
Cromosomopatias (Genética Médica)
46
CROMOSOMOPATÍAS:
-
Upload
bryan-fernando-reyes -
Category
Health & Medicine
-
view
1.788 -
download
9
description
Cromosomopatias (Genética Médica)
Transcript of Cromosomopatias (Genética Médica)

CROMOSOMOPATÍAS:
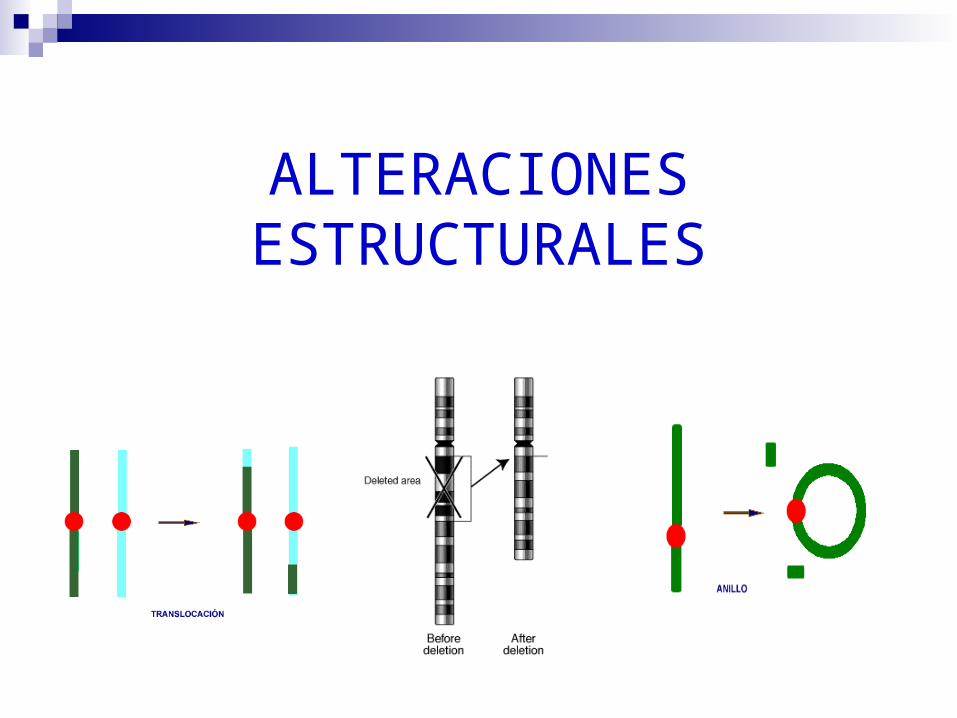
ALTERACIONES ESTRUCTURALES

DELECIONES
(MONOSOMÍAS PARCIALES AUTOSÓMICAS)

Síndrome de Cri du Chat: Alteración : pérdida parcial del
brazo corto del cromosoma 5 46,XX, (5)(p-) 46, XY, (5)(p-)
Cuadro clínico: Bajo peso al nacer, cara redonda, crecimiento lento, llanto como maullido de gato, retraso mental severo, microcefalia, hipotonía, estrabismo, orejas mal formadas y de implantación baja, fisuras palpebrales bajas, pliegue de simio, micrognatia, 30% presentan hernias inguinales, diastasis rectal, miopía, puede haber ausencia de bazo o riñón.
Diagnostico: amniocentesis, llanto como maullido de gato, pruebas citogenéticas, cariotipo.
Tratamiento: No hay tratamiento se aborda el retraso mental y asesoría a los padres.

Síndrome de Wolf-Hirschhorn: Alteración : pérdida parcial del
brazo corto del cromosoma 4 46,XX, (4)(p-) 46,XY, (4)(p-)
Cuadro clínico: retraso mental y del crecimiento, problemas del lenguaje graves, convulsiones, microcefalia, hipertonía en lactancia luego hipotonía, paladar hendido, clinodactilia del 5to dedo, frente prominente, nariz ancha, puente nasal deprimido, macroglosia, uñas hipoplásicas, orejas mal formadas y de implantación baja, estenosis aórtica pulmonar. Varones micro- pene y criptorquidia.
Diagnostico: amniocentesis, pruebas citogenéticas y cariotipo.
Tratamiento: No hay, se tratan los síntomas y se asesora a los padres.

MICRODELECIONES

Síndrome de Angelman o Síndrome de la Muñeca Feliz: Alteración: microdeleccion de la
región 15q11-13 = 46,XX,del (15) (q11-13).
Cuadro clínico: retraso mental, del desarrollo motor grave, ataque de risa inadecuados, carencia del habla, ataxia tipo marioneta, convulsiones, hiperactividad, disminución del pigmento del coroides y del iris, boca grande y lengua salida, dientes separados.
Diagnostico: hibridación in situ (Fish: análisis con sondas de ADN (fluorescencia).
Tratamiento: No hay; se tratan síntomas por separados y se asesora a los padre.
Impronta genómica: origen materno Disomía Uniparental: origen paterno

Síndrome de Prader Willi Alteración: microdeleccion 15q11-13
= 46,XX,del(15) (q11-13) o 46,XY,del (15) (q11-13).
Cuadro clínico: retardo en el aprendizaje, talla pequeña, obesidad morbosa con alimentación obsesiva por falta de control de la saciedad, hipotonía grave en la infancia, manos y pies pequeños, hipogonadismo, micropené, criptorquidia, hipopigmen- tación pueden ser rubios o cabello café claro con ojos azules, facies típicas, problemas de conducta a medida que crecen.
Diagnostico: hibridación in situ, Fish, análisis de sondas de ADN.
Tratamiento: se tratan los síntomas por separado, terapia temprana con testosterona ayuda al desarrollo del micropené, asesorar a los padres.
Impronta genómica: origen paterno Disomía Uniparental: origen materno

Síndrome de Williams Alteración: microdeleccion de la región
7q11.23 = 46,XX,del (7) (q11.23) o 46,XY,del (7) (q11.23). Relacionado con gen de elastina.
Cuadro clínico: cara de duendecillo o gnomo, estatura baja, retraso mental leve, reflujo, cólicos, hipercalsemia, sensibilidad al sonido (hiperacustica), voz ronca, función motora reducida, puente nasal aplanado, frente pronunciada y alta, filtro largo, labios prominentes con boca grande abierta, uñas hipoplásicas, hernias, mal oclusion dentaria, estenosis aórtica supravalvular, personalidad impulsiva y elocuente.
Diagnostico: Hibridacion in situ, Fish, Fluorescencia y prueba para comprobar la hipercalcemia.
Tratamiento: correccion quirurgica del problema cardiaco, si hay hipercalcemia evitar la vit D y el calcio de la dieta, integracion a la sociedad.

Síndrome de Di George o Sind. del Velo-Cardio-Facial.
Alteración: microdeleción de la región 22q11.2 = 46,XX,del(22) (q11.2) 0 46,XY,del(22) (q11.2).
Cuadro clínico: hipoplasia o ausencia del timo y las paratiroides lo cual causa hipocalcemia grave y convulsiones en la infancia, malformaciones del arco aórtico, aorta interrumpida, baja producción de células T, hipertelorismo, paladar hendido, anomalías del pabellón auricular, fisuras palpebrales inclinadas.Espasmos dolorosos, mayor frecuencia de infecciones, cardiopatías congénitas.
Diagnóstico: Hibridación in situ, Fish, fluorescencia, Cardiografías.
Tratamiento: terapia enfocada a corregir defectos en órganos y tejidos afectados, suplementos de calcio, reemplazo de hormona paratiroides.

Aniridia o Tumor de Wilms: Alteración: microdeleción de la región
11p13.
46, XX del (11)(p13)
46 ,XY del (11)(p13)
Cuadro clínico: Aniridia (ausencia del iris) , tumor de Wilms (,tipo de tumor renal más común) retardo mental, defectos genitales, malformaciones del tracto urinario, hemihipertrofia.
Diagnóstico: Análisis con pruebas citogenéticas avanzadas (Hibridación in situ, Fish), Análisis con sondas de ADN (fluorescencia).
Biopsias.
Tratamiento: Cirugía según sintomatología.

Síndrome de Miller Dieker Alteración cromo.: microdeleccion de
la región 17p13.3 = 46,XX del (17) (p13.3) o 46,XY del (17) (p13.3).
Cuadro clínico: lisencefalia (ausencia de las circunvoluciones de la corteza cerebral), retraso mental, microcefalia, frente prominente, nariz pequeña con narinas ante vertidas, labio superior prominente, erupción tardía de los dientes primarios, retraso psicomotor grave, hipotonía, cardiopatías, criptorquidia, clinodactilia, anomalías del iris, convulsiones.
Diagnostico: Resonancia nuclear magnética, patrón electroesenfalico, biopsia, estudio citogenética avanzado.
Tratamiento: No tratamiento curativo
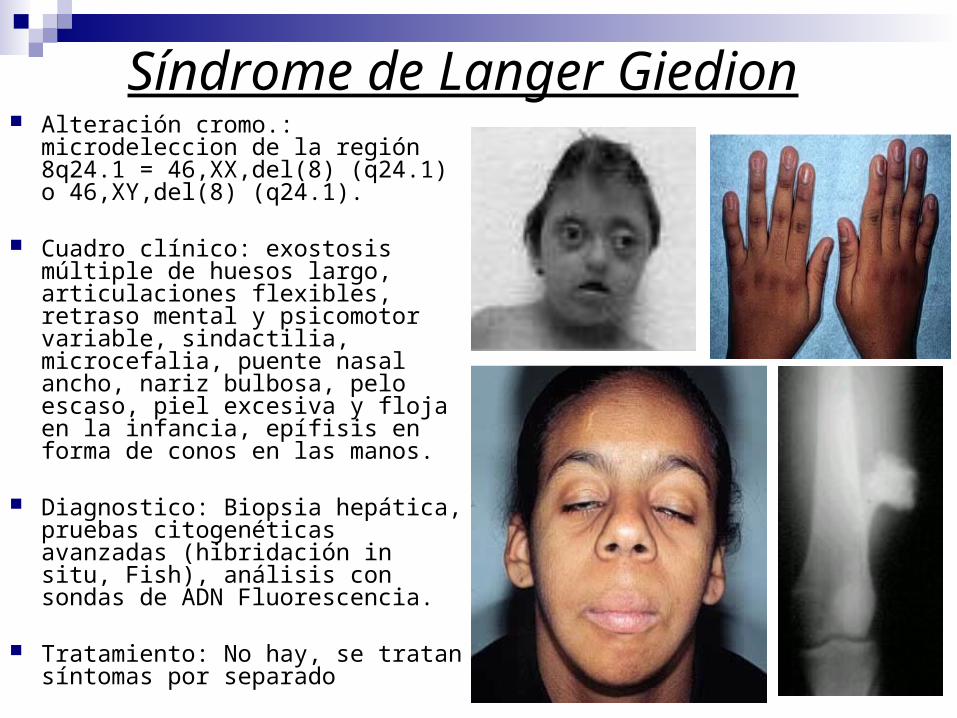
Síndrome de Langer Giedion Alteración cromo.: microdeleccion
de la región 8q24.1 = 46,XX,del(8) (q24.1) o 46,XY,del(8) (q24.1).
Cuadro clínico: exostosis múltiple de huesos largo, articulaciones flexibles, retraso mental y psicomotor variable, sindactilia, microcefalia, puente nasal ancho, nariz bulbosa, pelo escaso, piel excesiva y floja en la infancia, epífisis en forma de conos en las manos.
Diagnostico: Biopsia hepática, pruebas citogenéticas avanzadas (hibridación in situ, Fish), análisis con sondas de ADN Fluorescencia.
Tratamiento: No hay, se tratan síntomas por separado

Otras Alteraciones Estructurales:
TRANSLOCACIONES: Translocación robert- soniana más común S. Down
ISOCROMOSOMAS: S. Turner
CROMOSOMAS EN S. Turner
ANILLOS:

ALTERACIONES NUMÉRICAS

MONOSOMIAS GONOSÓMICAS

Síndrome de Turner Alteración cromo.: perdida del
cromosoma sexual x (45,X).
Cuadro clínico: disgenesia del ovario con hipoplasia, talla baja, obesas, implantación baja del cabello, cara triangular, tórax ancho y pezones separados, genitales externos infantiles labios menores hipoplásicos e hipertrofia del clítoris, estériles, escaso o nulo desarrollo de las mamas, pterigium colli, NO presentan retraso mental. OTROS SÍNTOMAS: anormalidades de las rodillas, displasia de los huesos, riñón en herradura, pelvis renal doble, malformaciones cardiacas.
Diagnostico: Fetal: linfedema de la cabeza, Al nacer: linfedema en manos y pies, implantación baja del cabello, Adulto: sintomatología, frotis bucal y cariotipo.
Tratamiento: estrógenos, hormonas del crecimiento, extirpación de las gónadas, y apoyo psicosocial.
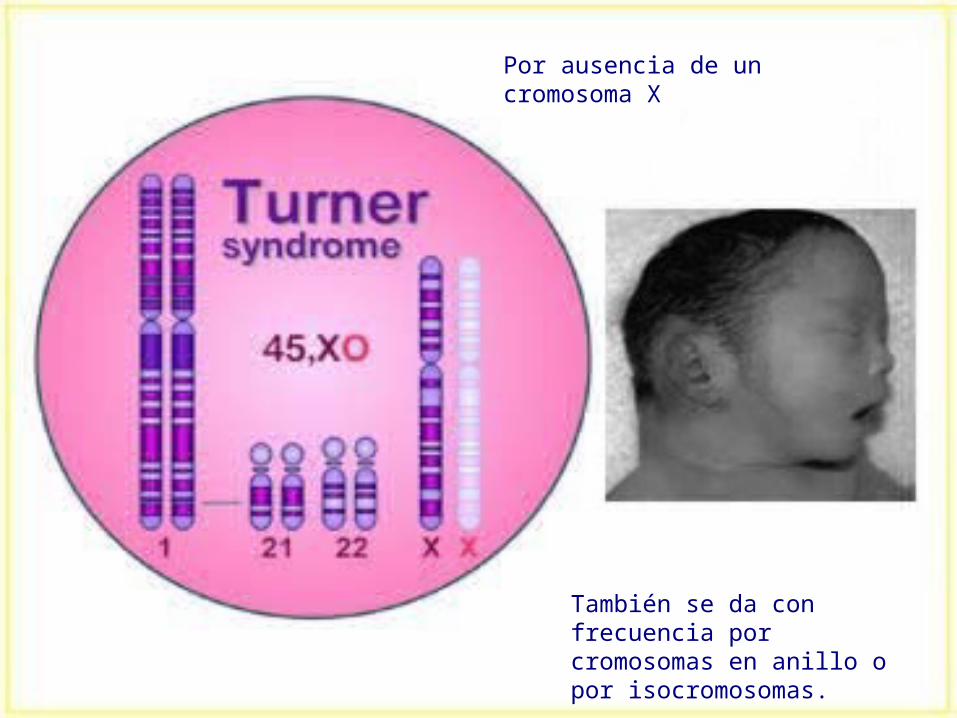
También se da con frecuencia por cromosomas en anillo o por isocromosomas.
Por ausencia de un cromosoma X


TRISOMÍAS AUTOSÓMICAS

Estos síndromes se presentan como:
Trisomías → 80 – 85 %
Translocaciones → 10 – 15 %
Mosaicos → 5 %

Trisomía 21 o Sindrome de Down Alteración cromo.: cromosoma 21
adicional :47,XX +21, 47,XY +21 o traslocado el 14;21 = 46,XX, (14;21) o 46,XY, t(14;21)
Cuadro clínico: retraso mental, perfil facial aplanado, pliegues palpebrales, pliegue palmar de simio, piel abundante en cuello, clinodactilia, manos cortas y anchas, orejas pequeñas y redondas, puente nasal aplanado, ausencia del reflejo de moro, gran espacio entre el 1er y 2do dedo del pie, manchas de BRUSHFIELD en iris, Varones con criptorquidia y estériles, puede presentar cardiopatías congénitas, leucemia, hipotiroidismo, Alzheimer en la adultez.
Diagnostico: Ecografías, Amniocentesis, cariotipo y prueba triple pre natal.
Tratamiento: cirugías cardiacas, asesoramiento a padres. Estimulación temprana disminuye defectos en desarrollo psicomotor.



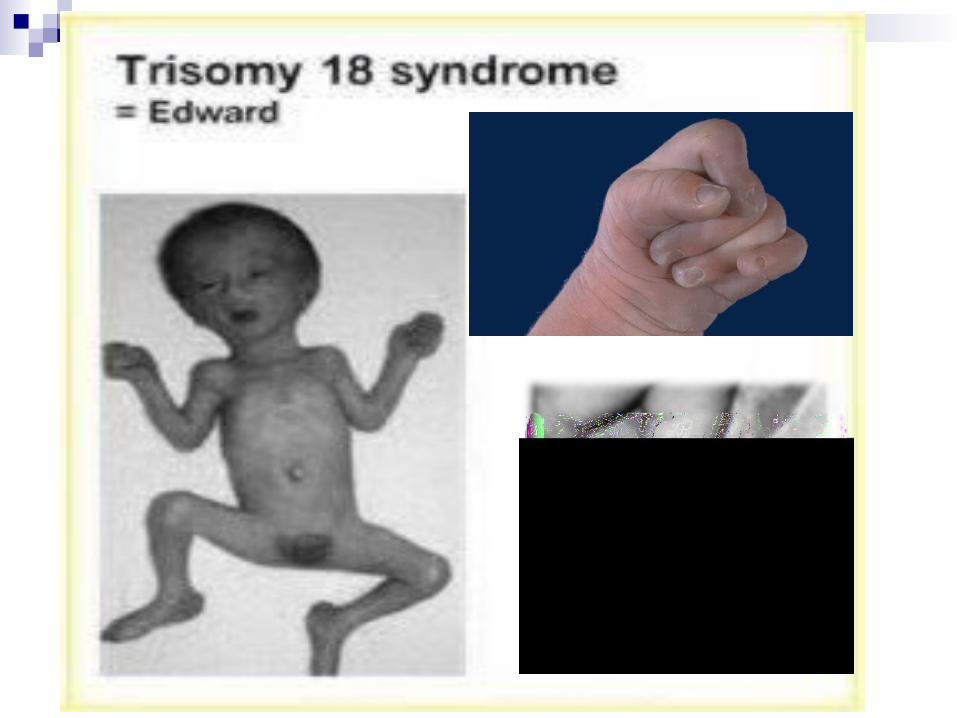
Trisomía 18 o Sindrome de Edwards Alteración cromo.: cromosoma 18
adicional (47,XX +18 o 47,XY +18)
Cuadro clínico: Bajo peso al nacer, retraso mental y del crecimiento, occipucio prominente, orejas hipoplásicas y de implantación baja, micro y retrognatia, esternón corto, pezones pequeños, mano empuñada, hipoplasia de las uñas, hipotonía general, abducción limitada de la cadera, pies en mecedora, criptorquidia en varones, hipertrofia de clítoris en mujeres, cardiopatías congénitas, riñón en herradura, páncreas ectópico, cuello corto, apnea, anormalidad en dermatoglifos
Mueren antes de los dos años de vida
Diagnostico: estudio de dermatoglifos,(huellas digitales) amniocentesis y cariotipo.
Tratamiento: No hay, asesoría a los padres

Trisonomia 13 o Síndrome de Patau Alteración cromo.: cromosoma 13
adicional (47,XX +13 o 47,XY +13).
Cuadro clínico: Bajo peso al nacer, holoprosencefalia, retraso mental grave, frente inclinada, orejas hipoplásicas y de implantación baja, micrognatia, microcefalia, lesiones posteriores del cráneo, labio leporino y paladar hendido, microftalmia, polidactilia posaxial, pies en mecedora, criptorquidia en varones, útero bicorne y vagina doble en mujeres, cardiopatías congénitas, riñón poliquístico en herradura.
Raramente llegan al año de vida.
Diagnostico: amniocentesis y cariotipo.
Tratamiento: No hay, asesoría a los padres.
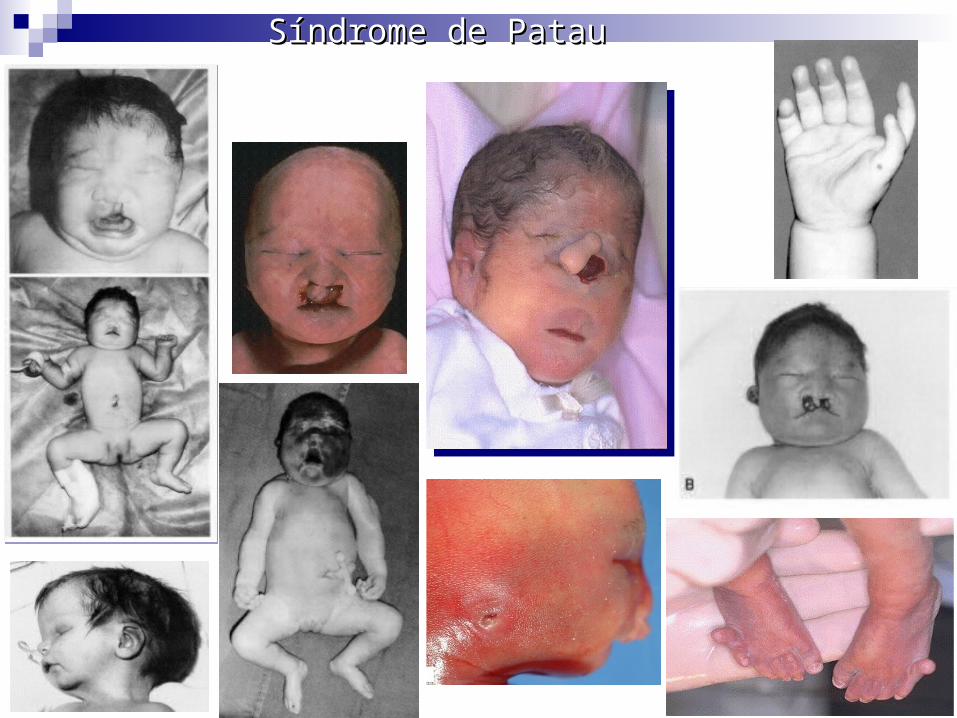
Síndrome de PatauSíndrome de PatauSíndrome de PatauSíndrome de Patau
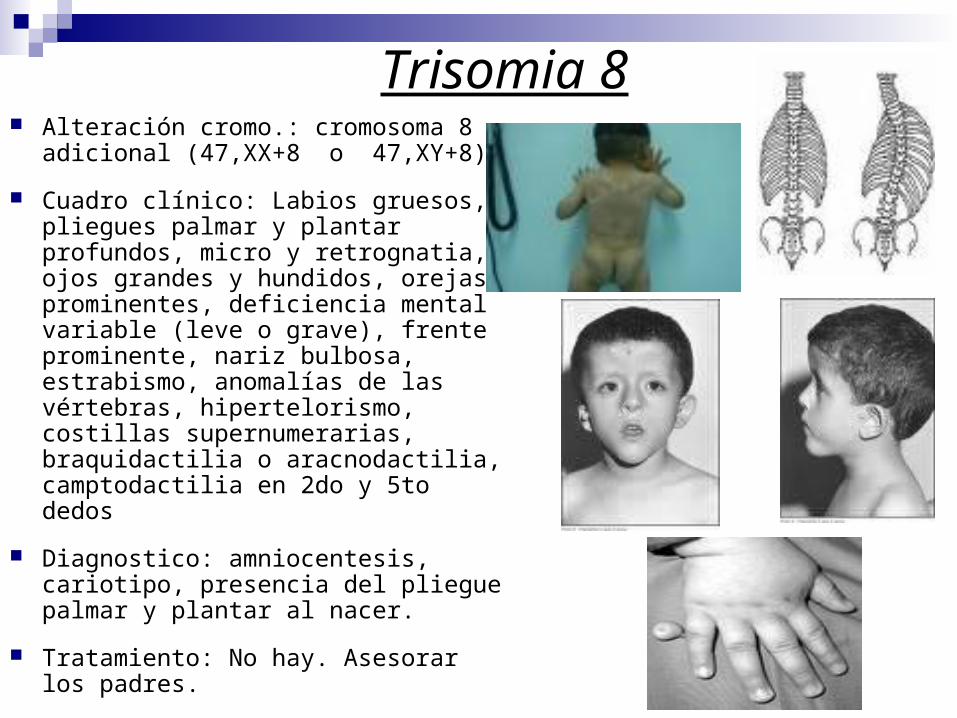
Trisomia 8 Alteración cromo.: cromosoma 8
adicional (47,XX+8 o 47,XY+8).
Cuadro clínico: Labios gruesos, pliegues palmar y plantar profundos, micro y retrognatia, ojos grandes y hundidos, orejas prominentes, deficiencia mental variable (leve o grave), frente prominente, nariz bulbosa, estrabismo, anomalías de las vértebras, hipertelorismo, costillas supernumerarias, braquidactilia o aracnodactilia, camptodactilia en 2do y 5to dedos
Diagnostico: amniocentesis, cariotipo, presencia del pliegue palmar y plantar al nacer.
Tratamiento: No hay. Asesorar los padres.
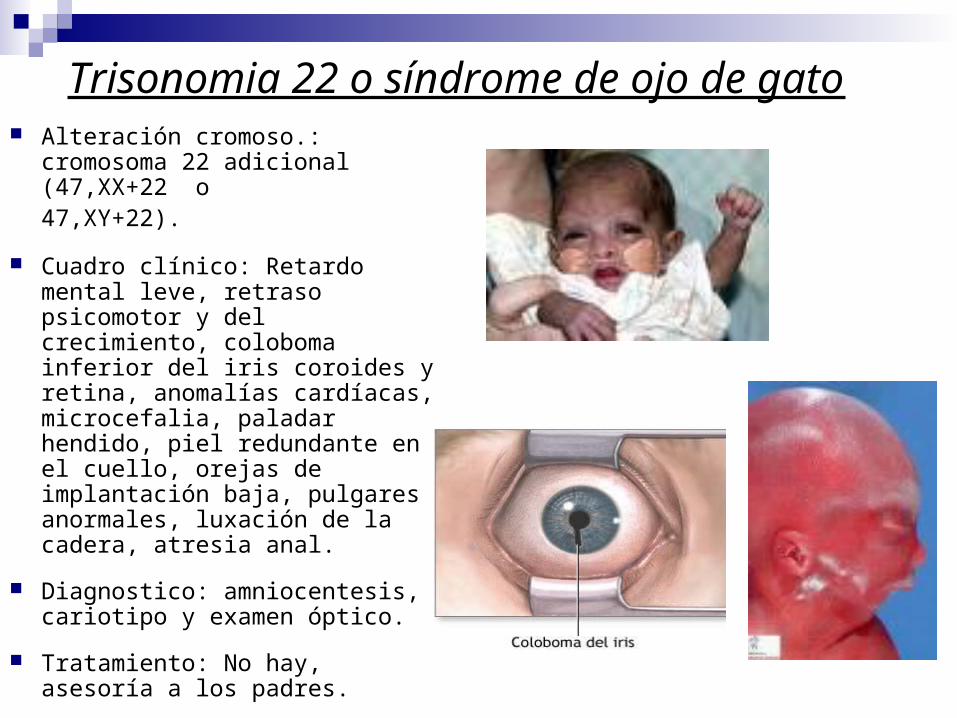
Trisonomia 22 o síndrome de ojo de gato Alteración cromoso.: cromosoma
22 adicional (47,XX+22 o 47,XY+22).
Cuadro clínico: Retardo mental leve, retraso psicomotor y del crecimiento, coloboma inferior del iris coroides y retina, anomalías cardíacas, microcefalia, paladar hendido, piel redundante en el cuello, orejas de implantación baja, pulgares anormales, luxación de la cadera, atresia anal.
Diagnostico: amniocentesis, cariotipo y examen óptico.
Tratamiento: No hay, asesoría a los padres.

TRISOMÍAS GONOSOMICAS

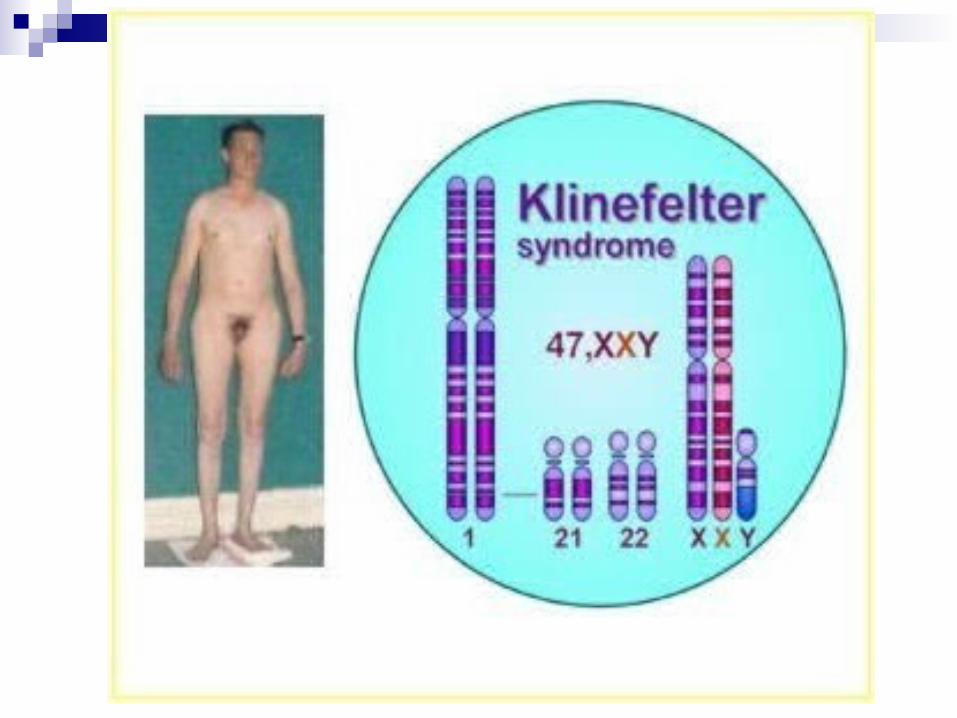
Síndrome de Klinefelter Alteración cromoso.: cromosoma X
adicional en un varón (47,XXY).
Cuadro clínico: Piernas anormalmente largas, altos y delgados, hipogonadismo, testículos pequeños y atróficos, pene pequeño, azoospermia, por lo general infértiles (se presentan excepciones en la adolescencia), virilización parcial e inadecuada, vello corporal escaso y fino, desarrollo ausente o bajo de características sexuales 2rias, ginecomastia, desarrollo intelectual variable, aunque la mayoría NO presenta retraso mental.
Diagnostico: infertilidad en la adolescencia, frotis bucal (cromatina positiva), medición de los niveles de testosterona y análisis de cariotipo.
Tratamiento: No hay para esterilidad, terapia con testosterona mejora el desarrollo de las características sexuales 2rias.
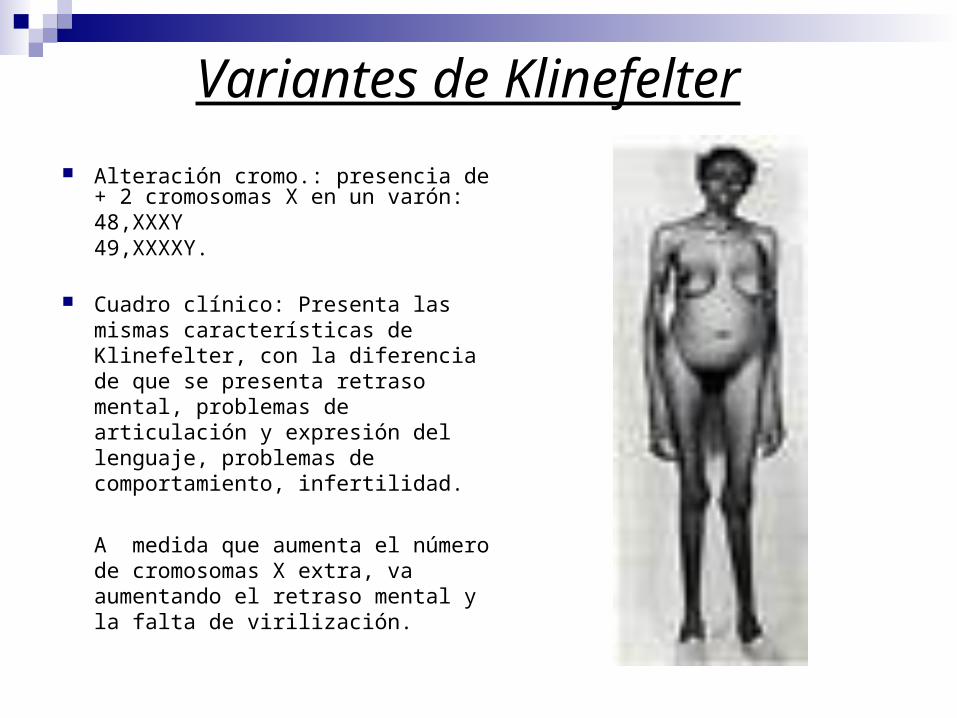
Variantes de Klinefelter
Alteración cromo.: presencia de + 2 cromosomas X en un varón: 48,XXXY49,XXXXY.
Cuadro clínico: Presenta las mismas características de Klinefelter, con la diferencia de que se presenta retraso mental, problemas de articulación y expresión del lenguaje, problemas de comportamiento, infertilidad.
A medida que aumenta el número de cromosomas X extra, va aumentando el retraso mental y la falta de virilización.

Trisomia XYY o síndrome de los súper machos
Alteración cromoso.: cromosoma Y adicional en un varón (47,XYY).
Cuadro clínico: Fenotipicamente casi normales, crecimiento acelerado a mitad de la niñez, pobre coordinación motora fina, presentan acné severo, dientas grandes, excesivamente altos, inteligencia casi normal (C.I. poco bajo pero no menor de 70), inmadurez emocional, comportamiento infantil, impulsividad, auto imagen devaluada, trastornos en el aprendizaje y en el lenguaje.
Diagnostico: suele detectarse en la adolescencia, análisis de cariotipo.
Tratamiento: terapia educacional ayuda por parte de los familiares y personal escolar, asesorar a los padres

Variante Polisómica del Síndrome de los Súper Machos
Alteración cromo.: presencia de mas de 2 cromosomas Y adicionales en un varón:48, XYYY)(49 XYYYY).
Cuadro clínico: Crecimiento acelerado a mitad de la niñez, retraso mental, trastornos neuromotores, del lenguaje y del aprendizaje, presentan acné severo, dientas grandes, excesivamente altos, comportamiento explosivo, violento y antisocial, comportamiento criminal.
A medida que aumenta el número de cromosomas Y extra, va aumentando el retraso mental, hay infertilidad y otras malformaciones.

Trisomia XXX o súper Hembras alteración cromoso.: cromosoma X
adicional en una mujer (47,XXX).
Cuadro clínico: No presentan anormalidades físicas importantes, disminución de 5 a 20 puntos en el coeficiente intelectual, el cromosoma adicional es de origen materno, mujeres más altas y de mayor peso que sus hermanas, menarquia tardía, trastornos neuromotores, del lenguaje y del aprendizaje, mala adaptación psicosocial.
Diagnostico: suele detectarse en la adolescencia por menarquia tardía, análisis de cariotipo, frotis bucal (doble cromatina positiva).
Tratamiento: terapia educacional, ayuda de los familiares y del personal escolar, asesorar a los padres.

Variante Polisómica del Síndrome de las súper hembras
Alteración cromo.: presencia de mas de 2 cromosomas X adicionales en una mujer:48,XXXX49 XXXXX.
Cuadro clínico: Mujeres más altas y de mayor peso que sus hermanas, menarquia tardía, retraso mental, trastornos neuromotores, del lenguaje y del aprendizaje, mala adaptación psicosocial al grado de ser peligrosas.
A medida que aumenta el número de cromosomas X extra, va aumentando el retraso mental, hay infertilidad, órganos genitales atrofiados, obesidad, voz pasiva.

TRASTORNOS EN LA DIFERENCIACIÓN SEXUAL
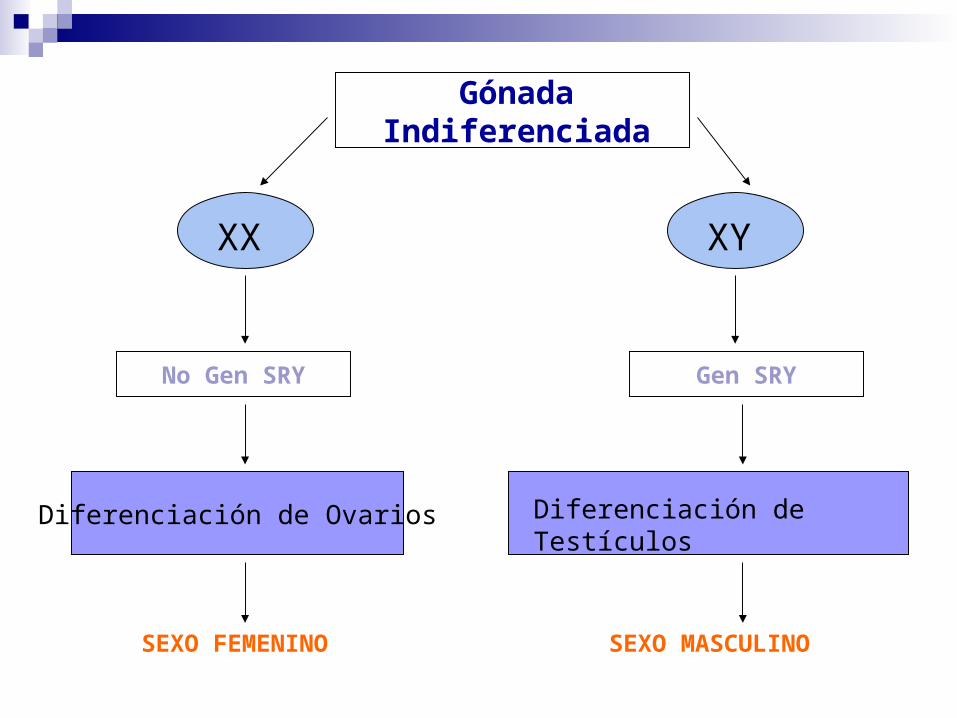
Gónada Indiferenciada
Diferenciación de Ovarios Diferenciación de Testículos
SEXO FEMENINO SEXO MASCULINO
XX XY
No Gen SRY Gen SRY
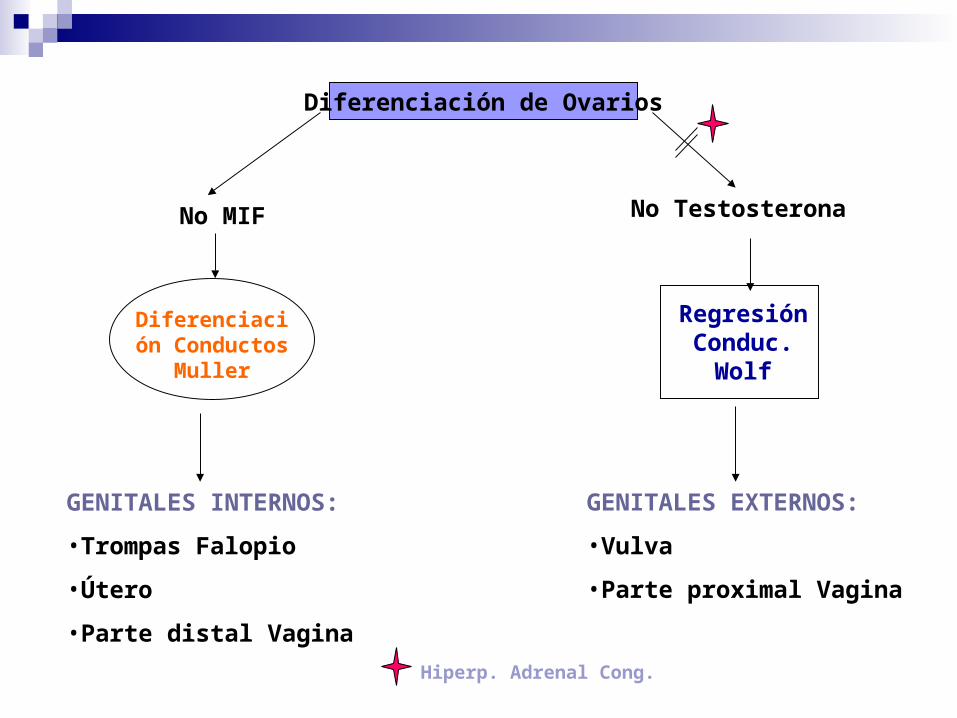
GENITALES INTERNOS:
•Trompas Falopio
•Útero
•Parte distal Vagina
GENITALES EXTERNOS:
•Vulva
•Parte proximal Vagina
Diferenciación
Conductos Muller
Regresión
Conduc. Wolf
Diferenciación de Ovarios
No MIF No Testosterona
Hiperp. Adrenal Cong.
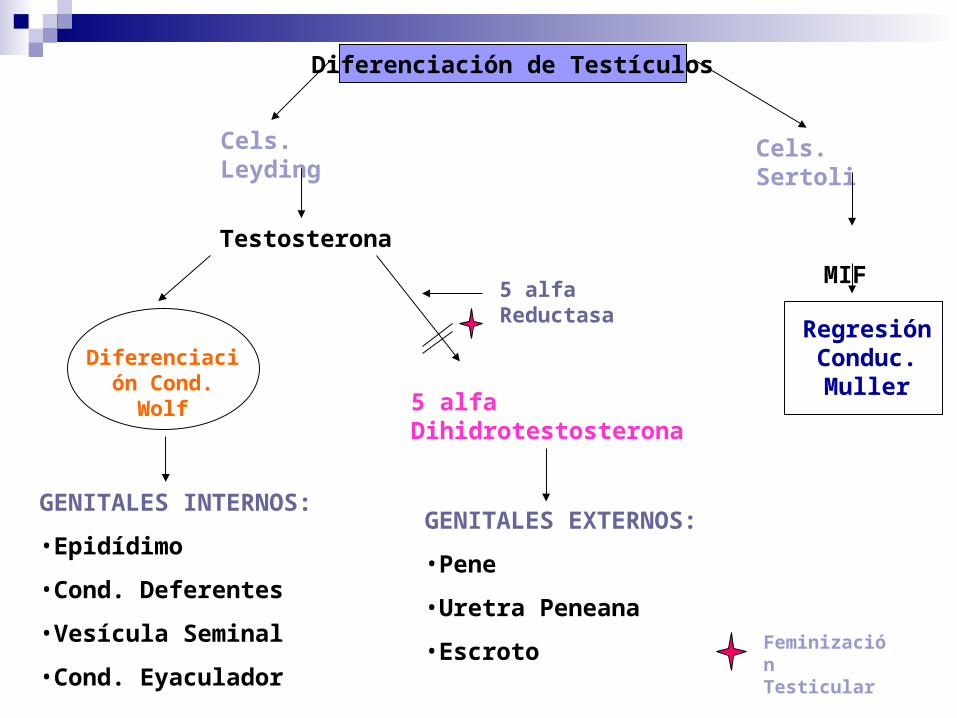
5 alfa Dihidrotestosterona
GENITALES INTERNOS:
•Epidídimo
•Cond. Deferentes
•Vesícula Seminal
•Cond. Eyaculador
GENITALES EXTERNOS:
•Pene
•Uretra Peneana
•Escroto Feminización Testicular
Diferenciación Cond.
Wolf
Regresión
Conduc. Muller
MIF
Testosterona
Diferenciación de Testículos
Cels. Leyding
Cels. Sertoli
5 alfa Reductasa
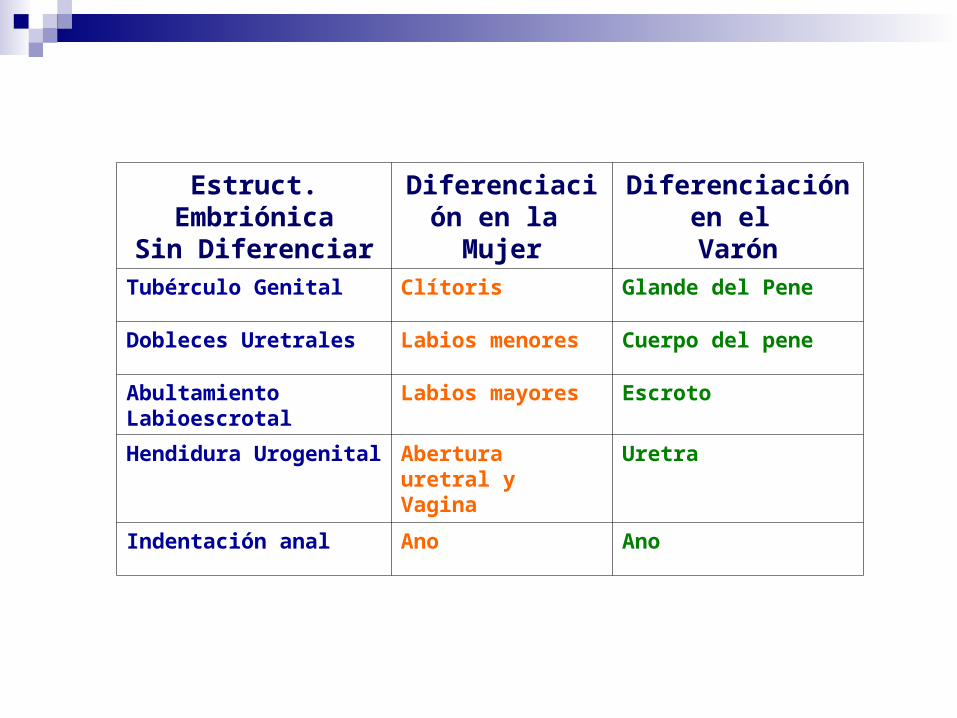
Estruct. EmbriónicaSin Diferenciar
Diferenciación en la Mujer
Diferenciación en el Varón
Tubérculo Genital Clítoris Glande del Pene
Dobleces Uretrales Labios menores Cuerpo del pene
Abultamiento Labioescrotal Labios mayores Escroto
Hendidura Urogenital Abertura uretral y Vagina
Uretra
Indentación anal Ano Ano

Pseudo Hermafroditismo Femenino Alteración cromo.: ambigüedad sexual por
falla de la enzima 21-hidroxilasa o 11-hidroxilasa. (46,XX).
Hiperplasia Suprarrenal Congénita = desorden más común
Cuadro clínico: desarrollo de genitales externos ambiguos o masculinizados, desarrollo de gónadas y genitales internos normales, produccion excesiva de andrógenos (causa virilacion) en mujeres, hipertrofia del clítoris, fusión parcial de labios mayores, presencia del seno urogenital, núcleos positivos en cromatina.Cuando ocurre en varones causa perdida de sal, maduración ósea inicial acelerada pero luego se detiene prematuramente.
Diagnostico: Ambigüedad sexual, cariotipo, frotis bucal, medición de niveles de 21-hidroxilasa o 11-hidroxilasa.
Tratamiento: tratamiento con hormonas para acentuar características femeninas, corrección quirúrgica de los defectos anatómicos, apoyo a la identidad del genero.

Pseudo Hermafroditismo Masculino Alteración cromo.: ambigüedad sexual por
falta de la 5 α-dihidrotestoaterona o en receptores de andrógenos, (46,XY).
Feminización Testicular o Insensibilidad a Andrógenos = Desordenes más comunes.
Cuadro clínico: genitales externos ambiguos o feminizados, no hay útero ni tropas, vagina ciega, los testículos se localizan en el abdomen, hipospadia, ginecomastia, agenesia o hipoplasia de las celulas de Leyding, No hay Menstruación.
Diagnostico: Ambigüedad sexual, frotis bucal (cromatina negativa), cariotipo, estudio de receptores de andrógenos, estudio de niveles plasmáticos de testosterona.
Tratamiento: androgenico sustitutivo , corrección quirúrgica y apoyo a la identidad del genero.

Hermafroditismo Verdadero Alteración cromo.: cariotipo 46,XX
en las 2/3 partes de los casos, los otros son mosaicos.
Cuadro clínico: presencia de tejido ovárico y testicular: un testículo y un ovario .
O pueden presentar ovoteste combinación de tejido ovárico y testicular en una solo gónada
Genitales externos ambiguos.
Diagnostico: ambigüedad sexual, por cariotipo.
Tratamiento: corrección quirúrgica si fuera posible, apoyo a la identidad del genero.


















