
EQUILIBRIOS DE PARTICIÓN Y ADSORCIÓN - geocities.ws · estacionaria son líquidos y lo que se...
20
E.E.T. N 0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos 1º 16º A - 1º 17º – Prof. Mariana Furuken Cromatografía – Página 1 0 0 C C R R O O M M A A T T O O G G R R A A F F Í Í A A - - E E Q Q U U I I L L I I B B R R I I O O S S D D E E P P A A R R T T I I C C I I Ó Ó N N Y Y A A D D S S O O R R C C I I Ó Ó N N 0 0 Dentro de las técnicas experimentales básicas de laboratorio de química se encuentra la cromatografía, que sirve para separar y purificar mezclas. La palabra cromatografía significa “Escribir en Colores” ya que cuando fue desarrollada los componentes separados eran colorantes. Se llama CROMATOGRAFÍA al proceso de separación que permite la resolución de mezclas por división de los componentes en zonas concentradas o en fases distintas de aquellas en que los componentes fueron presentados originariamente, independientemente de la naturaleza de las fuerzas que causan la movilización de los componentes entre dos fases. De estas dos fases una es Estacionaria y la otra es Móvil que pasa a través de la fase estacionaria. La técnica de cromatografía se basa en el principio general de distribución de un compuesto entre dos fases, una estacionaria (o fija) y otra móvil, que se mueven en forma relativa entre sí mientras permanecen en íntimo contacto. Reducida a su fundamento, se la puede ver como la remoción selectiva de los componentes de una mezcla por acción de una fase móvil que fluye a través de una fase estacionaria. La muestra es introducida en la fase móvil, y los componentes de la muestra se distribuyen entre las fases estacionaria y móvil. Los componentes estarán determinada cantidad de tiempo en la fase estacionaria, dependiendo de su estructura y la de las fases. Si alguno de los componentes está más tiempo en la fase móvil, se moverá junto con ella rápidamente; si estás más tiempo en la fase estacionaria, se moverá lentamente. Al igual que en un proceso de extracción, la cantidad de tiempo que permanece una muestra en cada fase dependerá del coeficiente de distribución, que está relacionado con los mismos factores que determinan la solubilidad de una muestra. El grado de separación de una muestra estará, por tanto, controlado por las diferencias entre los coeficientes de distribución de sus componentes. FASE ESTACIONARIA Æ Æ LECHO CROMATOGRÁFICO FASE MÓVIL Æ Æ ELUYENTE
Transcript of EQUILIBRIOS DE PARTICIÓN Y ADSORCIÓN - geocities.ws · estacionaria son líquidos y lo que se...

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
Cromatografía – Página 1
CCRROOMMAATTOOGGRRAAFFÍÍAA -- EEQQUUIILLIIBBRRIIOOSS
DDEE PPAARRTTIICCIIÓÓNN YY AADDSSOORRCCIIÓÓNN Dentro de las técnicas experimentales básicas de laboratorio de química se encuentra la cromatografía, que sirve para separar y purificar mezclas.
La palabra cromatografía significa “Escribir en Colores” ya que cuando fue desarrollada los componentes separados eran colorantes.
Se llama CROMATOGRAFÍA al proceso de separación que permite la resolución de mezclas por división de los componentes en zonas concentradas o en fases distintas de aquellas en que los componentes fueron presentados originariamente, independientemente de la naturaleza de las fuerzas que causan la movilización de los componentes entre dos fases. De estas dos fases una es Estacionaria y la otra es Móvil que pasa a través de la fase estacionaria.
La técnica de cromatografía se basa en el principio general de distribución de
un compuesto entre dos fases, una estacionaria (o fija) y otra móvil, que se mueven en forma relativa entre sí mientras permanecen en íntimo contacto. Reducida a su fundamento, se la puede ver como la remoción selectiva de los componentes de una mezcla por acción de una fase móvil que fluye a través de una fase estacionaria. La muestra es introducida en la fase móvil, y los componentes de la muestra se distribuyen entre las fases estacionaria y móvil. Los componentes estarán determinada cantidad de tiempo en la fase estacionaria, dependiendo de su estructura y la de las fases. Si alguno de los componentes está más tiempo en la fase móvil, se moverá junto con ella rápidamente; si estás más tiempo en la fase estacionaria, se moverá lentamente. Al igual que en un proceso de extracción, la cantidad de tiempo que permanece una muestra en cada fase dependerá del coeficiente de distribución, que está relacionado con los mismos factores que determinan la solubilidad de una muestra. El grado de separación de una muestra estará, por tanto, controlado por las diferencias entre los coeficientes de distribución de sus componentes.
FASE ESTACIONARIA LECHO CROMATOGRÁFICO
FASE MÓVIL ELUYENTE

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
Sólo líquidos y sólidos pueden servir como fase estacionaria mientras que la fase móvil puede estar representada por gases o líquidos. Aquellos métodos que emplean como fase fija un sólido, se denominan de adsorción; mientras que los que utilizan un líquido se denominan de partición. Por lo tanto, se pueden obtener cuatro combinaciones fundamentales tal como se señala a continuación:
Crom. en Papel
Crom. Líquido – Líquido con
solvente a Presión y Soporte sólido
Cromatografía Líquido – Líquido
Fase Móvil: Líquido
PARTICIÓN Fase Estacionaria
Líquida
PARTICIÓNADSORCIÓN Fase Estacionaria
Sólida
ADSORCIÓN
CCRROOMMAATTOOGGRRAAFFÍÍAA
Cromatografía Líquido – Sólido
Fase Móvil: Líquido
Crom. en Columna
Crom. en Placa Delgada
Crom. Gaseosa
Crom. de Adsorción en Geles (Sephadex)
Crom. de Afinidad
Crom. de Intercambio Iónico
Cromatografía Gas – Sólido
Fase Móvil: Gas
Crom. Gaseosa
Cromatografía Gas – Líquido
Fase Móvil: Gas
El procedimiento se basa en un equilibrio entre una fase móvil y una fase estacionaria. De acuerdo a la naturaleza de la fase estacionaria, la cromatografía puede ser de adsorción o de partición. En una cromatografía de adsorción, la mezcla se coloca sobre una fase estacionaria sólida sobre la cual se adsorberán los componentes. Luego, se hace pasar una fase móvil sobre el sólido (un solvente puro o mezcla de solventes), estableciéndose una competencia por las sustancias de la mezcla entre la fase estacionaria y la fase móvil. Dependiendo de las características de cada compuesto de la mezcla, algunos tenderán a quedarse más retenidos en el sólido, mientras que otros avanzarán con el paso del solvente. Así, por este simple procedimiento, la diferente estructura química de los componentes de una mezcla permiten la separación de los mismos. Si bien el comportamiento de los componentes depende de varios factores, en muchos casos se pueden hacer algunas predicciones:
• Para compuestos de peso molecular comparable, aquellos que tengan una polaridad mayor tienen menor tendencia a moverse del punto de partida.
• Para compuestos de polaridad comparable, quedan más retenidos aquellos que posean el peso molecular más grande.
En una cromatografía de partición, tanto la fase móvil como la fase estacionaria son líquidos y lo que se establece son pequeños equilibrios de distribución sucesivos entre dos fases líquidas. En este caso, además de una fase fija y una fase móvil debe existir un soporte adecuado para la fase fija. El caso más común es el de la cromatografía en papel: la celulosa actúa como soporte de
Cromatografía – Página 2

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken pequeñas proporciones de agua que actúan como fase estacionaria, estableciéndose el equilibrio de partición entre el agua que hay en la celulosa y el solvente que actúa como fase móvil.
Existen, además, numerosas TÉCNICAS CROMATOGRÁFICAS que fueron desarrolladas con el paso de los años. Entre ellas se puede mencionar las cromatografías de afinidad, por exclusión, de fase reversa, cromatografías líquidas de alta resolución y cromatografías gaseosas. Estas dos últimas requieren de equipo especializado y si bien son de uso corriente en investigación e industria, su implementación en establecimientos educativos resulta económicamente dificultosa.
La cromatografía puede llevarse a cabo de forma cualitativa o cuantitativa. Para la cromatografía cualitativa se utilizan placas cromatográficas que pueden prepararse con anterioridad al momento de su uso o bien cromatofolios que se pueden adquirir en comercios especializados. Los cromatofolios constan de un soporte de vidrio, plástico o aluminio sobre el cual se fija la fase estacionaria. En la cromatografía de adsorción la fase estacionaria puede estar constituida por diversos tipos de adsorbentes, siendo los más comunes la sílica y la alúmina. Estas fases son sensibles a la humedad, perdiendo como consecuencia de ésta su poder de separación: el agua de un ambiente húmedo se fija sobre el adsorbente, bloqueando los sitios de adsorción disponibles para las sustancias a separar.
En la placa cromatográfica se “siembra” la mezcla a analizar. Se coloca luego
en una cuba cromatográfica que contiene la fase móvil, la cual moja la parte inferior de la placa y asciende por capilaridad. Este proceso se denomina “desarrollo del cromatograma”.
El desarrollo termina cuando la fase móvil ha llegado hasta alrededor de 1 cm del borde superior de la placa. Esta línea que marca el solvente se denomina “frente del solvente”.
En el caso de que las sustancias sean coloreadas, se identifican a simple vista
las posiciones de los distintos pigmentos separados. En caso de ser incoloros se debe recurrir a un “revelador”. Existen varios tipos de reveladores y uno de los más utilizados es el vapor de iodo (es un revelador “universal”, dado que tiene amplia aplicabilidad y sólo excepcionalmente no puede ser utilizado).
Como el iodo tiende a volatilizarse, si colocamos unos cristales de iodo en una cámara cerrada, ésta se saturará con vapores de iodo. Al introducir la placa cromatográfica en dicha cámara, el iodo se adsorberá a la fase fija y lo hará con mayor intensidad allí donde hay un componente de la mezcla. Por este sencillo método pueden visualizarse las “manchas” correspondientes a componentes no coloreados.
La cromatografía analítica puede utilizarse como criterio de identificación para algunas sustancias. Para ello se define una propiedad de las sustancias que se denomina Rf o relación de frente.
solvente elpor recorrida
soluto elpor recorrida
distancia
distanciaRf =
Cromatografía – Página 3

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
Cuando se desarrolla el cromatograma, mientras el solvente alcanza una determinada altura en la placa los componentes de la muestra se moverán una distancia menor o igual a la del solvente. El cociente entre la distancia recorrida por el solvente y la recorrida por un determinado componente recibe el nombre de “relación de frente” y naturalmente es siempre menor o igual a 1. Si una determinada sustancia se cromatografía sobre el mismo tipo de adsorbente utilizando el mismo solvente de desarrollo, presentará siempre la misma relación de frente. Esto es útil en la identificación de componentes en el análisis de pesticidas, analgésicos, barbitúricos, etc., dado que se conoce previamente el tipo de sustancias a analizar y en los que los métodos cromatográficos están estandarizados. Sin embargo, a no ser en las circunstancias específicas mencionadas, la relación de frente no constituye un mecanismo de identificación, ya que muchas sustancias pueden tener la misma relación de frentes. La relación de frente puede ser utilizada como un criterio de identificación negativo: si dos sustancias tienen diferentes Rf, entonces no son el mismo compuesto, pero no se puede afirmar que dos sustancias que tienen idéntico Rf sean el mismo compuesto. La cromatografía cuantitativa se realiza utilizando columnas cromatográficas. Están constituidas por cilindros, generalmente de vidrio, que actúan como soporte del adsorbente. Tienen mayor capacidad de carga y se preparan en el momento de utilizarse. El equilibrio de partición entre dos fases líquidas implica poner en contacto dos líquidos inmiscibles entre sí (acetato de etilo y agua, por ejemplo) y permite separar sustancias que tengan preferencia para solubilizarse más en uno que en otro. De esta manera, un compuesto que se encuentra en una disolución acuosa en la cual es poco soluble, pasará a la fase de acetato de etilo con rapidez. Así, los componentes de una mezcla pueden redistribuirse utilizando el principio de que lo igual disuelve a lo igual: las sustancias polares tenderán a quedarse en la fase acuosa, mientras que las sustancias no polares pasarán a la fase orgánica. Sin embargo, debemos recordar que se trata de un equilibrio y por ende la separación no será total, es decir que no todo lo polar quedará en el agua ni todo lo no polar pasará al solvente orgánico. Siempre existirá parte de cada compuesto en ambas fases y es por eso que la operación debe repetirse varias veces para lograr una separación virtualmente completa. Este equilibrio, como todos, tiene asociada una constante (que depende, como todas, de la temperatura): la constante de distribución, KD, que se define como la concentración de una sustancia A en la fase orgánica dividida por la concentración de la misma sustancia en la fase acuosa. A esta constante se la denomina también constante de partición.
a
oD ]A[
]A[K =
Este procedimiento se conoce como separación o purificación de mezclas por extracción. También puede ocurrir que la distribución del componente A tenga lugar entre una fase sólida y una fase líquida. Este proceso también recibe el nombre de extracción y en particular se lo denomina “extracción sólido-liquido”.
Cromatografía – Página 4
Un producto sólido, seco, se pone en contacto con un solvente orgánico con el fin de lograr que cierto tipo de componentes del sólido pase a dicha fase líquida. Se concentra el solvente orgánico y se recuperan los productos deseados. Para que se realice la extracción debe haber un contacto superficial directo entre ambas fases y

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken por tanto es conveniente que el sólido esté finamente dividido y que el proceso de extracción se repita varias veces para incrementar su eficiencia. Cuando se realiza este tipo de extracción, sobre todo en la extracción de productos naturales, suelen utilizarse aparatos llamados de extracción continua o semicontinua que optimizan la extracción con un mínimo de solvente.
MMEECCAANNIISSMMOOSS DDEE RREETTEENNCCIIÓÓNN
11.. CCRROOMMAATTOOGGRRAAFFÍÍAA DDEE AADDSSOORRCCIIÓÓNN
Es una técnica útil para muestras solubles en solventes poco polares. La retención depende de la interacción entre los grupos polares con los de la fase estacionaria por medio de fuerzas electrostáticas originadas por dipolos permanentes o puentes de Hidrógeno. En este tipo de cromatografía la fase estacionaria es el adsorbente, superficie altamente polar y la fase móvil (o eluyente), obliga a las sustancias a migrar y esto produce una competencia entre la tendencia del soluto a quedarse retenido en la fase fija o correr con el eluyente. Las sustancias altamente polares tendrán mayor afinidad por la fase estacionaria (que es polar) y por lo tanto, eluirán en segundo término respecto a una sustancia menos polar que, al ser poco retenida por la fase estacionaria, será fácilmente arrastrada por el solvente. Los solventes altamente polares son agentes eluyentes más poderosos que los no polares.
CROMATOGRAFÍA DE ADSORCIÓN
- Lecho sólido altamente polar (adsorbente) - Materiales adsorbentes: Óxidos de aluminio, silicatos, carbón activo,
polisacáridos, ... - Los eluyentes altamente polares son más poderosos que los no polares - Interacciones reversibles inespecíficas entre soluto y fase estacionaria
Cromatografía – Página 5

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
TTÉÉCCNNIICCAASS DDEE LLAA CCRROOMMAATTOOGGRRAAFFÍÍAA DDEE AADDSSOORRCCIIÓÓNN
A. CROMATOGRAFÍA EN COLUMNA El proceso consiste en la separación de los componentes de una muestra en
solución que ha sido depositada en la parte superior de una columna, entre un adsorbente sólido (contenido en la columna) y un solvente o mezcla de ellos que, como fase móvil atraviesan la columna. La fase fija retendrá en mayor o menor grado las sustancias dependiendo de su polaridad relativa. Al descender por la columna, la fase móvil produce el desarrollo del cromatograma apareciendo zonas o bandas horizontales bien definidas que corresponden a cada uno de los compuestos de la mezcla. La disposición y tamaño de cada una de estas bandas está relacionada con la afinidad de los compuestos por el adsorbente o por el solvente de corrida. Las sustancias que tienen mayor afinidad por el adsorbente aparecerán en la parte superior de la columna, mientras que aquellas que son retenidas débilmente por la fase estacionaria, aparecerán como bandas más anchas y en la parte inferior de la columna.
Para una buena cromatografía en columna se debe tener en cuenta: 1. Elección del adsorbente adecuado: El adsorbente elegido debe poseer
ciertas características especiales como por ejemplo: debe tener una gran superficie por unidad de peso, debido a que la adsorción se produce a nivel de la superficie del sólido al depositarse sobre él una monocapa de la sustancia a cromatografiar y debe poseer una capacidad preferencial para adsorber un compuesto determinado. Según su naturaleza química los adsorbentes se dividen en inorgánicos (alúmina, carbonato de calcio, silicagel, etc.) y orgánicos (carbón, sacarosa, etc.).
2. Elección del solvente: se realiza experimentalmente basándose en una
guía denominada serie elutrópica, que ordena a los solventes en orden creciente de polaridad. El orden general para su uso está dado por: éter de petróleo, ciclohexano, tetracloruro de carbono, tricloroetileno, tolueno, benceno, éter etílico, acetato de etilo, acetona, n-propanol.
Cuando se utiliza la técnica en columna, la siembra puede efectuarse con
Cromatografía – Página 6

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
un solvente que será también el primer solvente de desarrollo y de elución. En columna, siempre los solventes de desarrollo se utilizan como eluyentes. El primer solvente de elución puede utilizarse para separar más de un soluto de la mezcla inicial, pero, generalmente se recurre a una secuencia de solventes de elución, con polaridades crecientes. Si el solvente es muy polar, será fuertemente adsorbido por los gránulos de la fase estacionaria y el desplazamiento o desorción de los componentes adsorbidos será inmediato. El resultado de esta desorción indiferenciada será que la mayoría de los solutos correrán con el frente del solvente, y no se logrará separación alguna. Por lo tanto, es una condición general empezar el desarrollo de la columna con solventes de baja polaridad, para luego aumentarla lentamente. Aun solventes poco polares logra eluir a sustancias más polares que ellos, debido a que el eluyente siempre está presente en gran exceso con respecto a la muestra y compite con ella por los sitios activos. Es fundamental, además, que se utilicen solventes anhidros y miscibles entre sí, de lo contrario, se afecta la resolución y las experiencias resultan no reproducibles.
La velocidad de elución de una columna, debe ser lenta, para
permitir que tienda a alcanzar el equilibrio adsorción-desorción, de esta manera, se lograrán bandas netas. Sin embargo, si las bandas permanecen demasiado tiempo dentro de la columna, por aplicarse una velocidad de flujo muy lenta, el fenómeno de difusión se hace importante. Si la velocidad de flujo es demasiado rápida, no se logran los equilibrios adsorción- desorción, y el frente de las bandas aparece deformado. El inconveniente que esto plantea tiene la misma índole que el planteado para el mal empaquetamiento del adsorbente.
3. Rellenado de la columna: Uno de los métodos más simples consiste en
preparar una papilla espesa del adsorbente que se empleará junto con el solvente de elución, verterla en el tubo y esperar que el solvente escurra. Otro método consiste en llenar la columna con el solvente y verter el adsorbente seco, observando que sedimente en forma pareja.
Una vez obtenido el cromatograma, es necesario identificar cada zona para poder obtener los compuestos a la salida de la columna por arrastre con el solvente, proceso que se conoce como elución. Para realizar la elución, es común en la práctica eluir las sustancias de la columna usando series de diferentes solventes en variadas concentraciones de forma tal que aumentan la polaridad de los mismos cada vez.
Si los compuestos son coloreados, las distintas bandas se pueden identificar y seguir su desplazamiento a lo largo de la columna sin problemas; pero si las sustancias que se separan son incoloras, se pueden emplear distintas técnicas para visualizarlas, especialmente para obtenerlas en fracciones independientes. Para ello se presentan distintas posibilidades:
a. Transformación en derivados coloreados. b. Utilizar adsorbentes con propiedades fluorescentes para observar los
compuestos que inhiben la fluorescencia o que absorben en otra longitud de onda.
c. Medir el índice de refracción de las sustancias eluidas o bien midiendo el pH del eluyente. Esto último solo es posible cuando las sustancias a cromatografiar poseen propiedades ácidas o básicas definidas.
Cromatografía – Página 7
E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
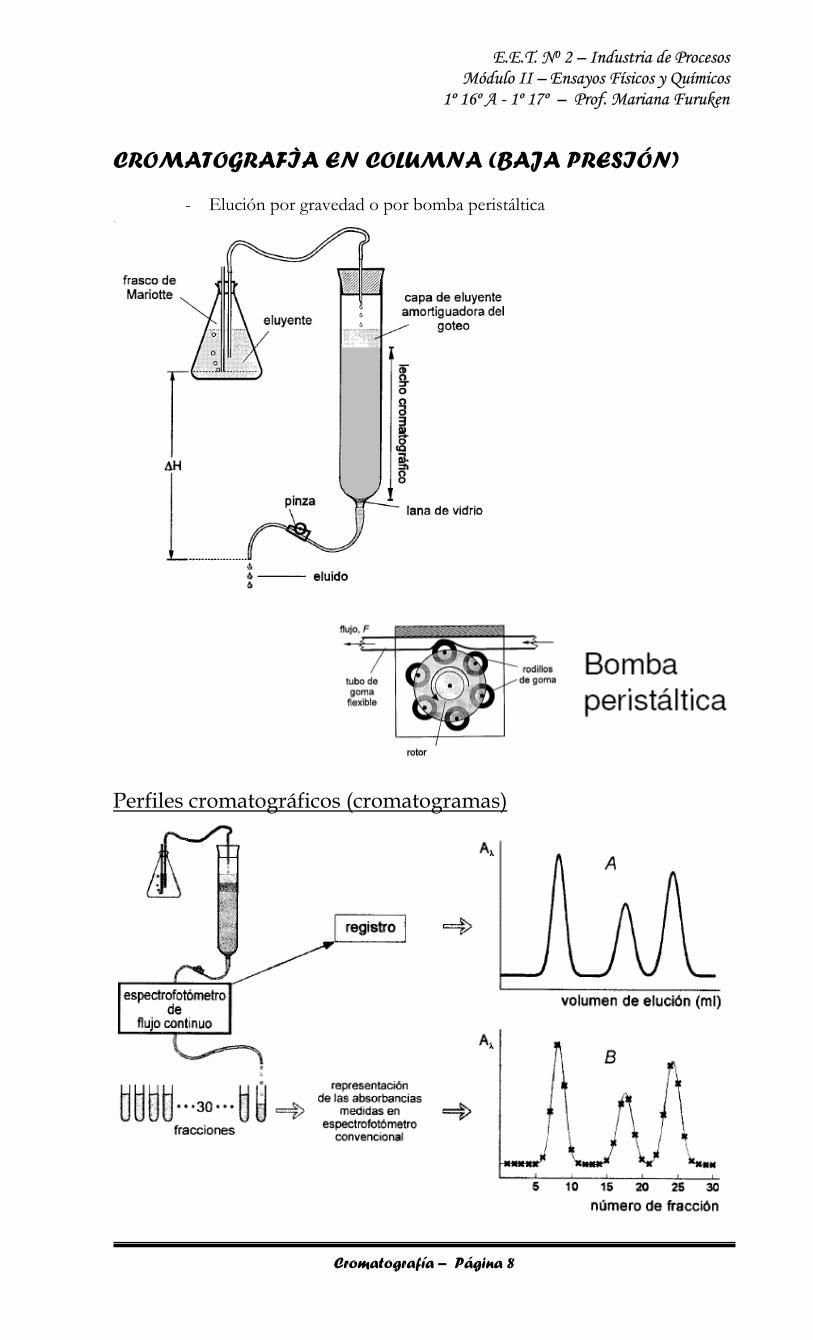
CROMATOGRAFÌA EN COLUMNA (BAJA PRESIÓN)
- Elución por gravedad o por bomba peristáltica
Perfiles cromatográficos (cromatogramas)
Cromatografía – Página 8

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
B. CROMATOGRAFÍA EN PLACA DELGADA (TLC)
Al igual que en columna, el fenómeno que rige la separación de los componentes entre la fase sólida depositada a manera de una fina capa sobre un vidrio y la fase móvil, que es un solvente líquido, es el de adsorción.
Esta técnica presenta ciertas ventajas apreciables:
a) Corto tiempo de desarrollo. b) Usa adsorbentes totalmente inertes frente a la acción de agentes corrosivos. c) Se pueden usar cantidades muy pequeñas de muestra.
La TLC constituye una herramienta excelente para la separación
micropreparativa de mezclas; para esto, lo mejor es usar placas de 0,5 a 1 mm de espesor y vidrios de 40 cm de ancho que permiten la separación de mezclas en cantidades desde 10 mg hasta 500 mg, dando suficiente material para la determinación de la composición química o de las constantes físicas de las sustancias separadas. Para ello, primeramente hay que extraer las sustancias de las placas en la zona que contiene la mancha (que corresponde a una sustancia).
Cromatografía – Página 9

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
Para recuperar las sustancias desde la placa, una vez separadas, hay varias formas de hacerlo; con una espátula se vuelca la porción de la placa que contiene la mancha directamente dentro de un tubo de centrífuga que contiene el solvente de extracción. Se centrífuga y el sobrenadante que contiene la sustancia pura está listo para investigar posteriormente.
Una aplicación cualitativa de esta técnica se obtiene calculando los Rf de las sustancias cromatografiadas.
solvente elpor recorrida distancia
soluto elpor recorrida distanciaRf = 1≤fR
El Rf es menor o igual que 1 y en determinadas condiciones experimentales es característico para cada sustancia. Un factor que gravita directamente sobre el valor del Rf es la saturación del recipiente de corrida con el solvente que se utiliza para el desarrollo del cromatograma. La saturación tiene como fin la concentración uniforme del vapor del solvente a cualquier altura del recipiente, para lograr el avance del frente del solvente en una línea recta y horizontal, y evitar los efectos de la evaporación del mismo durante la corrida.
La mayoría de los cromatogramas en TLC, utiliza como adsorbente a la silicagel G; aunque también se usan en menor grado silicagel con un 10% de yeso, alúmina, silicatos de magnesio y calcio, fosfatos de magnesio, hidróxido y sulfato de calcio o adsorbentes orgánicos como celulosa y poliamida.
Una vez obtenidos los componentes a analizar, se pueden evaluar cuantitativamente por medición espectrofotométrica directa de los cromatogramas o bien indirectamente transformándolos en derivados coloreados.
El Rf está relacionado con la movilidad de un compuesto en un proceso cromatográfico. Todas las siguientes variables influyen tanto en las técnicas de TLC como en la columna cromatográfica.
Las variables generales que afectan la movilidad en una cromatografía de
adsorción son las siguientes:
a. Adsorbente. b. Estructura del soluto. c. Solvente de desarrollo d. Temperatura e. Saturación de la cuba.
a. Adsorbente (fase fija): un orden decreciente de poder absorbente podría
generalizarse: − Carbón activado (de menor actividad) − Silicato de magnesio − Óxido de aluminio (alúmina) − Silicagel − Hidróxido de aluminio − Hidróxido de calcio − Carbonato de calcio − Talco y almidón
Cromatografía – Página 10

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
Las características de la fase fija son: − Actividad (o poder adsorbente): depende de su preparación. Los
adsorbentes para capa delgada tienen cantidades variables de ligante (por ejemplo, la sílica tiene 13% de yeso) que permite mayor adherencia al soporte. Este agregado puede alterar el poder adsorbente. Los dos adsorbentes más utilizado son la alúmina (Al2O3) y la sílicagel (SiO2 y sus hidratos); son especialmente sensibles al contenido de humedad. Ambos pueden servir para absorber prácticamente a todas las moléculas que presenten algún grupo funcional. Además al ser blancos permiten distinguir las zonas donde se localizan los compuestos, luego del desarrollo y revelado.
Alúmina: se consigue comercialmente con tamaños de partícula que oscilan entre 50 y 200 micrones. Se presenta en tres tipos: básica (pH=10), neutra (pH=7) y ácida (pH=4); y es importante asegurarse el empleo del tipo correcto, para evitar la catálisis de ciertas reacciones sobre los grupos funcionales de la muestra a separar. La alúmina presenta cinco grados de actividad según la fuerza de atracción que ejerza sobre grupos polares de la muestra, y según el número de sitios activos. El Grado I es el más activo y se obtiene al calentar la alúmina entre 300ºC y 400ºC varias horas. Grados de menor actividad (II a V) se obtienen por adición de determinadas cantidades de agua (II: 3-4%; III: 5-7%; IV: 25%; V: 38%). El grado se controla por el comportamiento cromatográfico de determinados pigmentos en estrictas condiciones de reproducibilidad. Sílica: También se la clasifica en grados según el porcentaje de agua incorporada. El Grado I se obtiene calentando varias horas la sílica a poco menos de 300ºC (II:5%; III: 15%; IV: 25%; V:38%). Si bien el pH de la sílica es 7, puede en ocaciones actuar como catalizador ácido. La disminución de la actividad o poder adsorbente frente a determinadas muestras orgánicas se debe a que numerosos “sitios activos” de su superficie están bloqueados por moléculas de agua. Por otro lado, la excesiva presencia de agua implicará fenómenos de partición simultáneos a los de adsorción, y esto complicará y restará al proceso cromatográfico.
− Inercia química: Debe tenerse cuidado con respecto a probables reacciones químicas entre los compuestos y el adsorbente. El adsorbente debe ser inerte con respecto a la muestra. La alúmina, por ejemplo, puede tener propiedades básicas y la sílica, ácidas. Si las muestras sembradas tienen algún componente ácido o básico, respectivamente, podrían quedar demasiado adsorbidas debido a una interacción eléctrica (formación de sales). Además, puede haber reacciones, como hidrólisis de ésteres, y condensaciones aldólicas (en alúmina), y lactonizaciones (en sílica). También hay que tener en cuenta que los adsorbentes en Grado I pueden deshidratar grupos funcionales lábiles de la muestra y, consecuentemente, modificar su estructura.
Cromatografía – Página 11

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
− Tamaño del gránulo: Es importante porque una gran superficie implica mayor adsorción, es decir mayor resolución. Cuando se usa el adsorbente de grano fino el espacio entre los gránulos es más pequeño y el equilibrio adsorción-desorción se establece más rápido. Este equilibrio nunca llega a establecerse completamente durante el proceso cromatográfico práctico, pero siempre se debe trabajar de tal forma de tender lo más posible a lograrlo. Cuanto más difícil resulte separar algunas sustancias, tanto más se deberá aproximar a dicho equilibrio para mejorar la resolución. Si el tamaño del gránulo es grande, el equilibrio adsorción-desorción será lento, debido a la lejanía entre sitios activos de los sucesivos gránulos adsorbentes. Esto implicará que el soluto necesitará estar solubilizado un cierto tiempo en el solvente de desarrollo o elución (hasta que se encuentre con el siguiente sitio activo) y en ese lapso puede difundir. El resultado de esta difusión del soluto es el ensanchamiento de las manchas observadas en la capa (respecto de sus puntos de siembra), y la formación de bandas en las cromatografías en columna. Debido a este fenómeno, resulta importante que los puntos o zonas de siembra sean pequeños (tanto en técnicas de capa como de columna).
− Empaquetamiento: Esto es la homogeneidad den el armado de la fase
fija. En las técnicas de capa es primordial un espesor parejo de la capa adsorbente. Una capa irregular ocasionará recorridos más rápidos (en zonas delgadas) o más lentos (en zonas más espesas), implicando un Rf aleatorio e irreproducible. En las técnicas en columna, la homogeneidad está dada por un asentamiento parejo de su relleno. Las inhomogeneidades implicarán un descenso rápido de las bandas por las zonas menos densas del relleno, y, en consecuencia, el frente de elución será desparejo y los compuestos saldrán mezclados.
b. Estructura del Soluto: La capacidad de un soluto para ser adsorbido depende
de su polarizabilidad (temporaria o permanente) y de su capacidad para formar puentes de Hidrógeno. Los grupos polares son los adsorbidos más fuertemente. El incremento de numerosos grupos funcionales no es siempre aditivo pero en general, el poder de ser adsorbido aumenta con el incremento de grupos funcionales unidos a la misma molécula.
La lista siguiente de grupos funcionales está ordenada según su poder decreciente de ser adsorbidos: ácidos y bases - alcoholes y tioles – aldehídos y cetonas – halogenuros de alquilo y ésteres – hidrocarburos insaturados – hidrocarburos saturados.
Para olefinas, la adsorción aumenta con el incremento del número de dobles enlaces (aumenta la polarizabilidad). El incremento de numerosos grupos funcionales no es siempre aditivo pero en general, el poder de ser adsorbido aumenta con el incremento de grupos funcionales unidos a la misma molécula, aunque existen casos en los cuales dos grupos funcionales presentes pueden formar uniones hidrógeno intramoleculares, y de esta forma, adsorberse menos (un ejemplo es el caso de los nitrofenoles).
Cromatografía – Página 12
c. Solvente de desarrollo: La elección correcta de los solventes de elución es vital para el éxito del proceso cromatográfico. La mezcla a separar se siembra a partir de una solución muy concentrada, es decir, disuelta en un solvente donde es muy soluble. Cuando se siembra en placa, se deja evaporar el solvente de siembra. El solvente (o mezcla de solventes) de desarrollo puede ser, o no, el mismo de siembra. En general, no lo es. El solvente se elige en forma experimental y según la naturaleza química de las sustancias a separar.

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
d. Temperatura: Como el proceso cromatográfico no es de equilibrio, se deben ajustar lo más posible todas las variables para que dicho proceso resulte reproducible. La temperatura afecta volatilidades y solubilidades, por lo tanto, debe mantenerse constante durante la experiencia y homogénea a lo largo de toda la placa o columna.
e. Saturación de la cuba: La saturación del recipiente de corrida con el solvente
que se utiliza para el desarrollo del cromatograma. La saturación tiene como fin la concentración uniforme del vapor del solvente a cualquier altura del recipiente, para lograr el avance del frente del solvente en una línea recta y horizontal.
22.. CCRROOMMAATTOOGGRRAAFFÍÍAA DDEE PPAARRTTIICCIIÓÓNN
Cuando se agita la solución de una sustancia con un disolvente inmiscible, el soluto se distribuye entre las dos fases, y cuando se llega al equilibrio, la relación:
B solvente elen ión concentracA solvente elen ión concentrac
=repC
es una constante que se denomina coeficiente de partición, de reparto o de distribución.
El principio fundamental de esta cromatografía se basa en este coeficiente de
partición y característica fundamental es que la fase estacionaria es el agua retenida sobre diferentes tipos de soporte (silicagel, celulosa, etc) y las fases móviles un líquido inmiscible en agua (pero saturado en agua), y la separación se basa en la distribución selectiva de los solutos en las fases, según las constantes de distribución particulares.
Es útil para muestras solubles en solventes polares o medianamente polares. El soluto se reparte entre la fase líquida que constituye la fase móvil y un líquido adsorbido o ligado al soporte, que constituye la fase estacionaria.
CROMATOGRAFÍA DE REPARTO
- Lecho líquido polar - Eluyente líquido apolar (hidrofóbico)
Cromatografía – Página 13

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
Existen diferentes tipos de cromatografía líquido-líquido, cromatografía líquida tradicional, donde la fase estacionaria es un líquido polar y la fase móvil es no polar es inmiscible en aquel; cromatografía líquida con fase químicamente unida o enlazada, donde la fase estacionaria está compuesta por sílice al cual se unen grupos como cianuro o amino que permiten la utilización de solventes acuosos y cromatografía líquido de fase reversa, donde la fase estacionaria es no polar y la fase móvil tiene mayor polaridad que la fase estacionaria. Como fase estacionaria se utiliza sílice unida a cadenas carbonadas. La polaridad de la fase estacionaria disminuye con el aumento del número de átomos de carbono empleados. Este tipo de cromatografía es empleada frecuentemente con productos farmacéuticos.
TTÉÉCCNNIICCAASS DDEE LLAA CCRROOMMAATTOOGGRRAAFFÍÍAA DDEE PPAARRTTIICCIIÓÓNN
A. CROMATOGRAFÍA EN PAPEL
Para poder explicar lo que ocurre aquí, es necesario examinar el estado en que se halla el papel saturado con agua. A medida que la celulosa adsorbe agua varían algunas de sus propiedades tales como densidad, calor de adsorción y velocidad de difusión, siendo estas propiedades cada vez más parecidas a las del agua, a medida que ésta se va adsorbiendo. Considerando la fase estacionaria como un complejo agua – celulosa; la sustancia cromatografiada se retendrá más o menos fuertemente sobre la fase estacionaria según sus propiedades hidrofílicas y la del solvente de elución (fase móvil).
Además de la partición, existe una verdadera adsorción sobre el papel, así como un intercambio iónico con los grupos carboxílicos libres que pudiera haber (el papel es similar a un intercambiador catiónico débil); pero si el tiempo de saturación del papel es despreciable.
Para obtener buenos cromatogramas de buena calidad sobre papel, se debe
considerar lo siguiente:
1) Selección y tratamiento del papel. 2) Elección del solvente. 3) Impregnación del papel.
4) Dosificación de la muestra. 5) Aparatos y métodos. 6) Visualización del cromatograma.
Cromatografía – Página 14

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
1) Selección y Tratamiento del Papel: Existen una gran variedad de papeles para cromatografiar que dan buenos resultados. Las instrucciones para su uso suelen encontrarse en los mismos papeles. Éstos deben ser manoseados lo menos posible y tomados por los bordes ya que toda secreción cutánea puede ocasionar sitios de disturbios en el cromatograma. Dichos papeles para cromatografía tienen 4 – 5 % de agua, son higroscópicos y en un sitio saturado con vapor de agua a 20º C se combina hasta alcanzar una composición de 20% en agua.
2) Elección del Solvente: Los solventes involucrados en la fase móvil no deben
reaccionar con las sustancias aplicadas. La elección del solvente está determinada sólo por las sustancias a ser separadas. Por ejemplo, para sustancias fuertemente polares (azúcares), se usan solventes fuertemente polares como propanol – agua, y para sustancias débilmente polares se usan solventes de débil polaridad. Los ácidos orgánicos se cromatografían con solventes neutros o débilmente básicos. Los solventes deben ser de alto grado de pureza ya que las impurezas en el solvente pueden reaccionar con las sustancias a separar y dejar manchas espúreas.
3) Impregnación del Papel: Se pueden distinguir dos modos de impregnación:
a. Con solventes o soluciones hidrofílicas se pueden usar soluciones salinas, agua o fomamida.
b. Con solventes o soluciones hidrofóbicas, por ejemplo: parafina.
4) Dosificación y Aplicación de la Muestra: Para la aplicación de la muestra se usan capilares o micropipetas graduadas especiales para cromatografía.
5) Métodos y Aparatos: Una vez realizada la siembra se deja secar la mancha y
se coloca en un recipiente cerrado que contiene en el interior el solvente orgánico para realizar la corrida que ha sido previamente saturado con agua para evitar la competencia entre el papel y el solvente orgánico por el agua retenida en las fibras del papel. Al igual que en la cromatografía en capa delgada, es importante que la cuba donde se realizará la corrida esté perfectamente saturada con el vapor del solvente de corrida.
a. Cromatografía Ascendente
Las sustancias a separar se siembran en forma de una gota en el punto de siembra que se halla aproximadamente a 1 cm del borde inferior de la tira de papel. Luego se coloca verticalmente en la cuba y el borde inferior del papel pesca justo por debajo del nivel del solvente de corrida. Posteriormente se deja que el solvente ascienda por capilaridad hasta 1 cm por debajo del borde superior del papel o del soporte por donde se lo tiene retenido.
En algunos casos se emplea la Cromatografía Bidimensional, donde la corrida se realiza en un sentido, luego el papel es extraído y secado y vuelto a correr pero en forma perpendicular a la primera corrida y con otro solvente. Las manchas se distribuyen así sobre todo el papel. Se utiliza cuando no se pueden lograr buenas resoluciones haciendo la corrida en un sólo sentido.
Cromatografía – Página 15

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
b. Cromatografía Descendente
El solvente de corrida está contenido en un recipiente especial en la parte superior de la cuba. La punta superior de la tira de papel pesca dentro del solvente, luego está apoyada sobre una varilla de vidrio y el resto cuelga libremente dentro del recipiente.
El punto de siembra está ubicado 1 cm por debajo de la varilla de vidrio. El solvente desciende por la fuerza de gravedad por el papel. Se llega al final del cromatograma cuando el frente del solvente está próximo al extremo inferior del papel.
Para las sustancias que tienen Rf muy pequeños se
usa una modificación que se conoce como “Cromatografía Nocturno”. En este caso no se detiene el desarrollo cuando el frente del solvente alcanza la otra punta del papel, se deja que siga corriendo, pudiéndose extender varios días. Como el frente viaja más allá del borde del papel los Rf no pueden calcularse, pero sí los Rst corriendo al mismo tiempo una sustancia conocida y siendo:
sembrado standard elpor recorrida distanciaadesconocid sustancia la por recorrida distancia
Rf =
Esta modificación se prefiere cuando las sustancias tienen pequeñas
velocidades de corrida y por lo tanto Rf semejantes, porque al incrementar las distancias recorridas, las diferencias entre los Rf se hacen más notables.
c. Cromatografía en Línea
Se usa para separar grandes cantidades de sustancias. Se pueden sembrar hasta varios decigramos. La muestra no se siembra como gotas sino como una línea continua a través de la línea de siembra.
Cromatografía – Página 16

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
d. Cromatografía Circular
Aquí se coloca el papel en forma horizontal, es un método rápido de separación. El papel de filtro circular está provisto de una mancha central axial que puede ser un hilo de algodón o una tirita del mismo papel. El centro está rodeado por varias marcas radiales y una marca situada cerca de la circunferencia externa. Una gota de la solución a separar es colocada sobre una de las marcas radiales y en las otras se siembran sustancias de referencia (patrones). Una vez que se ha hecho la siembra se deja secar el papel y se realiza luego la corrida en una placa de Petri que contiene el solvente de corrida, dentro del cual pesca la mecha. También se puede colocar el disco entre dos tapas de vidrio, teniendo la superior un orificio por donde penetra el solvente a través de la mecha que se ha proyectado hacia el orificio.
Una vez desarrollado el cromatograma, se seca inmediatamente el papel para evitar que las manchas se difundan.
6) Visualización del Cromatograma: Al igual que en la cromatografía en capa delgada, es necesario determinar las posiciones de las manchas para calcular su Rf. Si las sustancias son coloreadas se visualizan a simple vista. Si son incoloras pueden verse al U.V., o bien colorearse con agentes especiales, ya sea sumergiendo el papel o rociándolo. Los reactivos para coloración son en general específicos para determinados grupos químicos, producen coloración más o menos características que se pueden visualizar a simple vista o al U.V.
Cuando las sustancias investigadas dan coloraciones suficientemente estables es posible realizar la evolución cuantitativa. La muestra es sembrada en varios puntos de la línea de partida; es rociado un cromatograma exterior llamado “Cromatograma Guía”.
Así las manchas que no han sido tratadas pueden localizarse, cortar el papel y extraer las sustancia con un solvente adecuado. También se pueden analizar la manchas por fotocolorimetría con un densitómetro, haciendo el papel transparente a la adsorción colorimétrica por inmersión del papel durante 5 minutos en una mezcla α- bromonaftaleno y parafina líquida.
También existe un método de evaluación cuantitativa por comparación del tamaño de las manchas, que está basado en el hecho que el tamaño de las manchas es proporcional al logaritmo de la concentración de las sustancias disueltas.
Cromatografía – Página 17

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
B. CROMATOGRAFÌA EN COLUMNA (HPLC) - Elución por alta presión (hasta 500 atmósferas) - Alta resolución, rapidez y reproducibilidad - Purificación de moléculas biológicas de gran variedad de propiedades y tamaños
C. CROMATOGRAFÌA GASEOSA EN COLUMNA
- La fase móvil es un gas inerte que arrastrará los compuestos inyectados - la velocidad de migración que dependerá de la naturaleza de éstos, de la
naturaleza y velocidad de la fase móvil, de la naturaleza de la fase estacionaria y de la temperatura, sin interaccionar con los compuestos.
- requiere muestras volátiles, por lo que los compuestos que no lo son (azúcares, ácidos grasos, aminoácidos...) tienen que ser derivatizados, lo que significa transformar químicamente grupos químicos que originan puntos de presión bajas (punto de ebullición alto) en compuestos más volátiles
Cromatografía – Página 18
E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
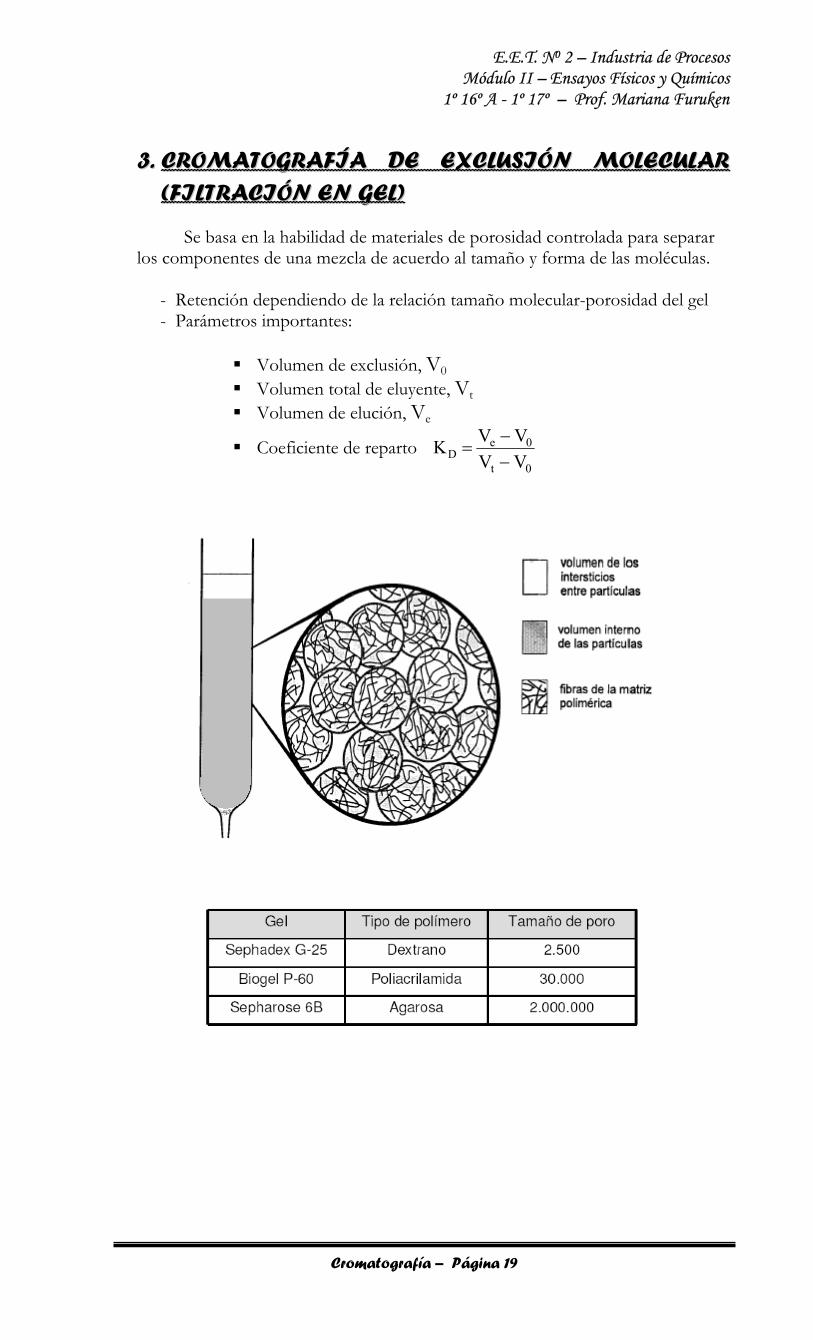
33.. CCRROOMMAATTOOGGRRAAFFÍÍAA DDEE EEXXCCLLUUSSIIÓÓNN MMOOLLEECCUULLAARR ((FFIILLTTRRAACCIIÓÓNN EENN GGEELL))
Se basa en la habilidad de materiales de porosidad controlada para separar los componentes de una mezcla de acuerdo al tamaño y forma de las moléculas.
- Retención dependiendo de la relación tamaño molecular-porosidad del gel - Parámetros importantes:
Volumen de exclusión, V0 Volumen total de eluyente, Vt Volumen de elución, Ve
Coeficiente de reparto 0t
0eD VV
VVK−−
=
Cromatografía – Página 19

E.E.T. N0 2 – Industria de Procesos Módulo II – Ensayos Físicos y Químicos
1º 16º A - 1º 17º – Prof. Mariana Furuken
44.. CCRROOMMAATTOOGGRRAAFFÍÍAA DDEE AAFFIIN DDAADD NII
Se basa en la especificidad de
algunas macromoléculas biológicas. Éstas se unen específicamente a la fase estacionaria y para separar dicha macromolécula, bastará con variar el pH una vez que la columna esté limpia y solo se encuentre la que interesa.
- Interacción específica y reversible con ligandos inmovilizados en la fase fija - Purificación de enzimas y anticuerpos
55.. CCRROOMMAATTOOGGRRAAFFÍÍAA DDEE IINNTTEERRCCAAMMBBIIOO IIÓÓNNIICCOO
Se basa en la afinidad de los iones en solución por los sitios de polaridad opuesta que se encuentran en la fase estacionaria.
- Interacción de moléculas ionizadas con una matriz cargada.
Gel cargado negativamente intercambio aniónico Gel cargado positivamente intercambio catiónico
Cromatografía – Página 20


















