de biorreguladores en Oryza Sativa L. con el fin de
generar una mejor respuesta y mayor tolerancia al déficit hídrico, para esto ellos
sometieron 160 semillas de Oryza Sativa L en un medio ausente de agua y
emplearon un homólogo de brasinoesteroides conocido como MH5 un tipo de
esteroides que favorecen el crecimiento y desarrollo en las plantas. Se encontró que el
MH5 posee grandes beneficios en concentraciones promedio de 0,01mg/L para
variedades susceptibles, ayudando a la planta en la respuesta ante el sometimiento
frente al estrés por sequía y determinando que este compuesto tiene un gran potencial
antiestres por carencia de agua.(Aymara García, et al. 2005) Por otro lado, estudios
frente a los altos índices de salinidad, sugieren que los efectos en plantas de arroz son
definitivos y en segundo lugar que este daño está muy relacionado con el estrés por
déficit hídrico, como lo demuestra Morales en el 2006, en donde se expusieron las
semillas de arroz a diferentes concentraciones de salinidad encontrando que hay una
relación directamente proporcional entre la concentración de sal y la perdida de agua
que se observa por la acumulación de materia seca, de igual forma, el crecimiento de
la planta disminuye aproximadamente a la mitad en altas concentraciones de sal en
comparación a las plantas cultivadas con condiciones normales de Na+, por otro lado,
también se encontró que pasado el periodo de estrés y sometidas a una fase de
control las plantas no se recuperaron de los daños, manifestando que los efectos
negativos ocasionados por la salinidad son irreversibles, aunque en la tapa de control
si se observó un incremento en la cantidad de agua en diferentes partes de la planta,
concluyendo que los efectos por salinidad restringe la absorción de agua a través de
las raíces generando a su vez estrés por déficit hídrico como modificaciones en el
crecimiento de la planta, además de verse la planta obligada a desarrollar un
mecanismo de ajuste osmótico (Morales et al. 2006).
Para entender mejor cómo funciona el proceso de respuesta a diferentes condiciones
de estrés abiótico se han analizado procesos celulares y estructurales de las plantas
que influyen en dicha tolerancia, algunos de los cuales involucran los compuestos
nitrogenados, que son parte fundamental de todo ser vivo. Las purinas y pirimidinas
están involucrados en diferentes proceso metabólicos primarios o secundarios que
favorecen el crecimiento y desarrollo de plantas; son precursores de los ácidos

10
nucleicos, participan en la síntesis de polisacáridos, fosfolípidos, glicolípidos, además
son precursores de vitaminas como riboflavina tiamina entre otros, pese a esto, pocos
estudios se han llevado a cabo en plantas en comparación con otros organismos
(Stasolla et al. 2003).
Las pirimidinas como las purinas poseen diferentes vías metabólicas como lo es la
síntesis de novo, catabolismo y salvamento de las pirimidinas y purinas entre otras
(Zrenner, et al. 2006). La vía catalítica de pirimidinas, centro de este estudio, se
encarga de la degradación de las bases nitrogenadas Timina y Uracilo mediante tres
reacciones en donde hasta la fecha se ha demostrado que en plantas superiores la vía
es regulada por tres enzimas (Stasolla et al. 2003). La DHPD primera enzima de la
ruta catalítica nombrada erróneamente como DHOD en el estudio realizado por Liu y
colaboradores en el 2009, es una proteína que se ha visto involucrada como
mecanismo de defensa frente al estrés abiótico en plantas de arroz, encontrando que
esta enzima sufre una sobre expresión al verse sometida a altos índices de salinidad o
sequía con el fin de mejorar las condiciones de resistencia en la planta, además, se
investigó la función de la proteína in vivo en células recombinantes de E. Colli
observando niveles altamente significativos de tolerancia bajo condiciones de salinidad
y tensión osmótica.
Bajo los estudios realizados sobre DHPD en diferentes especies, se ha caracterizado
la enzima principalmente en mamíferos, como en el caso de S. scrofa, en donde se
planteó que la estructura se divide en 5 dominios, los cuales presentan sitios de unión
a cofactores como Flavín Mononucleótido (FMN), Flavín adenin dinucleótido (FAD) y
clusters [4Fe-4S], que facilitan la transición de electrones al sustrato a partir de
Nicotinamina adenina dinucleótido fosfato (NADPH) (Dobritzsch et al. 2002), sin
embargo, en plantas se desconoce el dominio N-terminal que contiene la unión a
NADPH y que por tanto permite la transferencia de electrones, los cuales necesita la
enzima para la reducción de uracilo (Zrenner et al., 2006). A pesar de esto se han
sugerido posibles candidatos que cumplirían la función del transporte de electrones a
partir de estudios realizados en diferentes especies, como es el caso de la β
subunidad de GltS, la cual se alinea muy bien con la región N-terminal de DHPD de S.
scrofa (Rosenbaum et al. 1998). Otras especies como Pyrococcus y A. aeolicus
también presentan alineamientos con el N-terminal de DHPD (Vanoni et al. 1999).
Asimismo se ha mencionado la hipótesis de que una parte de la secuencia de A.
aeolicus codifica un Fd-GltS (Deckert et al. 1998), y que la β subunidad de dicha GltS

11
hace parte de una familia de NAD(P)H oxidoreductasas que requiere de clusters Fe/S
y FAD y se encuentra presente en la especie como dominio cedente de electrones que
podría ser parte complementaria de la enzima DHPD. Del mismo modo, estudios
relacionados proponen similitud entre las secuencias de la β subunidad de GltS de
Synechocystis sp., P. boryanum y A. aeolicus, por lo que posiblemente requieran de
esta subunidad para el intercambio electrónico en el complejo catalítico de DHPD
(Temple et al. 1998).
3.2 Oryza sativa L.
El arroz se clasifica tradicionalmente en dos subespecies principales, Indica y
Japónica, que difieren en su adaptación a diferentes condiciones climáticas,
geográficas y culturales (Zhao et al. 2010). Este cereal es cosechado desde hace
milenios según registros y el continente asiático fue el lugar donde tuvo sus inicios la
planta de Oryza sativa L (SAG, 2003). Sus primeras semillas se cultivaron en China
hace 5000 años a.c, luego en Tailandia 4500 a.c y finalmente paso a Vietnam,
Camboya y al sur de la India. Posterior a esto y alrededor de los 800 a.c la cosecha de
este cereal se extendió por Europa Occidental, mientras que por el continente Africano
nació la especie Oryza glaberrima desde Niger hasta Senegal entre los años 1500 y
800 a.c, no obstante, esta nunca creció en otro lugar diferente a este continente, caso
contrario de Oryza sativa L, el cual si expandió su cultivó hacia el continente Africano
gracias a los Arabes (Ibarguren, 2015).
Inicialmente Oryza sativa L era una planta que crecía únicamente en climas templados
es decir, en tierras de secano sin riego pues tenían una mejor disposición de luz, sin
embargo, hoy en día con las mejoras en el cultivo se ha evidenciado que el cereal
crece en unas temperaturas que oscilan entre 20 y 30 °C y fuera este rango retardan
la maduración del grano (Nakayama, 1974). Esta planta es de climas cálidos y
húmedos con excelentes rendimientos (SAG, 2003) ya que, bajo estas condiciones el
suelo cuenta con una buena disposición de agua permitiendo modificaciones
saludables en la riqueza nutritiva de este, lo cual brinda características físicas y
químicas de gran importancia para la germinación de este cereal (Degiovanni et al.
2010).
3.3 Taxonomía y Anatomía de Oryza sativa L.
A continuación se ilustra un cuadro con las especificaciones de la taxonomía de la
planta de arroz Oryza sativa L.

12
Tabla 1 Taxonomía de Oryza sativa L (Tascón & Garcfa, 1985).
Oryza sativa L. fanerogama
Tipo Espermatofita
Subtipo Angiosperma
Clase Monocotiledonea
Orden Glumiflora
Familia Graminea
Subfamilia Panicoideas
Tribu Oryzae
Subtribu Oryzineas
Genero
Oryza (Angladette,
Gonzále!, Porter)
3.3.1 Órganos Vegetativos
La planta de arroz se caracteriza por tener dos arquetipos de raíz una conocida como
seminales o temporales (figura 1) y el otro denominado adventicias o permanentes
(figura 2). Las raíces temporales están presentes en la primera etapa de germinación
las cuales se caracterizan por ser poco ramificadas y un poco gruesas y a medida que
la planta va creciendo estas se van alargando, siendo más delgadas y ramificadas
pasando así a ser las raíces permanentes (CIAT, 2005).
Figura 1. Imagen de Raíces seminales de Oryza sativa L., caracterizadas por ser más gruesas y poco ramificadas (CIAT, 2005).
Figura 2. Imagen de Raíces adventicias de Oryza sativa L., las cuales son más delgadas y ramificadas (CIAT, 2005).

13
El tallo está estructurado por nudos y entrenudos (figura 3) los cuales le proporcionan
la altura al tallo, pues entre mayor sea el número de entrenudos mayor será su
longitud y esta cantidad de entrenudos a su vez son dependientes de factores
ambientales.
Figura 3. Imagen con estructura del tallo evidenciándose los nudos y entrenudos de Planta de Oryza sativa L (CIAT, 2005) .
Las hojas de la planta presentan vello y pigmentación antociana; crecen de manera
alterna en cada nudo a lo largo del tallo; la hoja superior debajo de la panícula se
denomina bandera (figura 4). Las hojas están formadas por la vaina, la cual envuelve
el entrenudo; el cuello que se prolonga de la vaina está conformado por la lígula y
aurículas (figura 5), las cuales son características propias de esta planta permitiendo la
identificación entre malezas; por último la lámina, cuya forma alargada y angosta
depende tanto de las variedades como de las condiciones ambientales (CIAT, 2005).
Figura 4. Imagen de la hoja bandera de la planta Figura 5. Imagen de la lígula y Aurícula del cuello Oryza sativa L y al costado izquierdo la panícula (CIAT, 2005) de una hoja de la planta Oryza sativa L (CIAT, 2005)
3.3.2 Órganos Reproductivos
Las flores se encuentran agrupadas en una inflorescencia denominada panícula que
está situada sobre el nudo apical del tallo. Las panículas pueden clasificarse en
abiertas, compactas e intermedias dependiendo de la variedad y estas generan
ramificaciones secundarias de donde brotan las espiguillas (figura 6), las cuales, son la
unidad básica de la inflorescencia y se conforman de tres flores, pero solo una se

14
desarrolla. La flor consta de seis estambres y un pistilo. Los estambres mantienen las
anteras que a su vez contienen los granos de polen. En el pistilo se distinguen el
ovario, el estilo y el estigma.
Una parte fundamental de la planta de arroz es la semilla siendo un ovario maduro y
seco el cual, consta de una cáscara formada por la lemma y la palea, el embrión y el
endospermo, que provee alimento al embrión durante la germinación (CIAT, 2005).
Figura 6. Estructura de una espiguilla de Planta de Oryza sativa L Una espiguilla se caracteriza por tener dos lemmas esteriles, la raquilla y la florecilla, Las lemmas estériles envuelven la flor por debajo de la raquilla. La raquilla es el eje que sostiene la flor (CIAT, 2005).
El ciclo general de la planta consta de tres etapas, I) el período vegetativo en donde se
da la germinación de la semilla y la aparición del primordio floral en la base de la
planta; II) el período reproductivo, el cual inicia con la aparición de los primordios
florales hasta la apertura de la flor, que debido a su sistema de reproducción
autógamo generalmente ya está polinizada al abrir; y finalmentel III) el período de
maduración, que es aquel en el que se evidencia el llenado de granos, teniendo en
cuenta el aporte de carbohidratos generados por fotosíntesis en las hojas superiores,
especialmente la hoja bandera (Sotomayor, 2007).
3.4 Características Genéticas del arroz
Uno de los primeros genomas de cultivo en ser secuenciado fue el de plantas de arroz,
en este proyecto trabajaron cuatro grupos de investigación, El Instituto Beijing de
Genómica, en el cual trabajaron con la subespecie Indica; El grupo Syngenta quienes
estudiaron la subespecie Japónica ‘Nipponbare’; El Proyecto Internacional de
Secuenciamiento del Genoma del Arroz (International Rice Genome Sequencing
Project, IRGSP) que logró analizar la subespecie Japónica ‘Nipponbare’ y finalmente el
grupo Monsanto (Yu, 2002). El genoma del arroz se puede encontrar en los sitios web:
http://rice.plantbiology.msu.edu/ y http://rgp.dna.affrc.go.jp/.

15
El genoma de la planta es diploide, posee 24 cromosomas (n=12) y presenta una
constitución tipo AA con un tamaño significativamente pequeño de 430 millones de pb
(Soren et al. 2010) en comparación con otros miembros de la familia Gramineae. Se
ha sugerido que la subespecie Japónica contiene entre 32.000 y 50.000 genes
mientras que la Indica posee entre 46.022 y 55.615 genes (Goff et al. 2002, Yu, 2002).
3.5 Estrés Abiótico
Los factores ambientales del medio en donde se encuentra la planta son percibidos y
mediados por los diferentes órganos de esta, en donde una variedad de moléculas
intervienen en el proceso de señalización que activa respuestas de crecimiento
vegetativo, las cuales, una vez producidas limitarán el aporte de nutrientes, minerales
y carbohidratos, siendo la planta capaz de llegar a reprimir el desarrollo y
desencadenar mecanismos tanto de protección como de defensa si las condiciones se
vuelven adversas, asegurando su supervivencia. Las respuestas generadas durante el
estrés abiótico dependen del genotipo, del estado de desarrollo de la planta, de la
severidad, la duración y las condiciones que las provoquen (Vidal, 2010).
Existen distintos factores que generan estrés en la planta como la sequía, la salinidad,
las temperaturas extremas, la humedad, los fuertes vientos, entre otros. Estos factores
son la razón más importante de la reducción de los rendimientos en cultivos y de la
pérdida de cosechas en todo el mundo (Hidalgo, 2005), por lo que son de gran interés
en diversas investigaciones.
En las plantas de Oryza sativa L. la sequía y la salinidad limitan la cosecha de este
cereal puesto que altas concentraciones de sales originan una disminución en el
aporte de agua, un aumento en la producción de especies de oxígeno reactivas
(ROS), y carencia de nutrientes que a largo plazo conllevan al daño y posterior muerte
celular (Gill et al. 2010). La sequía reduce la floración de las plantas, la disposición de
almidón en el polen y aumento en el enrollamiento de las hojas. Asimismo estos
factores ambientales generan un alteración tanto en la parte estructural de las plantas
como en los procesos metabólicos de las mismas, esto con el fin de amortiguar
aquellas alteraciones del medio que los rodea. Uno de los mecanismos de defensa es
el almacenamiento de ciertos solutos metabólicamente compatibles, tales como,
prolina, glicina-betaína, pinitol, carnitina, manitol, sorbitol, polioles, trehalosa, sacarosa,
oligosachharides y fructanos. En segundo lugar la alta o baja expresión de ciertos

16
genes regulados mediante factores de transcripción, en el que su sobre expresión o
inhibición funcionan como señalizadores para mermar las consecuencias que conlleva
el estrés abiótico al cual se enfrentan (Soren et al. 2010).
3.5.1 Salinidad
La salinidad es un estrés que utiliza y modula el transporte iónico de una célula
vegetal, por tanto, se ha convertido en uno de los factores de estrés abiótico más
severo y en una problemática que afecta directamente la producción agrícola en el
mundo (Melorose et al. 2015). Se pueden distinguir dos tipos de salinización, la
primaria que se deriva de la acumulación de sales de origen natural y la secundaria
que se produce directamente por acción del hombre a través del agua de riego,
residuos de abono y fertilizantes químicos (Tanji, 2002). Este último tipo de
salinización es producido por un aporte escaso de agua que impide la adecuada
lixiviación de las sales del suelo favoreciendo su acumulación. A partir de esto, se
pueden producir diferentes tipos de estrés en la planta, tales como el estrés iónico,
que se da por la excesiva absorción de Na+ y Cl- incrementando el potencial osmótico
en la célula y generando toxicidad si no son secretados de forma adecuada; el estrés
osmótico-hídrico, ocasionado por grandes concentraciones de solutos en una zona
radicular además de la disminución del potencial hídrico resultando en plasmólisis; el
estrés nutricional, que afecta el crecimiento vegetal por la limitación en la absorción de
K+, NO3 y H2O; y finalmente el estrés oxidativo que es originado debido a los otros
estreses por aumento en la concentración de Ca+2 y especies reactivas de oxígeno
(Melorose et al. 2015).
La planta necesita acumular solutos para mantener el volumen celular y la turgencia, lo
cual realiza por medio del ajuste osmótico. Este se consigue mediante la acumulación
de osmolitos, permitiendo a la planta disminuir el potencial osmótico favoreciendo el
movimiento del agua hacia el interior de las células. Algunos osmolitos compatibles
son iones inorgánicos, pero en la mayoría son solutos orgánicos, especialmente
azúcares y ácidos orgánicos. Otros osmolitos son los alcoholes de azúcar (glicerol e
inositoles metilados), azúcares complejos (trehalosa, rafinosa y fructanos), derivados
de aminoácidos cuaternarios (prolina, glicina betaína, β-alanina betaína, prolina
betaína), aminas terciarias y compuestos de sulfónicos (Zhu, 2001).

17
Las especies vegetales han desarrollado diferentes mecanismos para evitar o evadir el
estrés salino, adaptarse a él o simplemente llegar a la tolerancia, algunos de estas
estrategias van desde el crecimiento limitado en épocas favorables, absorción
selectiva de iones, síntesis de solutos orgánicos, almacenamiento en los órganos
senescentes hasta el incremento de la halo-suculencia mediante la suculencia foliar o
del brote y disminución del número de hojas. La aplicación de alguno de los
mecanismos depende del tipo de planta, su facilidad de ser tolerante y de la
sensibilidad del genotipo (Vidal, 2010).
3.5.2 Sequía
La sequía hace referencia al estrés causado por déficit del potencial hídrico en la
planta. Los componentes primordiales del potencial hídrico son el potencial osmótico,
que se debe a la presencia de sustancias disueltas, el potencial de turgencia que
surge dada la presencia de la pared celular y el potencial matricial que se da por la
fuerza de absorción que ejercen las paredes sobre las moléculas de agua. Cuando
disminuye la disponibilidad de agua en la planta también se reduce el potencial de
turgencia y el potencial osmótico al aumentar la concentración de solutos (Vidal,
2010). Las plantas pueden responder a los cambios de disponibilidad hídrica en el
ambiente de cuatro formas: I) variando la superficie foliar, II) manipulando la pérdida
de agua mediante los estomas, III) transformando la conductividad hidráulica entre las
partes de la planta para minimizar las embolias y IV) ajustando el sistema radicular
para mejorar la captación de agua (Valladares et al. 2004). Estos mecanismos de
modificación que tienen lugar en la estructura y función de la planta le permiten
aumentar su posibilidad de supervivencia para crecer y reproducirse en determinado
ambiente, en pocas palabras acelerar su proceso de adaptación y tolerancia al estrés.
Por otro lado, también poseen relevancia gran cantidad de moléculas que podrían
actuar como mecanismos de resistencia desde el punto de vista biológico, ya que
muchas de estas presentan un carácter modulador de genes, lo que ha llamado la
atención en algunas investigaciones dirigidas al proceso de tolerancia al estrés
abiótico sobre todo a la sequía (Knight et al. 2001), puesto que esa resistencia es el
resultado de la activación o represión de varios genes, lo cual desencadena una
posible manipulación de los mismos para conservar cultivos. Esto ha conllevado a la
obtención de plantas transgénicas que sobreexpresan un gen particular (Ingram et al.
1996), siendo el caso de algunas especies como Solanum lycopersicum, Oryza sativa,
Nicotiana tabacum o Arabidopsis thaliana (Nakashima et al. 2009), que manifiestan

18
cierta tolerancia a la sequía, pero no presentan una resistencia total al estrés hídrico
como se espera desde el punto de vista agronómico (Robles, 2007).
Ya que la sequía se ha convertido en el factor de estrés más importante en la
producción agrícola a nivel mundial y que se espera un aumento notable en las
frecuencias de sequía durante los próximos 50 años (Houghton et al. 2001), se busca
continuamente implementar y mejorar los procedimientos dirigidos a la obtención de
cultivos resistentes a condiciones ambientales que imponen una limitación de agua
(Robles, 2007), generando nuevos proyectos de investigación orientados a obtención
de variedades capaces de resistir los cambios climáticos.
3.6 Nucleótidos de pirimidina
Las células tienen grandes requerimientos de nucleótidos púricos y pirimidínicos para
su desarrollo, funcionamiento y procesos metabólicos. Las pirimidinas desempeñan un
papel fundamental en la división celular ya que constituye parte de los ácidos
nucleicos formando el ADN y algunos compuestos de ARN (Itakura et al. 1981). Las
pirimidinas también cumplen funciones como intermediaros metabólicos y son
importantes en metabolismo primario, secundario, la expresión génica y en procesos
vitales de crecimiento celular (Zrenner et al. 2006).
Los nucleótidos de pirimidinas también presentan funciones esenciales como
precursores en las plantas, ya que UTP y UDP se encuentran implicados en la síntesis
y la degradación de la sacarosa. Por otro lado UDP-glucosa como nucleótido de
azúcar rico en energía, es el precursor para la síntesis de polisacáridos de celulosa,
glicoproteínas, y fosfolípidos, además actúa como donador de glucosilo en la
derivatización de los metabolitos secundarios y las hormonas con un gran número de
reacciones catalizadas por familia de proteínas de glicosiltransferasas UDP-glucosa
(Lim et al. 2004). El metabolismo de nucleótidos se puede dividir generalmente en la
síntesis de novo, la vía de reciclaje, ruta de degradación y reacciones de conversión
(Figura 8).

19
Figura 8. Metabolismo de pirimidinas con las rutas de síntesis de novo, la vía de reciclaje, ruta de degradación y
reacciones de conversión, los componentes metabólicos mostrados son: 5-fosforibosil-1-pirofosfato (PRPP ), ribose-
5-phosphate (R5-P ), glutamina (Gln), glutamato (Glu), adenosin trifosfato (ATP ), adenosin difosfato (ADP), fosfato
inorgánico (Pi ), carbamoil fosfato (CP ), carbamoil aspartato (CA), dihidro-orotato (DHO), ácido orotico (OA),
orotidina 5-monofosfato (OMP), uridina monofosfato (UMP), uridina difosfato (UDP), uridina trifosfato (UTP),
uridine difosfofato glucosa (UDP-glucosa), citosina monofosfato (CMP), citosina difosfato (CDP), citosina trifosfato
(CTP ), dihidrouracilo (DHU), β-ureidopropionato (β-UP ), β-alanina (β-ala), pirofosfato (PPi), glucosa-1-fosfato (G-
1P), aspartato (Asp) y adenosin monofosfato (AMP) (Zrenner et al. 2006).
3.7 Síntesis de novo de pirimidinas
La síntesis de Novo de pirimidinas hace referencia a la formación de UMP de
carbamoil fosfato, aspartato y 5-fosforribosil-1-pirofosfato; también se conoce como la
vía de orotato y es preservada en casi todas las especies, por lo que en plantas son
utilizadas las mismas reacciones, sin embargo, algunos organismos no tienen la vía de
síntesis de primidinas por ejemplo Cryptosporidium.

20
Figura 9. Imagen Vía de síntesis de novo de pirimidinas, la cual comprende 6 reacciónes para la obtención de Uridin
Monofosfato ( UMP) (Zrenner, 2005).
La vía está compuesta por seis reacciones enzimáticas (figura 9), la primera reacción
es llevada a cabo por Carbamoilfosfato sintasa (CPSII) y produce carbamoil fosfato, a
partir de dióxido de carbono (CO2), Adenosín trifosfato (ATP) y glutamina. La CPSII
está formada por dos subunidades diferentes, una subunidad pequeña glutamina
amidotransferasa (carA) que proporciona amoníaco a la subunidad grande y una
subunidad grande carbamilfosfato sintetasa (carB) que cataliza la reacción de CPS y
posee propiedades regulatorias (Giermann et al. 2005). Los siguientes tres pasos son
catalizados por las enzimas aspartato transcarbamilasa (ATC), dihydroorotasa (DHO)
y dihidroorotato deshidrogenasa (DHOD) (Giermann et al. 2005). ATC una enzima
exclusiva de la biosíntesis de pirimidinas cataliza la condensación de carbamilfosfato
con aspartato para formar carbamoil aspartato y la ciclación de este producto para
producir el anillo de pirimidina denominado dihidro-orotato es catalizada por la enzima
DHO. Subsecuentemente el dihidroorotato es oxidado por DHOD para dar orotato. Los
pasos quinto y sexto son catalizados por distintos dominios de una enzima única,
bifuncional llamada UMP sintasa (UMPS) (Giermann et al. 2005). El orotato es
condensado con fosforribosilpirofosfato a orotidina 5´monofosfosfato por orotato
phosphoribosyltransferase (OPRT), y por último orotidina 5´monofosfosfato también es
descarboxilado por orotidine-5'-fosfato carboxilasa (ODC) para formar uridina 5-
monofosfato (figura 9), el cual es fosforilado para producir uridina-5-trifosfato por

21
uridina monofosfato quinasa y nucleósido difosfato quinasa. Finalmente uridina-5-
trifosfato es aminado por citidin trifosfato sintasa para producir citidin 5-trifosfato
(Zrenner et al. 2006).
Es de gran interés resaltar que los pasos enzimáticos de la vía de síntesis de novo
son codificados por genes individuales, ninguno de los cuales parecen ser
funcionalmente reiterados, lo cual además de explicar por qué no se conocen
mutantes de dicha biosíntesis, también es un motivo de constantes estudios en esta
ruta (Zrenner et al. 2006).
3.8 Vía de reciclaje de pirimidinas
La síntesis de novo de pirimidinas requiere de grandes cantidades de energía, debido
a esto las células han desarrollado una estrategia de reutilizar nucleósidos y
nucleobases preformados a través de reacciones de reciclaje. La uridina 5-
monofosfato obtenida de la síntesis de novo de pirimidinas, es catabolizada a
nucleosidos de pirimidina por la expulsión de un grupo fosfato en una reacción
catalizada por uridina monofosfato hidrolasa (UMPH). Por otro lado, los nucleosidos
son convertidos en bases libres de pirimidinas por la remoción del grupo ribosa en una
reacción catabolizada por uridina ribohidrolasa (URH). Como plantas y animales no
poseen citosina deaminasa los nucleosidos de citidina son desaminados por citidina
deaminasa (CDA) a uridina. Los nucleósidos de uridina, citidina y timidina son
recuperados a sus respectivos nucleótidos por quinasas específicas como uridina
quinasa (UK) y timidina quinasa (TK), mientras que uracilo se obtiene directamente
con fosforribosil pirofosfato en UMP a través fosforribosiltransferasa uracilo (UPRT)
(figura 8) (Zrenner et al. 2006).
3.9 Vía de degradación de pirimidinas
La vía de degradación de pirimidinas no es una reversión de la vía de síntesis de novo
sino que conduce a la formación de β-alanina o β- aminobutirato. Se ha planteado que
la degradación del uracilo podría ser una fuente importante de ß-alanina como un
precursor para pantotenato y coenzima A (CoA) (Zrenner et al. 2009), además de que
este producto de degradación junto con betaína β-alanina pueden servir como
donantes de grupo amino a 2-oxoglutarato u otros aceptores. Por otro lado, las

22
pirimidinas también pueden ser metabolizadas a productos secundarios de las plantas
con funciones específicas de defensa (Zrenner et al. 2009).
Esta vía catalítica se da por la degradación de timina y uracilo, la cual es llevada a
cabo por tres enzimas, dihidropirimidina deshidrogenasa (DHPD), la cual es
dependiente de NAD(P)H; dihidropiriminidasa (DHP), que degrada hidrolíticamente
dihidrouracillo a N-carbamil-β-alanina o dihidrotimina a N-carbamil-β-aminobutirato y la
última enzima β-ureidopropionasa (β-UP), a partir de la cual se obtiene como producto
final β-aminobutirato o β-alanina (figura 10).
La relevancia de esta vía en el mantenimiento de la homeóstasis de pirimidina durante
el crecimiento y desarrollo de plantas o en respuesta a las condiciones adversas que
influyen sobre el metabolismo de nucleótidos aún es poco conocida. Asimismo, no
existe mucha información acerca de la localización subcelular de enzimas de la ruta
catabólica o de los mecanismos metabólicos y genéticos que regulan sus actividades
(Zrenner et al. 2009).
Figura 10. Vía de degradación de pirimidinas en Arabidopsis thaliana, en el costado izquierdo se observa la
degradación de uracilo hasta la obtención de ẞ -alanina, mientras que en el costado derecho se muestra la
degradación de timina hasta su producto ẞ- aminoisobutirato. La reacción subsecuente no se considerada parte de
la ruta sin embargo se ilustra con el fin de evidenciar como los átomos de nitrógenos de pirimidina son asimilados en
el metabolismo del nitrógeno en general (Zrenner et al. 2009).

23
3.9.1 Dihidropirimidina Deshidrogenasa
Dihidropirimidina Deshidrogenasa (DHPD, EC 1.3.1.2 ) es la primera enzima de la ruta
de degradación de pirimidinas y se encarga de la reducción de uracilo y timina en
dihidrouracilo y dihidrotimina respectivamente (Lohkamp et al. 2010). Se ha
evidenciado una relación filogenética entre las proteínas codificadas por cDNAs
homólogos y las enzimas DHOD de la biosíntesis de novo en algunas especies como
Arabidopsis thaliana, Lycopersicon esculentum, Homo sapiens, Drosophila
melanogaster, y Caenorhabditis elegans (Nara et al. 2000).
Figura 11. Subunidades superpuestas de DHPD Sus scrofa. El dominio I del N-terminal FeS-cluster se muestra en
verde, el dominio II de unión a FAD se muestra en amarillo, el dominio III de unión a NADPH se muestra en naranja,
el dominio IV de FMN se muestra en rojo y el dominio V de C-terminal FeS se muestra en azul. La estructura de la
subunidad de DHPD sin ligar se encuentra en negro (Dobritzsch et al. 2002).
Previos estudios han planteado que en Sus scrofa esta enzima de 220 kDa, está
compuesta por 5 dominios, los cuales tienen sitios de unión a cofactores como clusters
[4Fe-4S], Flavín adenin dinucleótido (FAD) y Flavín Mononucleótido (FMN), que
favorecen el transporte de electrones desde Nicotinamina adenina dinucleótido fosfato
(NADPH) hasta el sustrato y a pesar de que no se conoce la estructura del complejo
NADPH-DHPD se ha observado mediante cristalografía que el lugar de unión del
NADPH y sustrato están en sitios diferentes dentro de la proteína DHPD (figura 11)
(Dobritzsch, et al. 2002). Además de esto, Vanoni y colaboradores resaltan en el 2004

24
que a partir de bancos de secuencias se ha identificado alta identidad entre la β-
subunidad de NADPH-GltS y un fragmento de DHPD lo que establece como una
posible hipótesis que NADPH-GltS β-subunidad pueda ser el complemento necesario
para la óptima actividad de esta enzima (figura 12). Para el caso de plantas Zrenner
et al. en el 2006 reportaron que en Arabidopsis la DHPD carece de un dominio en el N-
terminal que presenta un sitio de unión NADPH, lo que sugiere que en las plantas se
requiere de una interacción de proteínas que se encargue de la transferencia de
electrones para su funcionamiento, pero esto no ha sido reportado para ninguna
especie vegetal.
Figura 12. En el costado izquierdo se encuentra la Glutamato sintasa (Subunidad α y β) modelo propuesto por
Vanoni en el 2005. La imagen derecha hace referencia a la interacción de DHPD con la subunidad β de Glutamato
sintasa (dependiente de FAD y NADPH) adapatada en el laboratorio de Dr. Zimmermann de (López, 2015 no
publicado).
En Oryza sativa L. Japónica (LOC4330657) Dihidropirimidina Deshidrogenasa tiene un
peso molecular de 45.31 kDa, posee 414 aminoácidos y 1242 nucleótidos, se
encuentra ubicada en el cromosoma 2 evidenciando 7 exones y 6 intrones (figura 13)
(NCBI, 2016).
Figura 13. Organización del gen de DHPD en Oryza sativa L. Japónica. Los exones se muestran en cajas y los intrones
lineales (Liu et al. 2009).
3.9.2 Gluatamato Sintasa
Glutamato Sintasa (GltS) es un complejo hierro-azufre flavoproteína que participa en el
proceso de asimilación de amoniaco a través de la transferencia del grupo amino de L-
glutamina al carbono de 2-oxoglutarato, produciendo dos moléculas de L-glutamato
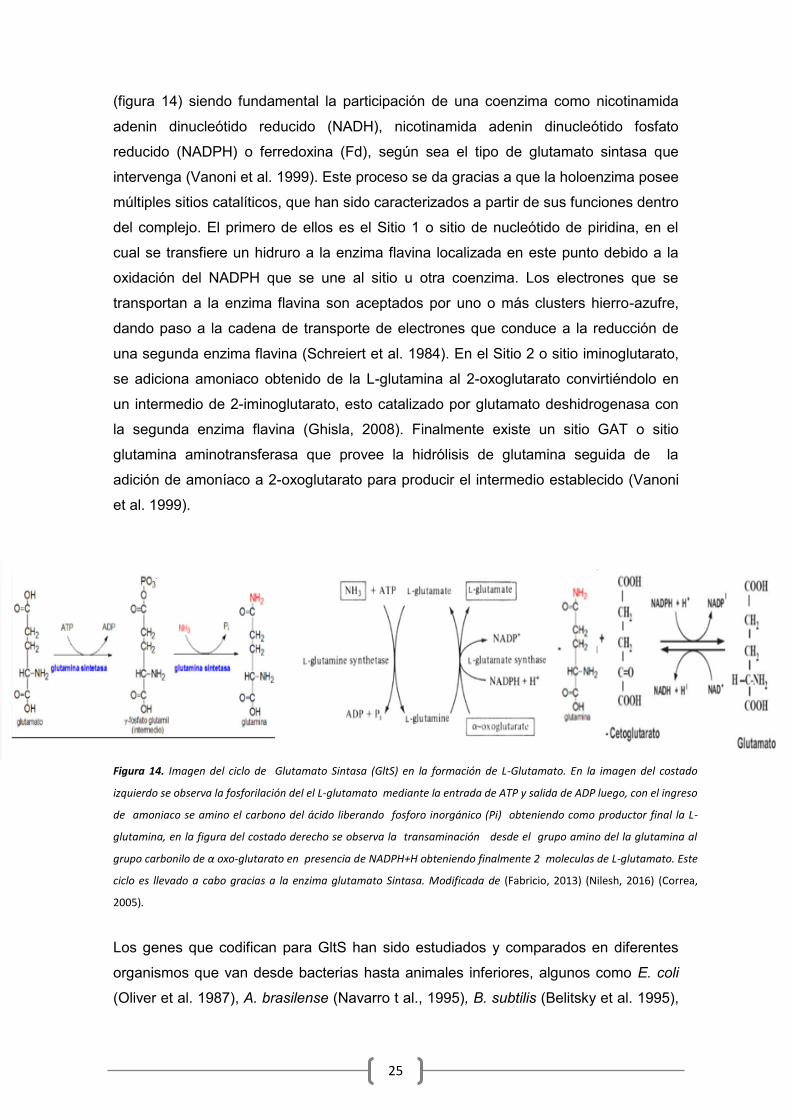
25
(figura 14) siendo fundamental la participación de una coenzima como nicotinamida
adenin dinucleótido reducido (NADH), nicotinamida adenin dinucleótido fosfato
reducido (NADPH) o ferredoxina (Fd), según sea el tipo de glutamato sintasa que
intervenga (Vanoni et al. 1999). Este proceso se da gracias a que la holoenzima posee
múltiples sitios catalíticos, que han sido caracterizados a partir de sus funciones dentro
del complejo. El primero de ellos es el Sitio 1 o sitio de nucleótido de piridina, en el
cual se transfiere un hidruro a la enzima flavina localizada en este punto debido a la
oxidación del NADPH que se une al sitio u otra coenzima. Los electrones que se
transportan a la enzima flavina son aceptados por uno o más clusters hierro-azufre,
dando paso a la cadena de transporte de electrones que conduce a la reducción de
una segunda enzima flavina (Schreiert et al. 1984). En el Sitio 2 o sitio iminoglutarato,
se adiciona amoniaco obtenido de la L-glutamina al 2-oxoglutarato convirtiéndolo en
un intermedio de 2-iminoglutarato, esto catalizado por glutamato deshidrogenasa con
la segunda enzima flavina (Ghisla, 2008). Finalmente existe un sitio GAT o sitio
glutamina aminotransferasa que provee la hidrólisis de glutamina seguida de la
adición de amoníaco a 2-oxoglutarato para producir el intermedio establecido (Vanoni
et al. 1999).
Figura 14. Imagen del ciclo de Glutamato Sintasa (GltS) en la formación de L-Glutamato. En la imagen del costado
izquierdo se observa la fosforilación del el L-glutamato mediante la entrada de ATP y salida de ADP luego, con el ingreso
de amoniaco se amino el carbono del ácido liberando fosforo inorgánico (Pi) obteniendo como productor final la L-
glutamina, en la figura del costado derecho se observa la transaminación desde el grupo amino del la glutamina al
grupo carbonilo de α oxo-glutarato en presencia de NADPH+H obteniendo finalmente 2 moleculas de L-glutamato. Este
ciclo es llevado a cabo gracias a la enzima glutamato Sintasa. Modificada de (Fabricio, 2013) (Nilesh, 2016) (Correa,
2005).
Los genes que codifican para GltS han sido estudiados y comparados en diferentes
organismos que van desde bacterias hasta animales inferiores, algunos como E. coli
(Oliver et al. 1987), A. brasilense (Navarro t al., 1995), B. subtilis (Belitsky et al. 1995),

26
encontrando propiedades bioquímicas definidas que difieren según el tejido donde
actúa, las subunidades que presente, los cofactores, la función metabólica, entre otros
(Temple et al. 1998). Estas investigaciones han arrojado datos importantes, como la
conformación de la enzima por dos subunidades α y β que permiten el flujo de
electrones de una a la otra, puesto que se ha definido que el primer sitio catalítico de
GltS hace parte de su β subunidad y por ende, Vanoni y colaboradores han propuesto
que la proteína actúa como una oxidoreductasa dependiente de FAD, que sirve para
transferir electrones a la subunidad α dando paso a la síntesis reductora de glutamato
(Vanoni et al. 1999). Por otro lado, en estudios expuestos con DHPD de cerdo se
encontró mediante Blast que la región N-terminal de DHPD posee una alta similitud
con la β subunidad de Glutamato Sintasa y que está subunidad caracterizada en
Azospirillum Brasilense evidenció que posee el sitio de unión a NADPH donde sufre
una oxidación mientras que FAD se reduce subsecuentemente, lo cual permite
proponer que en la DHPD de cerdo esta unión y oxidoreducción anteriormente
mencionada se encuentra presente en el N-terminal del complejo enzimático. Además
se encontró que en DHPD existe una zona rica en cisteínas que coinciden en el
alineamiento del N- terminal de la β subunidad de GltS y se ha sugerido que esta
región podría estar involucrada en la formación de los grupos 4Fe-4S para Glutamato
Sintasa deduciendo que para DHPD la sección de cisteínas estaría cumpliendo la
misma función (Rosenbaum et al. 1998). A partir de esta gran semejanza entre el N-
terminal de DHPD en cerdo y la β subunidad de GltS en el Laboratorio de Barbara
Zimmermann se planteó que la β subunidad de GltS es un buen candidato para
cumplir la función de transferencia de electrones región que es desconocida en
plantas.
Por otra parte, de acuerdo a la conformación estructural de GltS, se han señalado tres
tipos diferentes de la holoenzima: NADPH-GltS, Fd-GltS y NADH-GltS.
3.9.2.1 NADPH-Glutamato Sintasa
La forma bacteriana pesa aproximadamente 200 kDa y está conformada por una α-
subunidad (150 kDa) y una β-subunidad (50 kDa), además contiene flavin adenin
dinnucleotido (FAD), Flavin mononucleótido (FMN) y tres clusters diferentes de hierro-
azufre. Esta enzima ha sido caracterizada bioquímicamente en algunas especies como
Klebsiella aerogenes, Escherichia coli, Bacillus subtilis y Azospirillum brasilense
(Vanoni et al. 1999).

27
3.9.2.2 Fd-Glutamato Sintasa
Es un monómero que cataliza la asimilación de amoniaco, tiene un peso molecular de
150 kDa y se encuentra presente tanto en cianobacterias como en los plástidios de las
plantas, posee un contenido más bajo de hierro-azufre y FMN mientras que la
presencia de FAD es similar a NADPH-GltS (Vanoni et al. 2005). Se han realizado
estudios de su actividad en especies como Chlamidomonas, Sinechococcus,
Sinechocistis y más detalladamente en Spinacia oleracea (Temple et al. 1998). Otros
estudios como los realizados por Betti y colaboradores en el 2012 sugieren que
existen dos isoformas codificadas por genes distintos en plantas de Zea mays, pues la
enzima que se encuentra en la raíz presenta características inmunológicas distintas a
la enzima analizada en las hojas. Por otro lado, en Arabidopsis thaliana también se
reportan dos isoformas funcionales, una que sobresale en las hojas (Glu1) y otra
predominante en la raíz (Glu2) (Oliveira et al. 1997).
3.9.2.3 NADH-Glutamato Sintasa
Esta enzima existe en plantas superiores como monómero con un peso aproximado de
230kDa, en donde NADH actúa como donador de electrones para su funcionamiento,
ha sido purificada de especies como Medicago satia, Saccharomyces cereisiae y
recientemente de Bombyx mori (Hirayama et al. 1998), además han sido clonados y
secuenciados genes y ADNc en algunos eucariotas. Se ha reportado que Phaseolus
vulgaris parece poseer dos isoformas de esta enzima (I y II), las cuales aumentan de
manera notable su actividad durante el crecimiento y se expresa en mayor medida la
isoforma II (Chen et al. 1988). Por otra parte, investigaciones proponen a partir de
bancos de secuencia y pruebas bioquímicas, que la enzima de nucleótidos que
dependen de piridina está conformada por un polipéptido de 200 kDa que
aparentemente proviene de la fusión de polipéptidos de las subunidades α y β de GltS
bacterianas (Vanoni et al. 1999).
A partir de varios casos de secuenciación del N-terminal de GltS purificada o
secuenciación de un número de péptidos obtenidos por proteólisis, se ha podido
establecer la estructura primaria de la proteína y de esto se ha comparado la
conformación estructural en diferentes casos, concluyendo que en bacterias la α
subunidad es similar a Fd-GltS y tres cuartos similar al N-terminal de NADH-GltS,
además las subunidades β de distintas especies bacterianas son afines entre sí y un
cuarto parecidas al C-terminal de NADH-GltS (figura 15), esto junto con otros estudios

28
a cofactores, demuestran que un gran número de regiones de la enzima se conservan
en diferentes especies (Cavicchioli et al. 1996), permitiendo abrir campo a otras
investigaciones.
Figura 15. Propuesta de localización de los dominios funcionales en los diferentes tipos de glutamato sintasa (NADPH-
GltS, Fd-GltS y NADH-GltS) a partir de su secuencia. GAT, dominio amidotransferasa; FMN, región de unión; 3Fe4S,
región rica en cisteínas para la formación del cluster; ADP, sitio de unión; FeS, regiones ricas en cisteínas fuertemente
implicadas en la formación de los clusters 4Fe-4S; FAD, sitio de unión a ADP; NAD(P)H, región de unión a ADP; FAD-II,
segunda secuencia de consenso (Cavicchioli et al. 1996).
3.10 Vectores de Clonación
Los vectores de clonación son herramientas de gran ayuda y muy empleadas en
Biología Molecular. Estos vectores son hebras de ADN de cadena doble proveniente
de plásmidos bacterianos (figura 16), fagos, cosmidos entre otros capaces de aceptar
gen ajeno a él dentro de su secuencia denominándose en este punto vector
recombinante, la utilidad de este vector se da gracias a que este puede ser insertado
en una célula hospedera y replicarse muchas veces dentro de ella de manera
independiente y autónoma, por tanto la finalidad de un vector de clonación es la de
almacenar y obtener en gran cantidad secuencias de interés para diversos estudios (
Salazar A. et al. 2013)
Figura 16. Imagen de un plásmido empleado como vector de clonación. Todo Vector está conformado por un sitio de
origen (Ori) el cual es donde la polimerasa empieza la replicación, un sitio múltiple de clonación (SMC) lugar que

29
contiene sitios de reconocimiento por enzimas de restricción permitiendo así la inserción del gen exógeno y un
marcador de selección o resistencia a antibiótico que por lo general es ampicilina, el cual funciona para comprobar si
el vector entró a la célula hospedera (Salazar et al. 2013)
3.10.1 Vector de Clonación pET
El vector de Clonación pET proveniente de plásmido pBR322 es uno de los más
empleados para las técnicas moleculares gracias a sus características que benefician
tanto la clonación como expresión de genes de interés, ya que este vector cuenta con
una alta actividad de la polimerasa y alta eficiencia en la traducción regulada por los
T7. Para el caso de la clonación de un gen de interés este se hace corriente abajo del
promotor T7 y para la expresión de este se hace mediante la inducción con IPTG un
homólogo de la lactosa en la que la ARN polimerasa T7 se expresa a partir del
promotor lacUV5 (Technologies, 2016).
3.10.2 Vector de Clonación pGEM-T EASY
Este vector tiene una longitud de 3kb, contiene el gen de resistencia a ampicilina como
marcador selectivo y posee diversos sitios múltiples de clonación lo cual favorece la
inserción del gen en diferentes partes según lo que se requiera y para el caso de
amplificación por PCR del gen exógeno insertado se realiza con primers M13. No
obstante este plasmido no funciona muy bien como vector de expresión ya que, a
diferencia de otros vectores de expresión este no contiene dentro de su estructura la
sección conocida como IRES (Biological, 2015) el cual es un sitio que facilita la unión
ribosomal a la secuencia dado que esta secuencia IRES es complementaria con el
extremo 3´del ARN ribosmal 16s lo que disminuirá la eficiencia en la traducción de
proteínas (Salazar et al. 2013).

30
4. METODOLOGÍA
4.1 Descripción del área de estudio
El laboratorio del Grupo de Investigación de Bioquímica y Biología Molecular de
parásitos (BBMP), se encuentra en la Carrera 1. Nº 18A – 10 en la Universidad de los
Andes, edifico M-302. El laboratorio cuenta con equipos propios especializados para el
estudio en el campo de la biología molecular y bioquímica.
4.2 Procedimientos
4.2.1 Búsqueda de las secuencias en las bases de datos
Las secuencias de OsβGltS1 y OsβGltS2 putativa fueron tomadas de la base de datos
NCBI National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov),
publicadas con el número LOC4324398 y LOC4339561 respectivamente y fueron
usadas para el diseño de los primers.
4.2.2 Diseño de primers
Por medio de la página web ExPASy (http://expasy.org/), se ubicó el programa
Translate (http://expasy.org/tools/dna.html) que permitió identificar un marco abierto de
lectura dentro de la secuencia de nucleótidos que codifican para las isoformas de
OsβGltS1 y OsβGltS2. Se tomó un templado entre 25 y 26 nucleótidos
respectivamente desde el codón de inicio para el diseño del primer con sentido. Para
el templado de los primers sin sentido, se obtuvo el reverso complementario en
dirección 5´-3´ en el programa EMBOSS (http://emboss.bioinformatics.nl/cgi-
bin/emboss/revseq).
Se realizó un análisis de las estructuras secundarias de los primers en el servidor
DNAfold (http://mfold.rna.albany.edu/?q=mfold/DNA-Folding-Form), se calculó el Tm
que no debía diferir en más de dos grados entre los primers con sentido y sin sentido,
el porcentaje de guanina y citocina que debía estar entre 40%-60%. Para confirmar
estos datos, se usaron varios programas como OligoAnalyzer (IDT), Insilico Melting
Calculator y Oligo Calculator 3.26. Finalmente, se identificó una enzima de restricción

31
que no cortara dentro de la secuencia de las isoformas mediante CLC Sequence
Viewer, para adicionar el sitio de reconocimiento de la enzima a los primers.
4.2.3 Extracción de ARN
La extracción se realizó usando el kit de aislamiento de ARN de plantas UltraClean®
Plant DNA Isolation Kit (MO BIO Laboratories, Inc), se utilizó nitrógeno líquido para
macerar el tejido y asegurar el rompimiento de la pared celular, luego se transfirió
100mg del tejido a un tubo de 2mL y se homogenizó en 1mL de PMR1 y se dejó en
vortex por 30 minutos para después centrifugar durante 3 minutos a 10000g, se
traspasó 600µL del sobrenadante a un tubo de 2ml y se adicionaron 500µL de PMR2
y luego 250µL de PMR3 y se mezcló por inversión 5 veces y luego se incubó en hielo
por cinco minutos, posteriormente se centrifugó a 10000g por 10 minutos y se transfirió
el sobrenadante a un nuevo tubo de 2mL mediante inversión y a este se adicionaron
800µL de la solución PMR4 y se mezcló, de esta solución se transfirieron 650µL al
tubo con filtro y se centrifugó por un minuto a 10000g, se descartó el sobrenadante y
se repitió el proceso con el sobrante de la mezcla de 800µL, luego de completar este
paso se adicionan 500 µL al tubo con filtro y se centrifugó por un minuto a 10000g, se
descartó el sobrenadante y nuevamente se centrifugó por un minuto con el fin de
retirar alguna traza de la solución de lavado, posterior a esto el tubo con filtro se
transfirió a un nuevo tubo de 2mL y se adicionaron 30 µL de la solución PMR6 en todo
el centro de la membrana y se centrifugó por 1 minuto a 10000g, el sobrenadante se
vuelve a pasar por la membrana y se centrifugó nuevamente con el fin de asegurar la
elución de todo el ARN de este se tomaron 3 µL para correr en gel de agarosa y al
sobrante se le realizó tratamiento de DNAsa (MIOBIO, 2016).
4.2.4 Digestión con DNAsa
Para la eliminación de ADN genómico en las muestras de ARN se usó DNAsa I de la
casa comercial Ambion, se adicionó el Buffer DNAsa I 10X para una concentración
final 1X en la muestra de ARN y 1 μL de DNAsa I (2 U) para un máximo de 10μg de
ARN en una reacción de 30μL. Se incubó a 37°C durante 15 min y luego de esto se
inactivaron las DNAsas a 75°C por cinco minutos no sin antes adicionar EDTA con una
concentración final de 5mM con el fin de preservar el la integridad del ARN en esa
temperatura. Luego se colocó el ARN total extraído en hielo y se transfirió a tubos de
PCR para su almacenamiento a -80°C. Finalmente se realizó una cuantificación
usando el equipo NanoDrop 2000™ Spectrophotometer (Thermo Scientific), eligiendo
32
la opción ARN y el factor 40 (Una unidad de absorbancia A260 es igual a 40μg de
ARN de cadena sencilla).
Para determinar la integridad de la muestra de ARN, se realizó una electroforesis en
un gel de agarosa al 1,2% usando Tris-acetato-EDTA (TAE 1X. Anexo: preparación de
reactivos) preparado con agua con DEPC, donde se visualizaron dos bandas que
correspondieron a las subunidades 18S y 28S del ARN ribosomal. En cada pozo se
sirvió 3μL de muestra y 2μL del buffer de carga y se corrió el gel usando una fuente de
poder (BIO-RAD) a 100 V por 30 minutos.
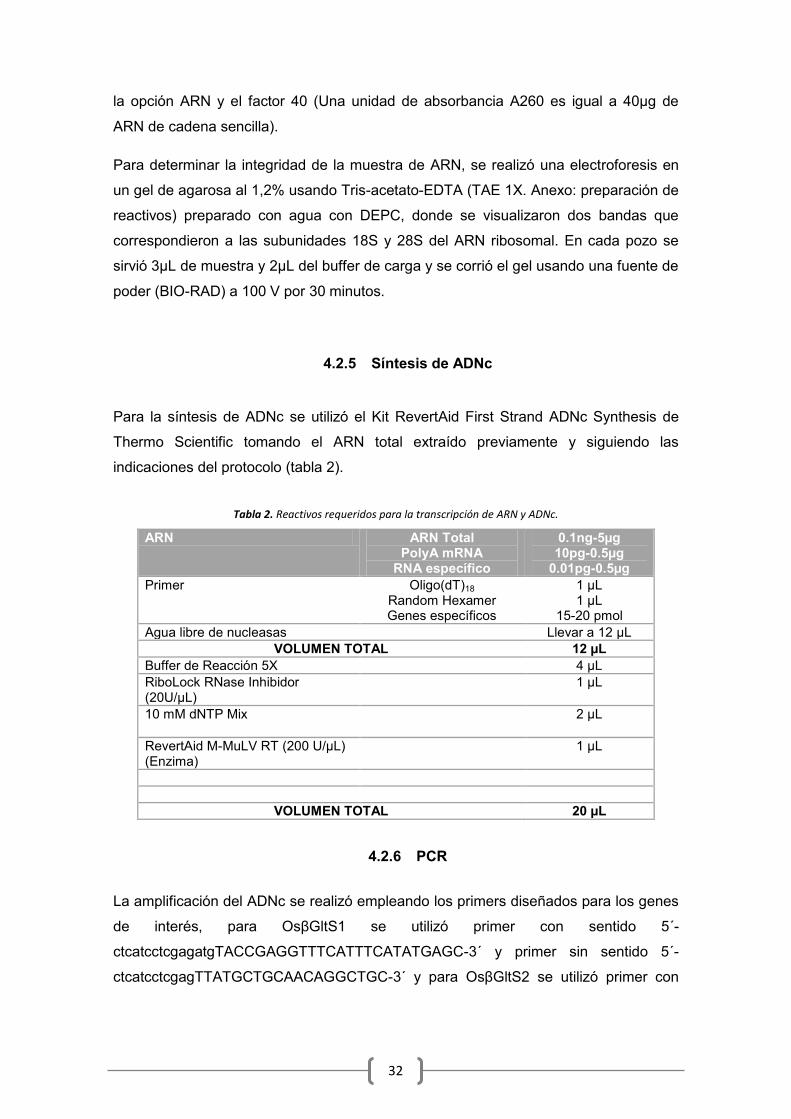
4.2.5 Síntesis de ADNc
Para la síntesis de ADNc se utilizó el Kit RevertAid First Strand ADNc Synthesis de
Thermo Scientific tomando el ARN total extraído previamente y siguiendo las
indicaciones del protocolo (tabla 2).
Tabla 2. Reactivos requeridos para la transcripción de ARN y ADNc.
ARN ARN Total PolyA mRNA
RNA específico
0.1ng-5µg 10pg-0.5µg
0.01pg-0.5µg
Primer Oligo(dT)18
Random Hexamer Genes específicos
1 µL 1 µL
15-20 pmol
Agua libre de nucleasas Llevar a 12 µL
VOLUMEN TOTAL 12 µL
Buffer de Reacción 5X 4 µL
RiboLock RNase Inhibidor (20U/μL)
1 µL
10 mM dNTP Mix 2 µL
RevertAid M-MuLV RT (200 U/μL) (Enzima)
1 µL
VOLUMEN TOTAL 20 µL
4.2.6 PCR
La amplificación del ADNc se realizó empleando los primers diseñados para los genes
de interés, para OsβGltS1 se utilizó primer con sentido 5´-
ctcatcctcgagatgTACCGAGGTTTCATTTCATATGAGC-3´ y primer sin sentido 5´-
ctcatcctcgagTTATGCTGCAACAGGCTGC-3´ y para OsβGltS2 se utilizó primer con

33
sentido 5´-ctcatcctcgagatgTACCGAGGTTTCATAAAATATGAGCG-3´ y primer sin
sentido 5´-ctcatcggatccCTATGCAACTACGGGTTGGACC-3´,.
Tabla 3. Reactivos requeridos para la PCR.
Se usaron las siguientes condiciones para la PCR: la denaturación inicial a 94°C por 3
minutos, la denaturación a 94°C por 30 segundos, una temperatura de anillage de
59°C para OsβGltS1 y 58°C para OsβGltS2 por 30 segundos, la extensión fue de
1minuto a 72°C, proceso que fue repetido 35 veces y finalmente un paso de extensión
final de 10 min a 72°C.
4.2.7 Preparación del vector para subclonación
4.2.7.1 Mini preparación de plásmidos: mini-prep
A partir de pET19-b congelado en céluas de E. coli DH5αF´ a -80°C, se tomó una
muestra y se incubó durante 16 horas en medio líquido LB con ampicilina en tubos
falcon de 50 mL. Posteriormente, se centrifugó por 10 minutos a 5500rpm, se descartó
el sobrenadante, se resuspendió el pellet en 100 μL de la solución 1, (50mM glucosa,
10mM de EDTA y 25mM de Tris-HCl a pH: 8. Anexo: 9.1.5.1), se transfirió la solución
a un tubo eppendorff y se incubó a temperatura ambiente por 5 minutos. Se adicionó
200 μL de la solución 2, (0.2N NaOH, 1%SDS. Anexo: 9.1.5.2) preparada en fresco
con el fin de lisar la bacteria y liberar el contenido genómico, se mezcló por inversión
varias veces y se adicionó 150μL de la solución 3 (3M Acetato de potasio, 5M de Ácido
acético glaciar en agua miliQ. Anexo: 9.1.5.3) se mezcló por inversión y se incubó a -
20°C por 10 minutos, luego se centrifugó por 30 minutos a 4°C y 13.000g. El
sobrenadante fue transferido a tubos eppendorf nuevos y se adicionó 1 mL de etanol
absoluto y se incubó a -20°C durante 2 horas, a continuación se centrifugó a 13.000g
Reactivo Concentración Inicial Concentración Final
TopTaq DNA Polymerase de QIAGEN
5 units/ μL 0.5 U/25 μL
PCR Buffer (contains 15 mM MgCl2)
10 X 1 X
MgCl2 25 mM 0.5 mM
dNTPs, 5 mM 0.2 mM
Primers 10 μM 0.2 μM
ADN 1 μg
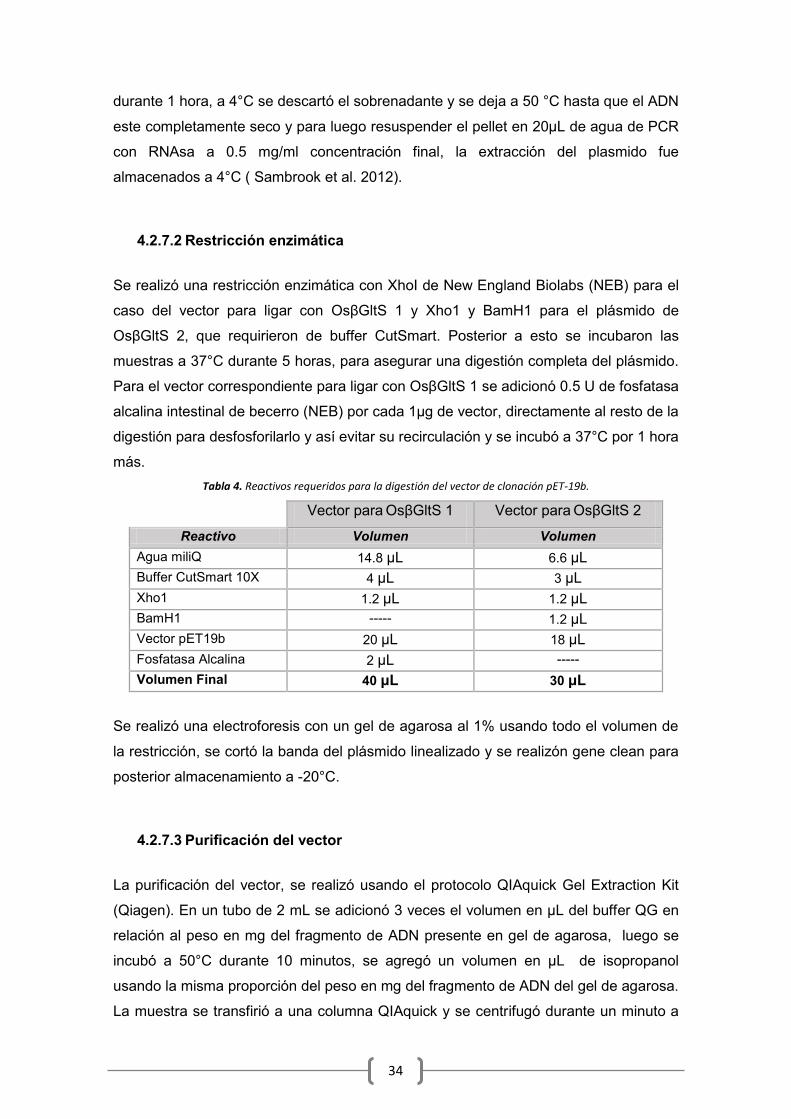
34
durante 1 hora, a 4°C se descartó el sobrenadante y se deja a 50 °C hasta que el ADN
este completamente seco y para luego resuspender el pellet en 20μL de agua de PCR
con RNAsa a 0.5 mg/ml concentración final, la extracción del plasmido fue
almacenados a 4°C ( Sambrook et al. 2012).
4.2.7.2 Restricción enzimática
Se realizó una restricción enzimática con XhoI de New England Biolabs (NEB) para el
caso del vector para ligar con OsβGltS 1 y Xho1 y BamH1 para el plásmido de
OsβGltS 2, que requirieron de buffer CutSmart. Posterior a esto se incubaron las
muestras a 37°C durante 5 horas, para asegurar una digestión completa del plásmido.
Para el vector correspondiente para ligar con OsβGltS 1 se adicionó 0.5 U de fosfatasa
alcalina intestinal de becerro (NEB) por cada 1μg de vector, directamente al resto de la
digestión para desfosforilarlo y así evitar su recirculación y se incubó a 37°C por 1 hora
más.
Tabla 4. Reactivos requeridos para la digestión del vector de clonación pET-19b.
Vector para OsβGltS 1 Vector para OsβGltS 2
Reactivo Volumen Volumen
Agua miliQ 14.8 μL 6.6 μL
Buffer CutSmart 10X 4 μL 3 μL
Xho1 1.2 μL 1.2 μL
BamH1 ----- 1.2 μL
Vector pET19b 20 μL 18 μL
Fosfatasa Alcalina 2 μL -----
Volumen Final 40 μL 30 μL
Se realizó una electroforesis con un gel de agarosa al 1% usando todo el volumen de
la restricción, se cortó la banda del plásmido linealizado y se realizón gene clean para
posterior almacenamiento a -20°C.
4.2.7.3 Purificación del vector
La purificación del vector, se realizó usando el protocolo QIAquick Gel Extraction Kit
(Qiagen). En un tubo de 2 mL se adicionó 3 veces el volumen en μL del buffer QG en
relación al peso en mg del fragmento de ADN presente en gel de agarosa, luego se
incubó a 50°C durante 10 minutos, se agregó un volumen en μL de isopropanol
usando la misma proporción del peso en mg del fragmento de ADN del gel de agarosa.
La muestra se transfirió a una columna QIAquick y se centrifugó durante un minuto a
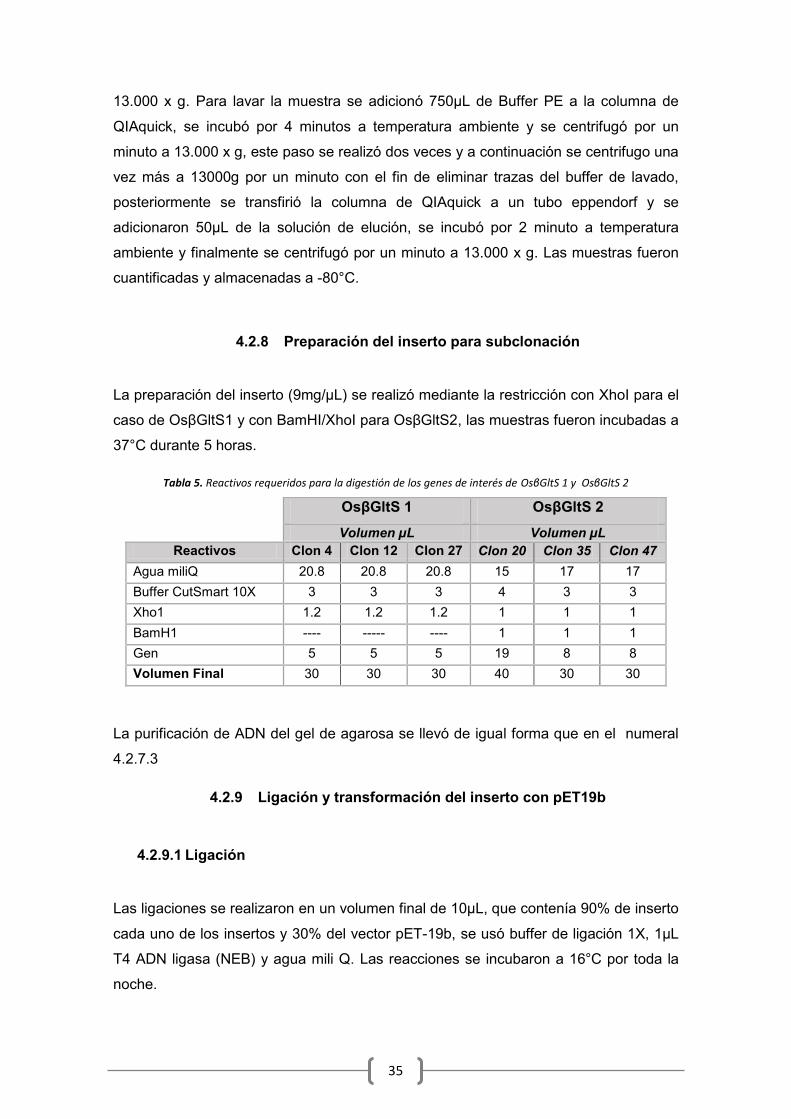
35
13.000 x g. Para lavar la muestra se adicionó 750μL de Buffer PE a la columna de
QIAquick, se incubó por 4 minutos a temperatura ambiente y se centrifugó por un
minuto a 13.000 x g, este paso se realizó dos veces y a continuación se centrifugo una
vez más a 13000g por un minuto con el fin de eliminar trazas del buffer de lavado,
posteriormente se transfirió la columna de QIAquick a un tubo eppendorf y se
adicionaron 50μL de la solución de elución, se incubó por 2 minuto a temperatura
ambiente y finalmente se centrifugó por un minuto a 13.000 x g. Las muestras fueron
cuantificadas y almacenadas a -80°C.
4.2.8 Preparación del inserto para subclonación
La preparación del inserto (9mg/µL) se realizó mediante la restricción con XhoI para el
caso de OsβGltS1 y con BamHI/XhoI para OsβGltS2, las muestras fueron incubadas a
37°C durante 5 horas.
Tabla 5. Reactivos requeridos para la digestión de los genes de interés de OsβGltS 1 y OsβGltS 2
OsβGltS 1 OsβGltS 2
Volumen µL Volumen µL
Reactivos Clon 4 Clon 12 Clon 27 Clon 20 Clon 35 Clon 47
Agua miliQ 20.8 20.8 20.8 15 17 17
Buffer CutSmart 10X 3 3 3 4 3 3
Xho1 1.2 1.2 1.2 1 1 1
BamH1 ---- ----- ---- 1 1 1
Gen 5 5 5 19 8 8
Volumen Final 30 30 30 40 30 30
La purificación de ADN del gel de agarosa se llevó de igual forma que en el numeral
4.2.7.3
4.2.9 Ligación y transformación del inserto con pET19b
4.2.9.1 Ligación
Las ligaciones se realizaron en un volumen final de 10µL, que contenía 90% de inserto
cada uno de los insertos y 30% del vector pET-19b, se usó buffer de ligación 1X, 1µL
T4 ADN ligasa (NEB) y agua mili Q. Las reacciones se incubaron a 16°C por toda la
noche.
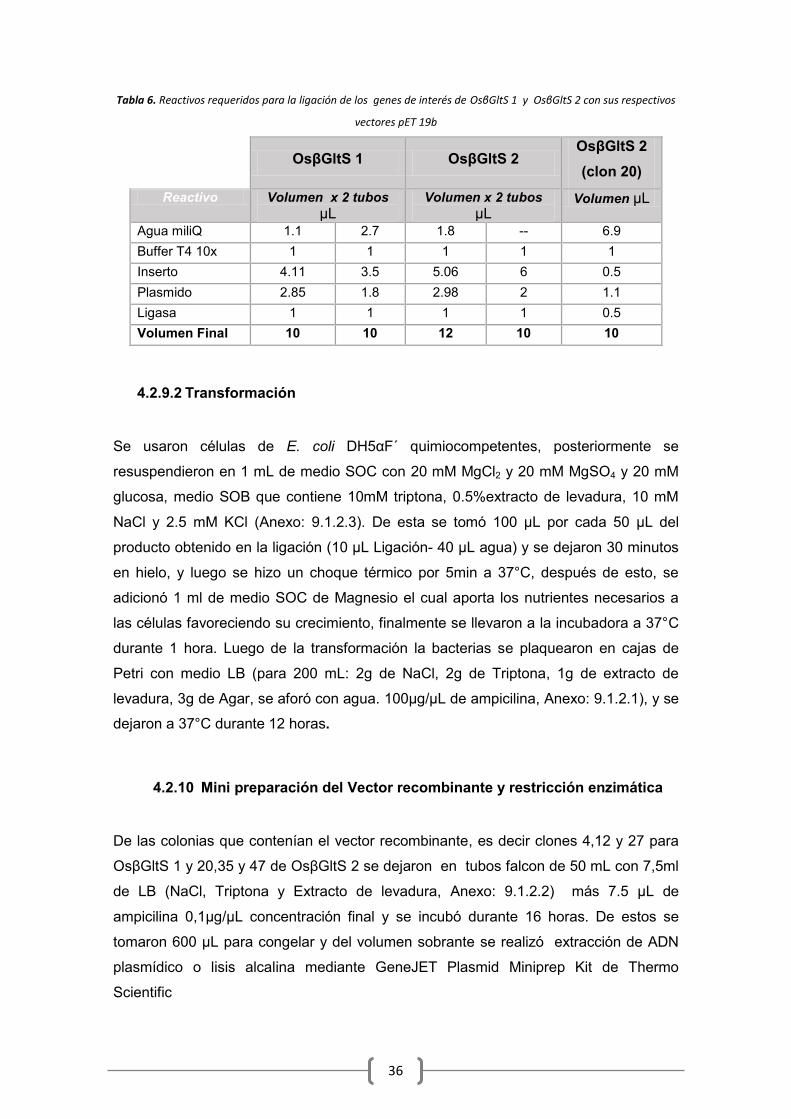
36
Tabla 6. Reactivos requeridos para la ligación de los genes de interés de OsβGltS 1 y OsβGltS 2 con sus respectivos
vectores pET 19b
OsβGltS 1 OsβGltS 2 OsβGltS 2
(clon 20)
Reactivo Volumen x 2 tubos
µL
Volumen x 2 tubos
µL Volumen µL
Agua miliQ 1.1 2.7 1.8 -- 6.9
Buffer T4 10x 1 1 1 1 1
Inserto 4.11 3.5 5.06 6 0.5
Plasmido 2.85 1.8 2.98 2 1.1
Ligasa 1 1 1 1 0.5
Volumen Final 10 10 12 10 10
4.2.9.2 Transformación
Se usaron células de E. coli DH5αF´ quimiocompetentes, posteriormente se
resuspendieron en 1 mL de medio SOC con 20 mM MgCl2 y 20 mM MgSO4 y 20 mM
glucosa, medio SOB que contiene 10mM triptona, 0.5%extracto de levadura, 10 mM
NaCl y 2.5 mM KCl (Anexo: 9.1.2.3). De esta se tomó 100 μL por cada 50 μL del
producto obtenido en la ligación (10 μL Ligación- 40 μL agua) y se dejaron 30 minutos
en hielo, y luego se hizo un choque térmico por 5min a 37°C, después de esto, se
adicionó 1 ml de medio SOC de Magnesio el cual aporta los nutrientes necesarios a
las células favoreciendo su crecimiento, finalmente se llevaron a la incubadora a 37°C
durante 1 hora. Luego de la transformación la bacterias se plaquearon en cajas de
Petri con medio LB (para 200 mL: 2g de NaCl, 2g de Triptona, 1g de extracto de
levadura, 3g de Agar, se aforó con agua. 100μg/μL de ampicilina, Anexo: 9.1.2.1), y se
dejaron a 37°C durante 12 horas.
4.2.10 Mini preparación del Vector recombinante y restricción enzimática
De las colonias que contenían el vector recombinante, es decir clones 4,12 y 27 para
OsβGltS 1 y 20,35 y 47 de OsβGltS 2 se dejaron en tubos falcon de 50 mL con 7,5ml
de LB (NaCl, Triptona y Extracto de levadura, Anexo: 9.1.2.2) más 7.5 μL de
ampicilina 0,1μg/μL concentración final y se incubó durante 16 horas. De estos se
tomaron 600 μL para congelar y del volumen sobrante se realizó extracción de ADN
plasmídico o lisis alcalina mediante GeneJET Plasmid Miniprep Kit de Thermo
Scientific
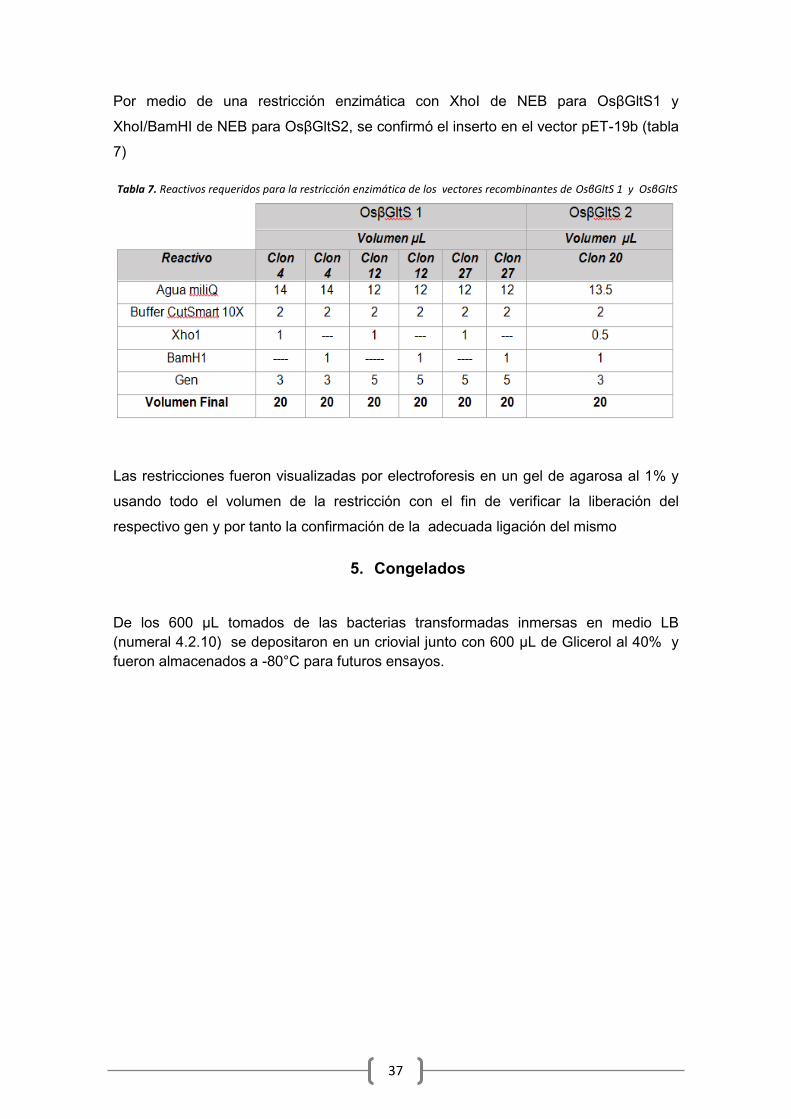
37
Por medio de una restricción enzimática con XhoI de NEB para OsβGltS1 y
XhoI/BamHI de NEB para OsβGltS2, se confirmó el inserto en el vector pET-19b (tabla
7)
Tabla 7. Reactivos requeridos para la restricción enzimática de los vectores recombinantes de OsβGltS 1 y OsβGltS
Las restricciones fueron visualizadas por electroforesis en un gel de agarosa al 1% y
usando todo el volumen de la restricción con el fin de verificar la liberación del
respectivo gen y por tanto la confirmación de la adecuada ligación del mismo
5. Congelados
De los 600 μL tomados de las bacterias transformadas inmersas en medio LB
(numeral 4.2.10) se depositaron en un criovial junto con 600 μL de Glicerol al 40% y
fueron almacenados a -80°C para futuros ensayos.

38
6. RESULTADOS
6.1 Búsqueda de las secuencias en las bases de datos
Las secuencias fueron localizadas en NCBI encontrando que Glutamato sintasa 1
[NADH] Oryza satiava Japonica (OsGltS1) hace parte del cromosoma 1, posee 22
exones y 21 intrones (figura 17), por otro lado, Glutamato sintasa 2 [NADH] Oryza
satiava Japonica (OsGltS2) se ubica en el cromosoma 5 y consta de 22 exones y 21
intrones (figura 18). También se obtuvieron las secuencias en nucleótidos de las β
subunidades de Glutamato sintasa 1 y Glutamato sintasa 2 (figura 19 y 20).
Figura 17. Exones e intrones de OsGltS 1 ubicado en el cromosoma 1, con 22 exones (azul) y 21 intrones (línea lila)
(MSU, 2016)
Figura 18. Exones e intrones de OsGltS 2 ubicado en el cromosoma 5, con 22 exones (azul) y 21 intrones (línea lila)
(MSU, 2016)

39
6.2 Diseño de primers
Se obtuvo la secuencia en nucleotidos y aminoácidos de la β subunidad de OsGltS 1 y
OsGltS 2 a través de Expasy Translate, con el fin de conocer el codón de inicio y de
parada de las secuencias (figuras 21 y 22) y de esta forma diseñar los primeros
correspondientes que permitan la amplificación de las isoformas empleadas para la
posterior clonación.
Figura 21. Codón de inicio y de parada de OsβGltS 1 en el marco de lectura 5’-3’ usando Expasy Translate.
Figura 19. Secuencia β subunidad OsGltS1
tomada de NCBI
Figura 20. Secuencia β subunidad OsGltS2
tomada de NCBI

40
Figura 22. Codón de inicio y de parada de OsβGltS 2 en el marco de lectura 5’-3’ usando Expasy Translate.
Se analizó el mapa del vector de clonación y de expresión pET-19b (figura 23) para
examinar las enzimas de restricción que se implementarían, seleccionando XhoI y
BamHI para el vector que se ligaría con OsβGltS2, ya que estas cortan en el plásmido
y no cortan dentro de la secuencia interés, sin embargo, para el caso de OsβGltS1
BamHI si corta internamente, por lo que se utilizó únicamente XhoI.
Figura 23. Vector de expresión pET-19b (Novagen, 2016).
Se diseñaron primers para la secuencia de OsβGltS 1 y OsβGltS 2 tomando como
templados los nucleótidos del extremo 5’ y del extremo 3’ de las secuencias (tabla 8).
Tabla 8. Secuencia de primers diseñados para OsβGltS 1 y OsβGltS 2. En rojo se encuentran señalados los sitios de
reconocimiento de la enzima XhoI y en azul el sitio de reconocimiento de la enzima BamHI.
Nombre Primer Secuencia Tm
(°C)
βGltS 1 F 5’ctcatcctcgagATGTACCGAGGTTTCATTTCATATGAGC’3 59.9
R 5’ctcatcctcgagTTATGCTGCAACAGGCTGC’3 59.2
βGltS 2 F 5’ctcatcctcgagATGTACCGAGGTTTCATAAAATATGAGCG’3 60.6
R 5’ctcatcggatccCTATGCAACTACGGGTTGGACC’3 60.5

41
Posteriormente implementando la plataforma DNAfold
(http://mfold.rna.albany.edu/?q=mfold/DNA-Folding-Form) se revisaron las estructuras
secundarias que podía formar cada primer (figura 24). A condiciones de temperatura
de 59°C para OsβGltS1 y de 60°C para OsβGltS2, utilizando 1mM [Na+] y 2.5mM de
[Mg++] no se observaron estructuras secundarias entálpicamente desfavorables.
Figura 24. Estructuras secundarias de los primers diseñados utilizando el programa DNAfold. 1. Estructura
secundaria del primer OsβGltS 1 forward con un ∆G= 0.19 kcal/mol a 57.6°C. 2. Estructura secundaria del primer
OsβGltS 1 reverse con un ∆G= 0.51 kcal/mol a 54.5 °C. Estructura secundaria del primer OsβGltS 2 forward con un
∆G=0.19 kcal/mol a 57.6 °C 4. Estructura secundaria del primer OsβGltS 2 reverse con un ∆G=0.85 kcal/mol a 52 °C.
Por último se compararon el Tm de los primers diseñados para cada una de las
secuencias en diferentes programas, con el fin de verificar los parámetros de los
mismos. De acuerdo a los resultados (tabla 9 y 10) se evidencia que los Tm están
balanceados, el porcentaje de G-C se encuentra entre 40-60% y la longitud de la
región homóloga no sobrepasa los 25 nucleótidos.
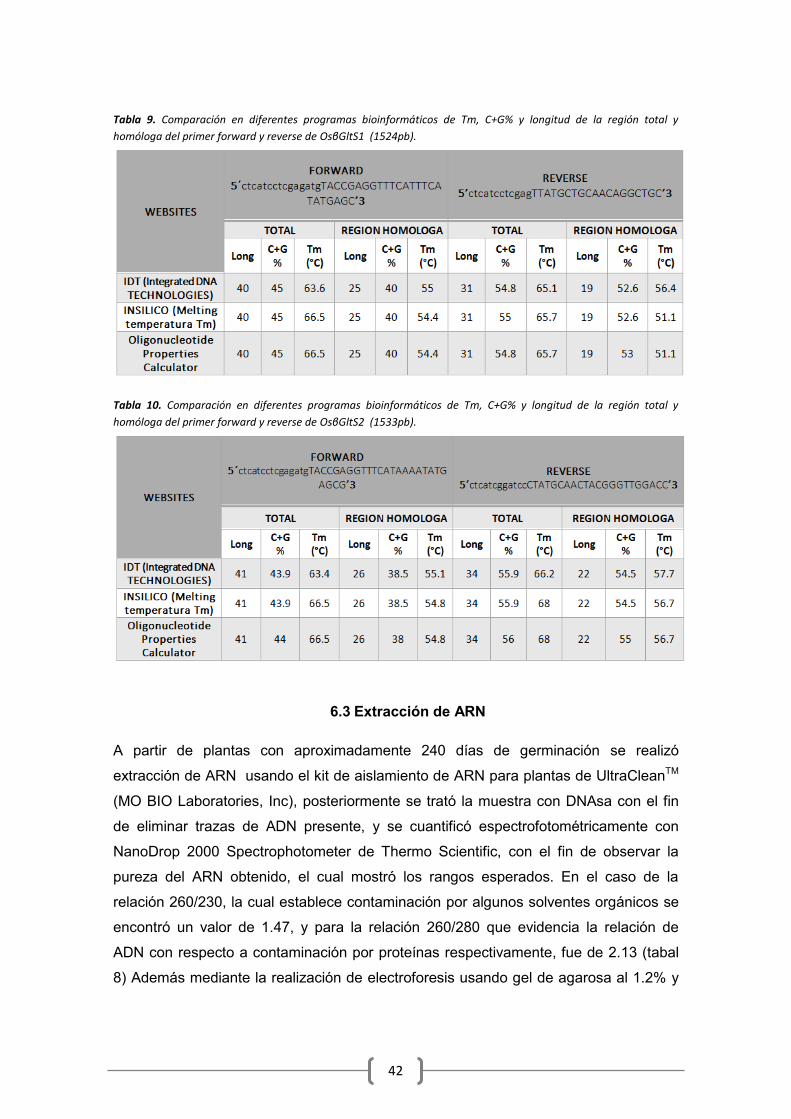
42
Tabla 9. Comparación en diferentes programas bioinformáticos de Tm, C+G% y longitud de la región total y
homóloga del primer forward y reverse de OsβGltS1 (1524pb).
Tabla 10. Comparación en diferentes programas bioinformáticos de Tm, C+G% y longitud de la región total y
homóloga del primer forward y reverse de OsβGltS2 (1533pb).
6.3 Extracción de ARN
A partir de plantas con aproximadamente 240 días de germinación se realizó
extracción de ARN usando el kit de aislamiento de ARN para plantas de UltraCleanTM
(MO BIO Laboratories, Inc), posteriormente se trató la muestra con DNAsa con el fin
de eliminar trazas de ADN presente, y se cuantificó espectrofotométricamente con
NanoDrop 2000 Spectrophotometer de Thermo Scientific, con el fin de observar la
pureza del ARN obtenido, el cual mostró los rangos esperados. En el caso de la
relación 260/230, la cual establece contaminación por algunos solventes orgánicos se
encontró un valor de 1.47, y para la relación 260/280 que evidencia la relación de
ADN con respecto a contaminación por proteínas respectivamente, fue de 2.13 (tabal
8) Además mediante la realización de electroforesis usando gel de agarosa al 1.2% y

43
HydraGreen™ Safe DNA Dye, 20,000X in Water, se observó la extracción y se
visualizó su pureza pues no se encontraron trazas de ADN genómico (figura 25).
Tabla 8. Cuantificación de ARN de plantas de Oryza sativa L.
Muestra Concentración
Ácidos
Nucleicos
Unidad Relación
260/280
Relación
260/230
Factor
1 1997 ng/µL 2.13 1.47 40
Figura 25. Extracción de ARN de O. sativa L. Se sembraron 3µL de muestra, se observan las bandas 28S y 18S
correspondientes a ARN ribosomal y la eliminación total de ADN genómico.
De igual forma, se realizó otra extracción de ARN a partir de plantas con
aproximadamente 120 días de germinación bajo las mismas condiciones mencionadas
anteriormente. El ARN extraído se visualizó en gel de agarosa 1.2% (figura 26)
demostrando que la muestra tenia alta pureza, siendo adecuada para posterior
síntesis de ADNc del cual se obtuvo el clon 20 de OsβGlts 2.
Figura 26. Extracción de ARN de O. sativa L. Se sembraron 3µL de muestra, se observan las bandas 28S y 18S
correspondientes a ARN ribosomal y la eliminación total de ADN genómico.

44
6.4 Síntesis de ADNc y PCR
Se sintetizó ADN complementario a partir de las muestras extraídas usando RevertAid
cDNA Synthesis Kit (Thermo Scientific). Para comprobar el estado del ARN extraído
mediante el ADNc obtenido, se realizó PCR usando como gen control Actina el cual es
un marcador para verificar la eficiencia de la extracción, debido a que es un gen que
se expresa continuamente, pero solo se evidenció la banda en la segunda PCR (figura
28), esto debido al poco volumen (5µL) que fue sembrado en el primer gel (figura 27).
La PCR para la amplificación de los genes se preparó con una temperatura de
anillamiento de 59°C durante 35 ciclos. El tamaño del gen fue comprobado por
electroforesis en un gel de agarosa al 1% HydraGreen™ Safe DNA Dye, 20,000X en
agua evidenciando para OsβGlts 1 una banda muy intensa con un tamaño de 1524pb
(figura 27 pozo 2), pero la banda de OsβGlts 2 es casi imperceptible indicando su baja
concentración (figura 27), por otro lado, para la segunda PCR realizada se observa la
amplificación de ambos genes con bandas perceptibles y de mismas intensidad (figura
28).
Figura 27. PCR de OsβGltS1 y OsβGltS2 a partir del ADNc sintetizado. Se sembraron 2µL de muestra de izquierda a
derecha: 1-2. OsβGltS1 con un peso aproximado de 1524 pb, 3. marcador de peso molecular de 10000 pb (GeneRuler TM
), 4. OsβGltS2 con un peso aproximado de 1533 pb y 5. Actina con un peso aproximado de 164pb.

45
Figura 28. PCR de OsβGltS1 y OsβGltS2 a partir del ADNc sintetizado. Se sembraron 2µL de muestra de izquierda a
derecha: 1. Actina con un peso aproximado de 164pb, 2. OsβGltS1 con un peso aproximado de 1524 pb, 3. marcador
de peso molecular de 10000 pb (GeneRuler TM
) y 4. OsβGltS2 con un peso aproximado de 1533 pb.
De las PCR realizadas, se unieron las muestras que amplificaron de cada gen y se
realizó GeneClean con QIAquick Gel Extraction Kit de QIAGEN para limpiar de
residuos no deseados cada muestra, y con el fin de confirmar la descontaminación y el
estado de cada gen se corrió electroforesis en gel de agarosa 1% HydraGreen™
Safe DNA Dye, 20,000X en agua obteniendo bandas intensas para el caso de los dos
genes (figura 29).
Figura 29. GeneCelan de OsβGltS1 y OsβGltS2 a partir de PCR. Se sembraron 2µL de muestra de izquierda a derecha:
1. marcador de peso molecular de 10000 pb (GeneRuler TM
), 2. OsβGltS1 con un peso aproximado de 1524 pb y 3.
OsβGltS2 con un peso aproximado de 1533 pb.
Por otro lado, se realizó PCR del segundo ADNc obteniendo únicamente amplificación
de OsβGltS 2 bajo una temperatura de anillamiento de 59°C, lo cual fue comprobado
en electroforesis con gel de agarosa 1% e HydraGreen™ Safe DNA Dye, 20,000X in
Water observando bandas intensas en todos los casos (figura 30). Las tres muestras
obtenidas se unieron y se implementó el kit de Gene Clean para retirar residuos de
contaminación de la PCR y proceder con la restricción del gen.

46
Figura 30. PCR OsβGltS2 a partir de ARN extraído. Se sembraron 2µL de muestra de izquierda a derecha: 1,3 y 4.
OsβGltS2 con un peso aproximado de 1533 pb, 2. marcador de peso molecular de 3000 pb (GeneRuler TM
).
6.5 Restricción Enzimática
A partir de las PCR se prosiguió a realizar restricción enzimática de cada gen y del
vector de expresión al cual se liga, el cual fue pET19b. Para el caso de OsβGlts 1 se
realizó restricción únicamente con enzima XhoI, mientras que para OsβGlts 2 se cortó
con enzimas BamHI y XhoI luego de este proceso se realizó nuevamente Gene Clean
mediante QIAquick Gel Extraction Kit de QIGEN Con el fin de limpiar las muestras de
las enzimas de restricción y demás contaminantes y posterior a esto se corrió
electroforesis en gel de agarosa 1% y HydraGreen™ Safe DNA Dye, 20,000X in
Water como tinción, obteniendo para el caso de la primera extracción bandas intensas
de OsβGlts 1 mientras que para OsβGlts 2 la banda fue muy tenue (figura 31), de la
segunda extracción se evidencia una banda fuerte para OsβGlts 2 (figura 32).
Figura 31. Restricción de OsβGltS1 y OsβGltS2 con XhoI y BamHI/XhoI respectivamente. Posterior del gene clean se
sembraron 2µL de muestra de izquierda a derecha: 1. OsβGlts 1/XhoI con un peso aproximado de 1524pb, 2.

47
3000
1000
1500
750
2000
6000
OsβGltS2/BamHI-XhoI con un peso aproximado de 1533 pb. 3. marcador de peso molecular de 10000 pb
(GeneRulerTM
).
Figura 32. Restricción de OsβGltS2 BamHI/XhoI. Posterior del gene clean se sembraron 2µL de muestra de izquierda
a derecha: 1. marcador de peso molecular de 3000 pb (GeneRulerTM
) y 2 OsβGltS2/BamHI-XhoI con un peso
aproximado de 1533 pb.
De igual forma para el vector de expresión empleado para ambos genes de interés el
cual fue pET-19b se obtuvo a partir de lisis alcalina y posterior a esto se llevó a cabo la
restricción enzimática, en donde parte de este plásmido se cortó con XhoI para la
ligación con OsβGltS 1 y dado que este solo es cortado con una enzima, se requirió de
adición de fosfatasa alcalina con el fin de evitar la recircularización del vector al
eliminar grupos fosfatos en los extremos y evitando la formación del enlance
fosfodiester entre este, por otro lado, el vector que se ligara con OsβGlts 2 se cortó
con las mismas enzimas del gen (BamHI y XhoI), Posterior a esto se realizó gene
clean y para ratificar la restricción se realizó electroforesis en un gel de agarosa al 1%
con tinción HydraGreen™ Safe DNA Dye, 20,000X en agua observándose las bandas
alrededor de 5717pb (figura 33 y 34).
1 2 3

48
Figura 33. En la parte izquierda se observa la Restricción de pET 19 para OsβGltS1 y pET 19b para OsβGltS2 con Xho1
y BamH1/Xho1 respectivamente. Posterior del gene clean se sembraron 2µL de muestra de izquierda a derecha: 1.
pET 19b/BamHI-XhoI con un peso aproximado de 5717pb, 2. marcador de peso molecular de 10000 pb (GeneRuler TM
), 3. pET 19b/-XhoI con un peso aproximado de de 5717 pb igualmente . En el sector derecho se encuentro el mapa
del vector de expresión de pET-19b con los diferentes lugares en los que corta las enzimas de restricción (Novagen,
2016).
Figura 34. Restricción de pET 19 para OsβGltS2 con XhoI y BamHI/XhoI. Posterior del gene clean se sembraron 2µL
de muestra de izquierda a derecha: 1. marcador de peso molecular de 3000 pb (GeneRuler TM
) y 2. pET 19b/BamHI-
XhoI con un peso aproximado de 5717pb.
6.6 Clonación en pET 19b
Luego de la restricción se ligó el plásmido con el gen respectivo mediante enzima T4
ADN ligasa la cual fue empleada para la transformación en E. Coli DH5α-F´
quimiocompetentes.
A partir de la transformación se realizó PCR de diferentes colonias transformadas
tanto con el gen ligado OsβGltS1 como OsβGltS2. Para el primer caso, debido a la
bidireccionalidad del gen se emplearon primers Forward T7 y Reverse el
correspondiente OsβGltS1, esto con el fin de confirmar la ligación de manera correcta,
es decir, de 5´- 3´, para el segundo caso colonias con el gen OsβGltS2 se emplearon
primers Forward y Reverse T7 únicamente ya que este es unidireccional, lo cual indica
que solo se puede unir de manera correcta. Para evidenciar las colonias
transformadas con el gen de interés se corrió un gel de agarosa 1% y tinción
HydraGreen™ Safe DNA Dye, 20,000X in Water. Para el caso de OsβGlts 1, se
obtuvieron tres clones 4, 12 (figura 35) y 27 (figura 36) confirmándose por la banda
intensa alrededor de 1624pb, las cuales corresponden a 1524pb del gen y cerca de
100pb del vector, dado por la alineación del primer T7 antes del lugar donde se
encuentra inserto el gen. Para el caso de OsβGlts 2 se obtuvieron solo dos clones 35 y
47 (figura 37) probándose por la banda intensa en los 1733pb, donde 1533pb
corresponde al gen y 200pb del vector, debido a que solo se emplearon primers T7
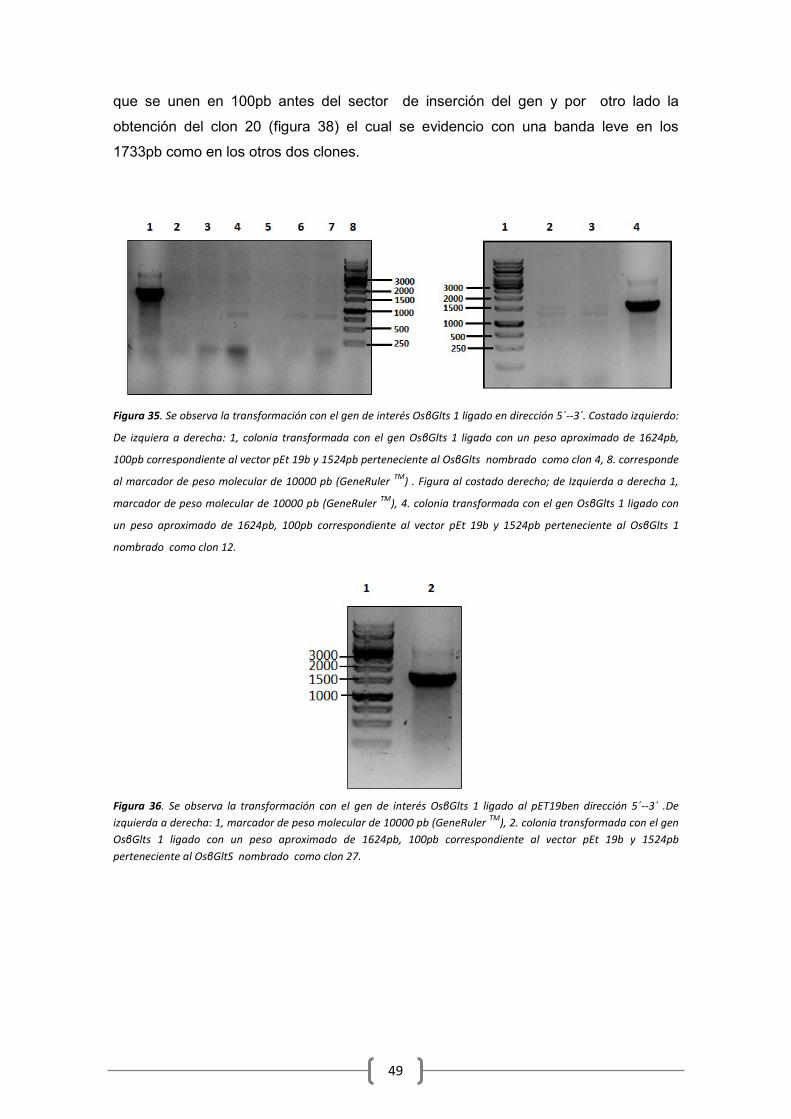
49
que se unen en 100pb antes del sector de inserción del gen y por otro lado la
obtención del clon 20 (figura 38) el cual se evidencio con una banda leve en los
1733pb como en los otros dos clones.
Figura 35. Se observa la transformación con el gen de interés OsβGlts 1 ligado en dirección 5´--3´. Costado izquierdo:
De izquiera a derecha: 1, colonia transformada con el gen OsβGlts 1 ligado con un peso aproximado de 1624pb,
100pb correspondiente al vector pEt 19b y 1524pb perteneciente al OsβGlts nombrado como clon 4, 8. corresponde
al marcador de peso molecular de 10000 pb (GeneRuler TM
) . Figura al costado derecho; de Izquierda a derecha 1,
marcador de peso molecular de 10000 pb (GeneRuler TM
), 4. colonia transformada con el gen OsβGlts 1 ligado con
un peso aproximado de 1624pb, 100pb correspondiente al vector pEt 19b y 1524pb perteneciente al OsβGlts 1
nombrado como clon 12.
Figura 36. Se observa la transformación con el gen de interés OsβGlts 1 ligado al pET19ben dirección 5´--3´ .De
izquierda a derecha: 1, marcador de peso molecular de 10000 pb (GeneRuler TM
), 2. colonia transformada con el gen
OsβGlts 1 ligado con un peso aproximado de 1624pb, 100pb correspondiente al vector pEt 19b y 1524pb
perteneciente al OsβGltS nombrado como clon 27.
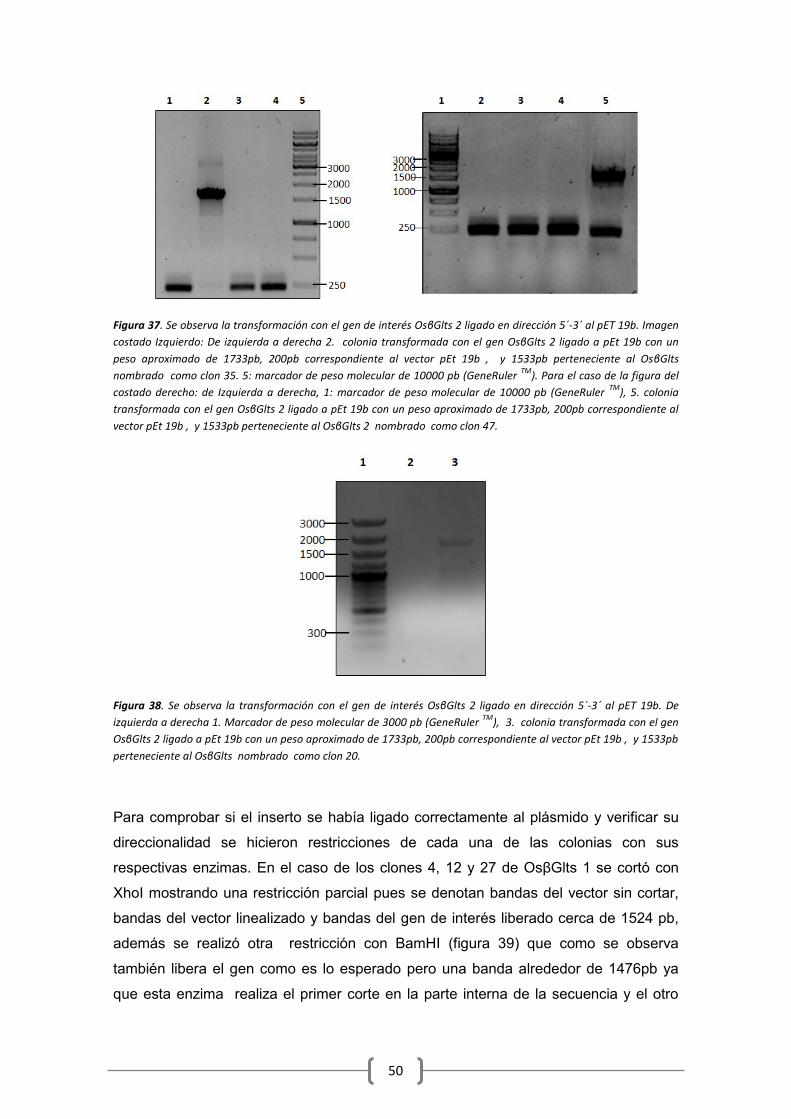
50
Figura 37. Se observa la transformación con el gen de interés OsβGlts 2 ligado en dirección 5´-3´ al pET 19b. Imagen
costado Izquierdo: De izquierda a derecha 2. colonia transformada con el gen OsβGlts 2 ligado a pEt 19b con un
peso aproximado de 1733pb, 200pb correspondiente al vector pEt 19b , y 1533pb perteneciente al OsβGlts
nombrado como clon 35. 5: marcador de peso molecular de 10000 pb (GeneRuler TM
). Para el caso de la figura del
costado derecho: de Izquierda a derecha, 1: marcador de peso molecular de 10000 pb (GeneRuler TM
), 5. colonia
transformada con el gen OsβGlts 2 ligado a pEt 19b con un peso aproximado de 1733pb, 200pb correspondiente al
vector pEt 19b , y 1533pb perteneciente al OsβGlts 2 nombrado como clon 47.
Figura 38. Se observa la transformación con el gen de interés OsβGlts 2 ligado en dirección 5´-3´ al pET 19b. De
izquierda a derecha 1. Marcador de peso molecular de 3000 pb (GeneRuler TM
), 3. colonia transformada con el gen
OsβGlts 2 ligado a pEt 19b con un peso aproximado de 1733pb, 200pb correspondiente al vector pEt 19b , y 1533pb
perteneciente al OsβGlts nombrado como clon 20.
Para comprobar si el inserto se había ligado correctamente al plásmido y verificar su
direccionalidad se hicieron restricciones de cada una de las colonias con sus
respectivas enzimas. En el caso de los clones 4, 12 y 27 de OsβGlts 1 se cortó con
XhoI mostrando una restricción parcial pues se denotan bandas del vector sin cortar,
bandas del vector linealizado y bandas del gen de interés liberado cerca de 1524 pb,
además se realizó otra restricción con BamHI (figura 39) que como se observa
también libera el gen como es lo esperado pero una banda alrededor de 1476pb ya
que esta enzima realiza el primer corte en la parte interna de la secuencia y el otro
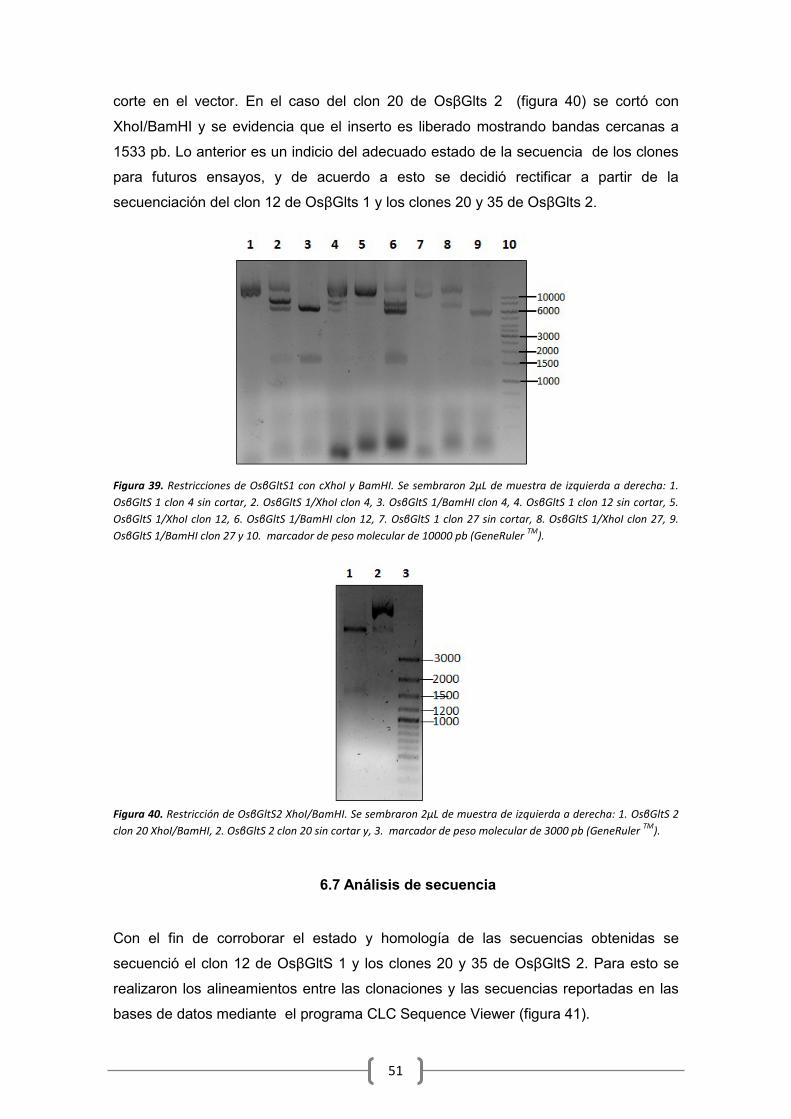
51
corte en el vector. En el caso del clon 20 de OsβGlts 2 (figura 40) se cortó con
XhoI/BamHI y se evidencia que el inserto es liberado mostrando bandas cercanas a
1533 pb. Lo anterior es un indicio del adecuado estado de la secuencia de los clones
para futuros ensayos, y de acuerdo a esto se decidió rectificar a partir de la
secuenciación del clon 12 de OsβGlts 1 y los clones 20 y 35 de OsβGlts 2.
Figura 39. Restricciones de OsβGltS1 con cXhoI y BamHI. Se sembraron 2µL de muestra de izquierda a derecha: 1.
OsβGltS 1 clon 4 sin cortar, 2. OsβGltS 1/XhoI clon 4, 3. OsβGltS 1/BamHI clon 4, 4. OsβGltS 1 clon 12 sin cortar, 5.
OsβGltS 1/XhoI clon 12, 6. OsβGltS 1/BamHI clon 12, 7. OsβGltS 1 clon 27 sin cortar, 8. OsβGltS 1/XhoI clon 27, 9.
OsβGltS 1/BamHI clon 27 y 10. marcador de peso molecular de 10000 pb (GeneRuler TM
).
Figura 40. Restricción de OsβGltS2 XhoI/BamHI. Se sembraron 2µL de muestra de izquierda a derecha: 1. OsβGltS 2
clon 20 XhoI/BamHI, 2. OsβGltS 2 clon 20 sin cortar y, 3. marcador de peso molecular de 3000 pb (GeneRuler TM
).
6.7 Análisis de secuencia
Con el fin de corroborar el estado y homología de las secuencias obtenidas se
secuenció el clon 12 de OsβGltS 1 y los clones 20 y 35 de OsβGltS 2. Para esto se
realizaron los alineamientos entre las clonaciones y las secuencias reportadas en las
bases de datos mediante el programa CLC Sequence Viewer (figura 41).

52
Figura 41. Alineamiento de la secuencia OsβGltS1 Clon 12 y la secuencia putativa LOC4324398 de NCBI en el programa
CLC Sequence Viewer. En el N-terminal y C-terminal se observan los sitios de restricción de XhoI adicionados a los
primers diseñados. La secuencia obtenida presentó 8 cambios (circulo) de nucleótido con respecto a la secuencia
reportada.
XhoI
XhoI

53
Asimismo, con los resultados de la secuenciación se realizó un BLAST en NCBI con el
fin de corroborar la similitud con la isoforma de la β-subunidad de Glutamato Sintasa 1
[NADH] de Oryza Sativa L. (figura 42)
Figura 42. Blast de la secuencia de OsβGltS 1 clonada, reportando una homología del 99% con Oryza sativa Japonica
Group glutamate synthase 1 [NADH], chloroplastic (LOC4324398), transcript variant X1, mRNA, (NCBI). Esta
secuenciación se realizó con primer Forward correspondiente a OsGlts 1, T7 y Reverse T7.

54
Figura 43. BlastX de la secuencia en proteína de OsβGltS 1 clonada, reportando una correlación del 99% con Oryza
sativa Japonica Group glutamate synthase 1 [NADH], chloroplastic (LOC4324398), transcript variant X1, mRNA,
(NCBI).

55
Figura 44. Alineamiento de las secuencias putativas de OsβGltS1 y OsβGltS2 en el programa CLC Sequence Viewer.

56
Figura 45. Blast Global alignment entre la secuencias putativas de OsβGltS 1 y OsβGltS 2, reportando una
correlación del 82% (NCBI).

57
7. DISCUSIÓN
Dado que el arroz es uno de los cereales más producidos a nivel mundial,
corroborándose por la FAO con valores en el 2016 de 2571 millones de toneladas,
ligeramente por encima de lo previsto en octubre y con 1.5% más de lo producido en
2015 (FAO, 2016), es de vital importancia salvaguardar y conservar su cultivo y
producción ante las constantes amenazas que sufre la planta Oryza sativa L. frente a
los cambios climáticas severos en los que hoy se ve inmerso el planeta. Ante esto se
han desarrollado diversas investigaciones en relación al comportamiento de la planta
frente a los factores ambientales a los cuales se ve sometida. Uno de estos estudios
llevados a cabo, está relacionado con la ruta de pirimidinas, donde previas
investigaciones demuestran que esta vía juega un papel importante frente a la síntesis
de ADN y ARN, asimismo la ruta es precursora de la síntesis de celulosa, fosfolípidos
y glicoproteínas, síntesis y degradación de sacarosa y además se conoce que los
productos secundarios obtenidos a partir de su metabolismo pueden ser importantes
en el mecanismo de defensa de las plantas (Ross,1981; Zrenner et al. 2006).
Para la ruta catalítica de pirimidinas, se ha reportado que podría encontrarse
relacionada con la tolerancia al estrés por sequía y salinidad, en especial la primera
enzima DHPD nombrada erróneamente como DHOD (enzima correspondiente a la
ruta anabólica de pirimidinas) en el artículo públicado por Liu y colaboradores en el
2009 y corregida como DHPD en el Laboratorio de Barbara Zimmermann mediante
técnicas moleculares y alineamientos múltiples realizados con diferentes DHPD de
distintas familias de plantas las cuales presentan una correlación por encima del 80%
esto con el fin de comparar estas dos enzimas (DHPD y DHOD), concluyendo que en
la publicación la proteína que realmente se analizó fue la DHPD mencionada
erróneamente como DHOD.
Partiendo de la enzima correcta se investigó sobre DHPD en plantas encontrando que
para estas se reporta tan solo una parte del tamaño de todo el complejo enzimático en
comparación a la DHPD de mamíferos, en el que se desconoce el dominio donde FMN
y NADPH se unen y se da la transferencia de electrones (Zrenner et al., 2006) y donde
la β-subunidad de NADPH-GltS es un posible candidato en la complementación de
este complejo proteico en especie vegetales, seleccionado desde bancos de
secuencias, ya que, arroja una alta identidad entre la β-subunidad de NADPH-GltS y
un dominio de DHPD (Vanoni et al. 2004)

58
Por tanto y a partir de estas investigaciones se clonaron las isoformas de la β-
subunidad de NADH-GltS a partir de la extracción de ARN de plantas de Oryza sativa
L. con aproximadamente 240 y 120 días de germinación con el fin de obtener ADNc y
amplificar los genes correspondientes a las isoformas con primers específicos para
cada gen, esperando un fragmento con 1524pb para OsβGltS 1 y 1533pb para
OsβGltS 2. Posterior a la obtención de las isoformas se clonó en pET-19b en
repetidas ocasiones, puesto que, en el momento de corroborar la transformación el
vector de expresión estaba vacío, es decir, sin la secuencia de interés ligado,
asimismo, para la OsβGltS 1 se obtuvieron tres clones con anterioridad en vector de
clonación pGEM-T Easy, no obstante a la hora de confirmar su inserción mediante
restricción enzimática el gen nunca se liberó lo que generó indicios de que la
secuencia no se encontraba en condiciones pertinentes para futuros estudios, motivos
por los cuales se realizó todo el proceso desde la extracción de ARN nuevamente,
obteniendo los clones 4, 12 y 27 de OsβGltS 1 y aparentemente los clones 35 y 47 de
OsβGltS 2.
Para los clones de OsβGltS 1 se comprobó la correcta direccionalidad a partir del
juego de primers Forward T7 y Reverse correspondiente a OsβGltS 1 (figura 35 y 36),
y también mediante restricción enzimática con XhoI y BamHI. Para XhoI se observa la
banda de la secuencia liberada del vector alrededor de los 1524pb, mientras que para
BamHI se obtuvo una banda cerca de 1476pb (figura 39), esto debido a que la enzima
corta inicialmente en la parte interna de la secuencia más exactamente a las 53pb del
N-Terminal de OsβGltS 1 y el segundo corte a 5pb después de la secuencia por el C-
Terminal, lo que indica que el inserto está ligado correctamente, dado que, si se
hubiera ligado al vector de manera invertida, al hacer el corte la banda liberada sería
de 58pb aproximadamente, es decir, no se apreciaría en la electroforesis.
Con el fin de asegurar la adecuada clonación de la isoforma de OsβGltS 1 se
secuenció el clon 12. La secuencia muestra una homología del 100% en las primeras
618pb según el alineamiento con la secuencia putativa reportada en la base de datos
NCBI, la secuencia restante presento 8 cambios de los cuales 5 fueron gaps
correspondientes a inserciones de nucleótidos en la clonación, 2 transiciones y 1
transversión (figura 41). De igual forma, para la secuencia depurada se llevó acabo
un blast mediante Nucleotide BLAST en NCBI encontrando una correlación del 99%
con Oryza sativa Japonica Group glutamate synthase 1 [NADH], chloroplastic
(LOC4324398), transcript variant X1, mRNA (figura 42), siendo uno de los resultados
esperados en esta investigación. Además, se realizó un BLASTx obteniendo una

59
semejanza del 96% con la secuencia proteica de Glutamato sintasa 1 [NADH],
cloroplástico X1 isoforma Oryza sativa japónica, encontrando 282 aminoácidos iguales
entre las secuencias comparadas (figura 43), sugiriendo que este clon puede ser
empleado en futuros ensayos de interacción proteica y actividad enzimática βGltS 1-
DHPD de Oryza sativa L.
Para OsβGltS 2 se seleccionaron los clones 20 y 35 para su secuenciación
correspondiente con primers T7, no obstante, al momento de realizar el alineamiento
con la secuencia putativa mediante CLC Sequence Viewer, los resultados arrojaron
que el fragmento clonado no correspondía a la isoforma esperada, ya que, se mostró
una alineación de pocas pares de bases desfavoreciendo la caracterización del clon
como OsβGltS 2. Igualmente se corroboró la secuenciación obtenida a través de la
herramienta nucleotide BLAST de NCBI en donde la identidad reportada se presentó
como la isoforma de OsβGltS 1 anteriormente analizada, sin embargo, esta homología
no es representativa para tomarla como base en futuras investigaciones contrario a lo
que mostraron las amplificaciones en donde siempre se obtuvo una banda del tamaño
en pb correspondientes a la isoforma de OsβGltS 2 y la restricción enzimática del clon
20 con XhoI/BamHI arrojó que el segmento liberado del plásmido era alrededor de
1533pb (figura 40), siendo un falso positivo dentro de todo el estudio.
En la amplificación se aseguró la obtención de las dos isoformas, ya que los primers
eran específicos para cada secuencia aunque tuvieran un tamaño en pares de bases
similares con una diferencia de 9pb de más para OsβGltS 2, lo anterior causó que no
se diferenciara la clonación de cada una de las isoformas por medio de electroforesis.
A partir de los inconvenientes presentados en los resultados se alineó OsβGltS 1 y
OsβGltS 2 en CLC Sequence Viewer (figura 44) indicando una alta correspondencia
entre las isofórmas y ratificándose por la base de datos NCBI, encontrando en esta
una semejanza del 82% (figura 45), en donde la similitud más alta se presentó al inicio
de las secuencias. Aunque los primers eran específicos para cada secuencia, entre
estos existía una diferencia mínima de pares de bases principalmente en el forward,
ocasionando que este se lograra unir tanto en una isoforma como en la otra, que para
el caso del estudio se dio en OsβGltS 1, debido a que se encuentra aparentemente en
mayor proporción. Para el caso del reverse de OsβGltS 1 y OsβGltS 2, se observó
una diferencia pronunciada que no fue eficiente en la temperatura a la que se llevaron
a cabo los anillamientos, ya que al ser medianamente parecidos el primer puede
formar híbridos permitiendo la unión con ambas secuencias. Debido a lo anterior y

60
semejanza entre los primers, se sugiere que para próximas amplificaciones se
aumente la temperatura de fusión (Tm), puesto que entre mayor sea la temperatura
mayor será la especificidad de unión entre el primer y la secuencia a clonar evitando
la formación de híbridos en el primer.

61
8. CONCLUSIONES
Se determinaron los parámetros requeridos para clonar un fragmento de
1524pb de β-subunidad de Glutamato Sintasa [NADH] 1 a partir de ADNc
generado de ARN de plantas de arroz.
Las secuencias de las β-subunidad de Glutamato Sintasa [NADH] 1 fue
subclonada en un vector de expresión pET-19b y su confirmación se realizó a
través de PCR, restricción enzimática y secuenciación.
La secuenciación del clon 12 de OsβGltS1 presentó una alta identidad al
compararse con la secuencia putativa, además el fragmento clonado se
comparó en bases de datos bioinformáticas confirmándose la similitud con la β-
subunidad de Glutamato Sintasa [NADH] 1, lo cual ratifica el uso de este clon
para futuros estudios de interacción proteína-proteína entre OsβGltS1 y la
DHPD.
La clonación de la isoforma de la β-subunidad de Glutamato Sintasa [NADH] 1
contribuirá al estudio del complejo enzimático de la Dihidropirimidina
Deshidrogenada en la especie vegetal y al papel que juega ésta en la
tolerancia al estrés abiótico.
La clonación de la β-subunidad de Glutamato Sintasa [NADH] 2 no fue posible
bajo los criterios estipulados, requiriendo un análisis más detallado en los
parámetros necesarios para la adecuada obtención de las secuencia a partir
del ADNc generado de ARN de plantas de arroz.

62
9. ANEXOS
9.1 Preparación de reactivos
9.1.1 Agua con DEPC
Para 1 litro de la solución se adiciona 1mL de Dietil pirocarbonato (DEPC) para que
quede a una concentración final de 0.1% y se afora con agua miliQ, se incuba a
temperatura ambiente durante toda la noche en la campana de extracción y en
agitación constante, se autoclava la solución antes de su uso 3 veces para inactivar el
DEPC.
9.1.2 Medios de Cultivo
9.1.2.1 Medio LB agar (12 cajas de petri)
Se prepara el medio y se autoclava, posteriormente se adiciona 1µL de ampicilina (100
mg/µL) por cada 1mL de medio y se adicionan 20mL a cada caja de petri.
Reactivo Cantidad
Triptona 2.4 g
NaCl 2.4 g
Extracto de levadura 1.2 g
Agar 3.6 g
H2O X
Volumen final 240 mL
9.1.2.2 Medio líquido LB (250 mL)
Después de preparar el medio se autoclava y luego se adiciona 1µL de ampicilina (100
mg/µL) por cada 1mL de medio.
Reactivo Cantidad
Triptona 2.5 g
NaCl 2.5 g
Extracto de levadura 1.25 g
63
H2O X
Volumen final 250 mL
9.1.2.3 Medio líquido SOB-Mg (50 mL)
Al realizarse se deja en agitación hasta la disolución completa de los sólidos, se
autoclava, luego se adicionan 500 µL de MgCl2+MgSO4 (2 M) para obtener una
concentración final de 20 mM y 1µL ampicilina (100 mg/µL) por cada 1 mL de medio.
Reactivo Concentración final Cantidad
Triptona 10 mM 1 g
NaCl (5M) 10 mM 100 µL
Extracto de levadura 0.5 % 0.25 g
KCl (0.5M) 2.5 mM 2.5 mL
H2O X
Volumen final 50 mL
9.1.2.4 Medio SOC
Se prepara el medio SOB-Mg se autoclava, la solución de MgCl2+MgSO4 y glucosa se
adicionan en fresco.
Reactivo Concentración final Cantidad
SOB-Mg Líquido 98 % 100 µL
MgCl2+MgSO4 (2M) 20 mM 500 µL
Glucosa (0.5M) 20 mM 2 mL
Volumen final 50 mL
9.1.3. Geles de agarosa 1%
Para un gel de 15 pozos se adiciona 1 g de agarosa en 100 mL de TAE 1X, se calienta
la solución con el fin de disolver la agarosa y se agregan 5 µL de tinción HydraGreen™
Safe DNA Dye, 20,000X in Water.

64
9.1.3.1 TAE 10X
Se diluye el TAE 10X a una concentración final de 1X para utilizarlo en la preparación
de geles de agarosa y como buffer de corrido.
Reactivo Cantidad
Tris 48.4 g
Ácido acético glacial 11.42 mL
EDTA 0.6M pH:8 20 mL
H2O X
Volumen final 1 L
9.1.4. Células quimiocompetentes
9.1.4.1 TFB (1L)
Se ajusta a pH 6.2 antes de aforar.
Reactivo Concentración final Cantidad
K-MES (0.5M) 10 mM 20 mL
KCl 100 mM 7.4g
MnCl2.4H2O 45 mM 8.9 g
CaCl2.2H2O 10 mM 1.5 g
HACoCl3 3 mM 0.8 g
Volumen final 1 L
9.1.4.2 DnD sol
En 9 mL de DMSO (Dimetilsulfóxido), adicionar 1.53 g de DTT (Ditiotreitol) y 100 µL de
acetato de potasio a 1 M previamente esterilizado y filtrado, ajustar el pH a 7.5 y
alicuotar 0.25 mL en tubos eppendorf. Finalmente se almacena a -80°C.
9.1.5 Soluciones de lisis alcalina
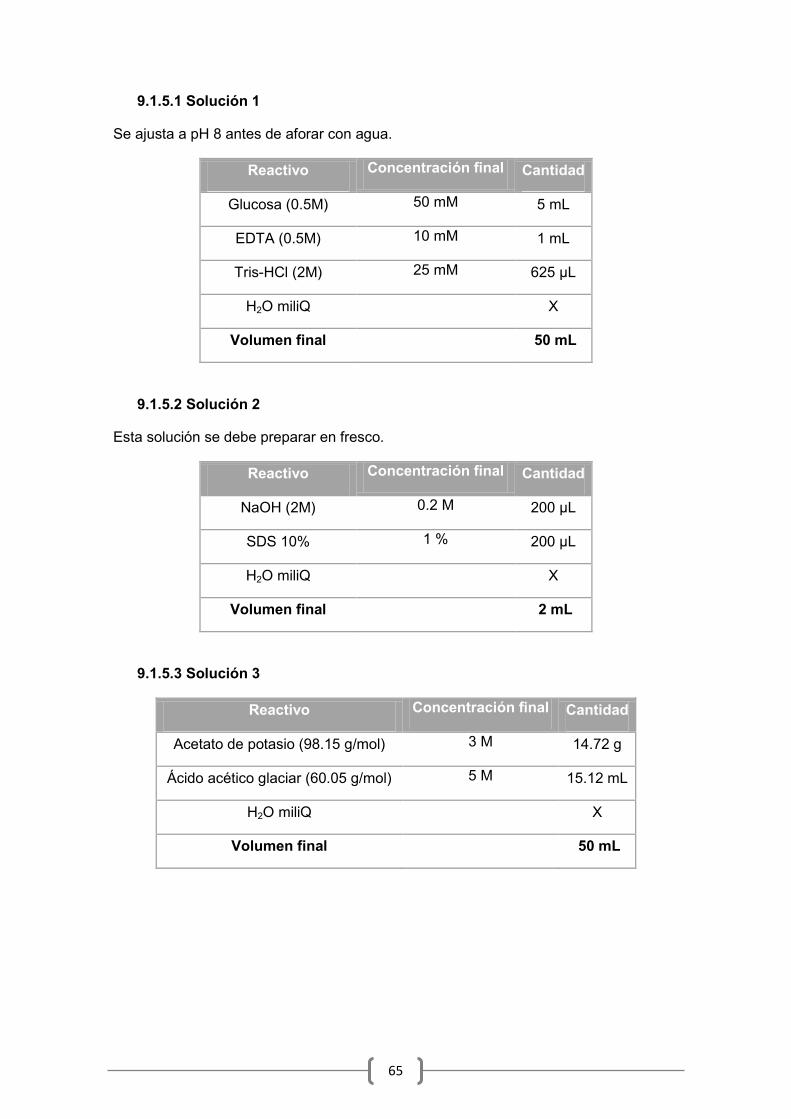
65
9.1.5.1 Solución 1
Se ajusta a pH 8 antes de aforar con agua.
Reactivo Concentración final Cantidad
Glucosa (0.5M) 50 mM 5 mL
EDTA (0.5M) 10 mM 1 mL
Tris-HCl (2M) 25 mM 625 µL
H2O miliQ X
Volumen final 50 mL
9.1.5.2 Solución 2
Esta solución se debe preparar en fresco.
Reactivo Concentración final Cantidad
NaOH (2M) 0.2 M 200 µL
SDS 10% 1 % 200 µL
H2O miliQ X
Volumen final 2 mL
9.1.5.3 Solución 3
Reactivo Concentración final Cantidad
Acetato de potasio (98.15 g/mol) 3 M 14.72 g
Ácido acético glaciar (60.05 g/mol) 5 M 15.12 mL
H2O miliQ X
Volumen final 50 mL

66
10. REFERENCIAS BIBLIOGRÁFICAS
g
Adriana María Salazar Montes, Ana Soledad Sandoval Rodríguez, J. S. A. B. (2013). Vectores de Clonación y Expresión. In Biología Molecular. Fundamentos y aplicaciones en las ciencias de la salud (p. capitulo 15).
Alexis Lamz Piedra, I. D. C. M. C. G. C. (2013). La salinidad como problema en la
agricultura: la mejora vegetal una solución inmediata. Scielo, vol.34 no.(0258–5936). Retrieved from http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0258-59362013000400005
Aymara García , Tania Rodríguez, E. H. y M. N. (2005). EFECTO DEL ANÁLOGO DE
BRASINOESTEROIDE MH-5 EN EL CRECIMIENTO In Vitro DEL ARROZ (Oryza sativa L.) EN CONDICIONES DE DÉFICIT HÍDRICO. Cultivos Tropicales, 26, 89–93. Retrieved from https://www.researchgate.net/profile/Miriam_Nunez/publication/228348787_Efecto_del_analogo_de_brasinoesteroide_MH-5_en_el_crecimiento_in_vitro_del_arroz_Oryza_sativa_L_en_condiciones_de_deficit_hidrico/links/5462bde40cf2cb7e9da654f0.pdf
Belitsky, B. R., Janssen, P. J., & Sonenshein, A. L. (1995). Sites Required for GltC-
Dependent Regulation of Bacillus subtilis Glutamate Synthase Expression, 177(19), 5686–5695.
Biological, S. (2015). Vector pGEM-T: Information and Usage Suggestion (Plasmid
map only for reference). Retrieved from http://www.sinobiological.com/Vector-pGEM-T-a-1636.html
Borras-hidalgo, O. (2005). Molecular aspects of abiotic stress in plants, (January). Bronwyn J. Barkla, R. V.-E. E. B. y O. P. (2007). Mecanismos de tolerancia a la
salinidad en plantas. Cavicchioli, R., Kolesnikow, T., Chiang, R. C., & Gunsalus, R. P. (1996).
Characterization of the aegA Locus of Escherichia coli : Control of Gene Expression in Response to Anaerobiosis and Nitrate, 178(23), 6968–6974.
Centro Internacional de Agricultura Tropiclar CIAT. (2005). MorfologÌa de la Planta de
Arroz. Retrieved from http://www.doc-developpement-durable.org/file/Culture-plantes-alimentaires/FICHES_PLANTES/riz/Morfologia_planta_arroz.pdf
Chen, F., & Cullimore, J. V. (1988). Isoenzymes of NADH-dependent Glutamate, (27),
1411–1417. CIAT. (2005). Morfología de la Planta de Arroz, 16. Cornelius, S., Witz, S., Rolletschek, H., & Möhlmann, T. (2011). Pyrimidine degradation
influences germination seedling growth and production of Arabidopsis seeds. Journal of Experimental Botany, 62(15), 5623–5632. http://doi.org/10.1093/jxb/err251
Correa, H. (2005). Aspectos clave del ciclo de la urea con relación al metabolismo
energético y proteico en vacas lactantes. Retrieved from

67
http://albeitar.portalveterinaria.com/noticia/3487/articulos-rumiantes-archivo/aspectos-clave-del-ciclo-de-la-urea-con-relacion-al-metabolismo-energetico-y-proteico-en-vacas-lactantes.html
Deckert, G., Warren, P. V, Gaasterland, T., Young, W. G., Lenox, A. L., Graham, D. E.,
… Swanson, R. V. (1998). The complete genome of the hyperthermophilic bacterium Aquifex aeolicus, 392(March).
Degiovanni B., V., Martinez R., C. P., & Motta O., F. (2010). Producción Eco’Eficiente
del Arroz en América Latina: Tomo I. Produccion eco-eficiente del arroz en América latina. Retrieved from http://ciat-library.ciat.cgiar.org/articulos_ciat/2010_Degiovanni-Produccion_eco-eficiente_del_arroz.pdf
Division, F. F. and N. (2004). El Arroz y la Nutrición Humana. Food and Agriculture
Organization of the United Nations. Roma. Retrieved from http://www.fao.org/rice2004/es/f-sheet/hoja3.pdf
Dobritzsch, D., Ricagno, S., Schneider, G., Schnackerz, K. D., & Lindqvist, Y. (2002).
Crystal Structure of the Productive Ternary Complex of Dihydropyrimidine Dehydrogenase with NADPH and 5-Iodouracil, 277(15), 13155–13166. http://doi.org/10.1074/jbc.M111877200
Fabricio, P. (2013). Produccion del Metabolismo Nitrogenado. Retrieved from
http://biologia-4to.wikispaces.com/Produccion+del+Metabolismo+Nitrogenado FAO. (2016). Situación Alimentaria Mundial. FAO, O. de las naciones unidas para la alimentacion y la agricultra. (2015). La
agricultura es la mayor afectada por los desastres, según un nuevo informe. Fernández, L. C. S. y R. R. (2010). Mecanismos de transducción de señales en
plantas afectadas por salinidad y sequía, 106 (3),15, 185–286. Retrieved from http://www.aida-itea.org/aida-itea/files/itea/revistas/2010/106-3/ITEA_106-3.pdf#page=3
Ghisla, S. (2008). Dehydrogenation Mechanism in Flavoprotein Catalysis, 133–142. Giermann, N., Zrenner, R., Schröder, M., Giermann, N., & Zrenner, R. (2005).
Functional analysis of the pyrimidine de novo synthesis pathway in solanaceous species. Plant Physiology, 138(4), 1926–38. http://doi.org/10.1104/pp.105.063693
Gill, S. S., & Tuteja, N. (2010). Reactive oxygen species and antioxidant machinery in
abiotic stress tolerance in crop plants. Plant Physiology and Biochemistry, 48(12), 909–930. http://doi.org/10.1016/j.plaphy.2010.08.016
Goff, S. A., Ricke, D., Lan, T.-H., Presting, G., Wang, R., Dunn, M., … Briggs, S.
(2002). A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science (New York, N.Y.), 296(5565), 92–100. http://doi.org/10.1126/science.1068275
Hirayama, C., Saito, H., Konno, K., & Shinbo, H. (1998). Purification and
characterization of NADH-dependent glutamate synthase from the silkworm fat body (Bombyx mori). Insect Biochemistry and Molecular Biology, 28(7), 473–482. http://doi.org/10.1016/S0965-1748(98)00019-8

68
Houghton JT, Y, D., DJ, G., M, N., PJ, van der L., X, D., … C, J. (2001). Climate
Change 2001: The Scientific Basis. Climate Change 2001: The Scientific Basis, 881. http://doi.org/10.1256/004316502320517344
Ibarguren Leandra. (2015). ARROZ - Oryza sativa, 1–8. Ingram, J., & Bartels, D. (1996). the Molecular Basis of Dehydration Tolerance in
Plants. Annual Review of Plant Physiology and Plant Molecular Biology, 47, 377–403. http://doi.org/10.1146/annurev.arplant.47.1.377
Itakura, M., Sabina, R. L., Heald, P. W., & Holmes, E. W. (1981). Basis for the control
of purine biosynthesis by purine ribonucleotides. Journal of Clinical Investigation, 67(4), 994–1002.
Josep Peñuelas, Santi Sabaté, I. F. Y. C. G. (2004). CAPÍTULO 15 Efectos del cambio
climático sobre los ecosistemas terrestres : observación , experimentación y simulación *. Ecologia del bosque mediterraneo en un mundo cambiante.
Joseph Sambrook, & Green, M. R. (2012). Molecular Cloning (Vol. 1). Knight, H., & Knight, M. R. (2001). Abiotic stress signalling pathways: Specificity and
cross-talk. Trends in Plant Science, 6(6), 262–267. http://doi.org/10.1016/S1360-1385(01)01946-X
Labrín Sotomayor, Y. N. (2007). Estudio de la resistencia en variedades de arroz
(Oryza sativa L.) venezolanas al virus de la hoja blanca, 84. Lim, E.-K., & Bowles, D. J. (2004). A class of plant glycosyltransferases involved in
cellular homeostasis. The EMBO Journal, 23(15), 2915–22. http://doi.org/10.1038/sj.emboj.7600295
Liu, W. Y., Wang, M. M., Huang, J., Tang, H. J., Lan, H. X., & Zhang, H. S. (2009). The
OsDHODH1 gene is involved in salt and drought tolerance in rice. Journal of Integrative Plant Biology, 51(9), 825–833. http://doi.org/10.1111/j.1744-7909.2009.00853.x
Lohkamp, B., Voevodskaya, N., Lindqvist, Y., & Dobritzsch, D. (2010). Insights into the
mechanism of dihydropyrimidine dehydrogenase from site-directed mutagenesis targeting the active site loop and redox cofactor coordination. Biochimica et Biophysica Acta - Proteins and Proteomics, 1804(12), 2198–2206. http://doi.org/10.1016/j.bbapap.2010.08.014
Martinez Covaleda, H., & Acevedo Gaitán, X. (2004). Caracteristicas y Estructura de la
Cadena de Arroz en Colombia. Ministerio de Agricultura Y Desarrollo Rural. Retrieved from https://books.google.com.co/books?id=IkqcUqIvndAC&pg=PA2&lpg=PA2&dq=En
+el+a�o+2002,+el+arroz+en+Colombia+se+cultiv�+en+468.906+hect�reas+(H
a)+que+rindieron+2.347.917+toneladas+m�tricas+(Tm)+de+paddy+y+aproximadamente+1.526.146+Tm+de+arroz+blanco.+El+arroz
Melorose, J., Perroy, R., & Careas, S. (2015). No Title No Title. Statewide Agricultural
Land Use Baseline 2015, 1. http://doi.org/10.1017/CBO9781107415324.004 MIOBIO. (2016). UltraClean Plant RNA Isolation Kit.

69
Morales, D.; Bolarín, María del C.; Cayuela, E. (2006). RESPUESTA DE PLANTAS DE
ARROZ (Oryza sativa L.) A LA APLICACIÓN DE DIFERENTES NIVELES DE NaCl. I. CRECIMIENTO Y RELACIONES HÍDRICAS. Instituto Nacional de Ciencias Agricolas. Cuba, 27, 27–32. Retrieved from http://www.redalyc.org/pdf/1932/193215912004.pdf
MSU, R. G. A. P. (2016). Glutamate synthase Oryza sativa Japonica. Nakashima, K., Ito, Y., & Yamaguchi-Shinozaki, K. (2009). Transcriptional regulatory
networks in response to abiotic stresses in Arabidopsis and grasses. Plant Physiology, 149(January), 88–95. http://doi.org/10.1104/pp.108.129791
Nakayama. (1974). Panicle senescence in rice plant. Bulletin of the Hokiriku National
Agricultural Experimental Station, 16, 15–17. Nara, T., Hshimoto, T., & Aoki, T. (2000). Evolutionary implications of the mosaic
pyrimidine-biosynthetic pathway in eukaryotes. Gene, 257(2), 209–222. http://doi.org/10.1016/S0378-1119(00)00411-X
Navarro, F., Candau, P., & Florencio, F. J. (1995). and gltS genes, 753–767. NCBI. (2016). Dihydropyrimidine dehydrogenase (NADP(+)), chloroplastic [Oryza
sativa Japonica Group]. Nelson, G. C., Rosegrant, M. W., Koo, J., Robertson, R., Sulser, T., Zhu, T., … Lee, D.
(2009). POLÍTICA ALIM E N T A R I A Cambio Climático. Instituto Internacional de Investigación Sobre Políticas Alimentarias IFPRI.
Nilesh. (2016). Ammonia Assimilation. Novagen. (2016). pET19b Vector. Oliveira, I. C., Lam, H. M., Coschigano, K., Melo-Oliveira, R., & Coruzzi, G. (1997).
Molecular-genetic dissection of ammonium assimilation in Arabidopsis thaliana. Plant Physiology and Biochemistry, 3(35), 185–198.
Oliver, G., Gosset, G., Sanchez-pescador, R., Lozoya, E., Becerril, B., Valle, F., … Ku,
L. M. (1987). for the glutamate synthase structural genes of Escherichia, 60. R. Serraj, A. Kumar, K. L. McNally, I. Slamet-Loedin, R. B., & R. Mauleon, J. Cairns,
and R. J. H. (2009). Improvement of Drought Resistance in Rice. Retrieved from https://www.researchgate.net/profile/Rachid_Serraj/publication/229088442_Chapter_2_Improvement_of_Drought_Resistance_in_Rice/links/542966740cf26120b7b6e5fe.pdf
Ritchie, J., & Smucker, A. J. M. (2000). LA INTERCEPCIÓN DE LUZ EN FRIJOL
COMÚN 1, 9(2), 1–8. Robles, A. A. C. (2007). Sobrevivir al estrés: cómo responden las plantas a la falta de
agua. Biotecnologia, 14, 253–262. Rosenbaum, K., Jahnke, K., Curti, B., Hagen, W. R., Schnackerz, K. D., & Vanoni, M.
A. (1998). Porcine Recombinant Dihydropyrimidine Dehydrogenase : Comparison of the Spectroscopic and Catalytic Properties of the Wild-Type and C671A Mutant,

70
17598–17609. Schreiert, H. J., & Bernlohr, R. W. (1984). Purification and Properties of Glutamate
Synthase from Bacillus licheniformis, (42), 591–599. Singh, A. K., Ansari, M. W., Pareek, A., & Singla-Pareek, S. L. (2008). Raising salinity
tolerant rice: Recent progress and future perspectives. Physiology and Molecular Biology of Plants, 14(1–2), 137–154. http://doi.org/10.1007/s12298-008-0013-3
Soren, K. R., Ali, K., Tyagi, V., & Tyagi, A. (2010). Recent advances in molecular
breeding of drought tolerance in rice (Oryza sativa L.). Indian Journal of Biotechnology, 9(3), 233–251.
Stasolla, Claudio, Riko Katahira, Trevor A. Thorpe Ashihara, H. (2003). Purine and
pyrimidine nucleotide metabolism in higher plants. Journal of Plant Physiology. Retrieved from http://sci-hub.cc/10.1078/0176-1617-01169
Stephen J. Temple, Carroll P. Vance, S. G. (1998). Glutamate synthase and nitrogen
assimilation.pdf, 3(2), 51–56. Tanji, kenneth. (2002). Salinity in the Soil Environment. Salinity: Environment - Plants -
Molecules, (1995), 21–51. Tascón, E., & Garcfa, E. (1985). Arroz: Investigación y Producción (pp. 47–48).
Retrieved from http://ciat-library.ciat.cgiar.org/Articulos_Ciat/Digital/SB191.R5_A7_C3_Arroz_Investigación_y_producción_Referencia_de_los_cursos_de_capacitación_sobre_ar.pdf#page=57
Technologies, A. (2016). pET Expression Systems - Details & Specifications. Retrieved
from http://www.genomics.agilent.com/article.jsp?pageId=472&_requestid=672836
Unidas, O. de las N. U. para la alimentación y la A. (2014). Cultivar maíz, arroz y trigo
de forma más sostenible. Retrieved from http://www.fao.org/news/story/es/item/273338/icode/
Valladares, F., Vilagrosa, A., Peñuelas, J., Ogaya, R., Julio, J., Corcuera, L., & Sisó, S.
(2004). CAPÍTULO 6 Estrés hídrico : ecofisiología y escalas de la sequía. Water, (OCTOBER 2014), 163–190.
Vanoni, M. A., & Curti, B. (1999). Glutamate synthase: A complex iron-sulfur
flavoprotein. Cellular and Molecular Life Sciences, 55(4), 617–638. http://doi.org/10.1007/s000180050319
Vanoni, M. A., Dossena, L., Van Den Heuvel, R. H. H., & Curti, B. (2005). Structure-
function studies on the complex iron-sulfur flavoprotein glutamate synthase: The key enzyme of ammonia assimilation. Photosynthesis Research, 83(2), 219–238. http://doi.org/10.1007/s11120-004-2438-z
Vidal, a M. (2010). Respuestas fisiológicas de los cítricos sometidos a condiciones de
estrés biótico y abiótico. Aspectos comunes y específicos. TDX (Tesis Doctorals En Xarxa), 213.
Y, S. de A., & (SAG), G. (2003). MANUAL TÉCNICO PARA EL CULTIVO DE ARROZ.

71
(ORYZA SATIVA) (PARA EXTENSIONISTAS Y PRODUCTORES). Comayagua, Honduras. Retrieved from https://curlacavunah.files.wordpress.com/2010/04/el-cultivo-del-arroz.pdf
Yanelis ReyesI, D. C. L. M. M. y D. C. M. N. (2008). Aspectos fisiológicos y
bioquímicos de la tolerancia del arroz al estrés salino y su relación con los brasinoesteroides. Scielo, v.29 n.4. Retrieved from http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0258-59362008000400010
Yu, J. (2002). A Draft Sequence of the Rice Genome (Oryza sativa L. ssp. indica).
Science, 296(5565), 79–92. http://doi.org/10.1126/science.1068037 Zhao, K., Wright, M., Kimball, J., Eizenga, G., McClung, A., Kovach, M., … McCouch,
S. R. (2010). Genomic diversity and introgression in O. sativa reveal the impact of domestication and breeding on the rice genome. PLoS ONE, 5(5). http://doi.org/10.1371/journal.pone.0010780
Zhu, J. K. (2001). Plant salt tolerance. Trends in Plant Science, 6(2), 66–71.
http://doi.org/10.1016/S1360-1385(00)01838-0 Zrenner, R., Riegler, H., Marquard, C. R., Lange, P. R., Geserick, C., Bartosz, C. E., …
Slocum, R. D. (2009). A functional analysis of the pyrimidine catabolic pathway in Arabidopsis. New Phytologist, 183(1), 117–132. http://doi.org/10.1111/j.1469-8137.2009.02843.x
Zrenner, R., Stitt, M., Sonnewald, U., & Boldt, R. (2006). Pyrimidine and Purine
Biosynthesis and Degradation in Plants. Annual Review of Plant Biology, 57(1), 805–836. http://doi.org/10.1146/annurev.arplant.57.032905.105421


















