
Gestión y Aseguramiento de Calidad Analítico Para Laboratori_unlocked
91
-
Upload
piedad-garcia -
Category
Documents
-
view
81 -
download
4
description
herramienta muy util para los bateriologos en su laboratorio clinico
Transcript of Gestión y Aseguramiento de Calidad Analítico Para Laboratori_unlocked


2 | P á g i n a
INSTITUTO NACIONAL DE SALUD
PROGRAMA DE EVALUACIÓN EXTERNA DEL DESEMPEÑO EN QUIMICA
CLINICA Y HEMATOLOGÍA
GESTIÓN Y ASEGURAMIENTO DE CALIDAD ANALÍTICO PARA LABORATORIOS
CLÍNICOS
Bogotá, Agosto de 2014

3 | P á g i n a
TABLA DE CONTENIDO
1. VOCABULARIO DE TÉRMINOS DE METROLOGÍA PARA EL LABORATORIO CLÍNICO
REVISIÓN (2012) ______________________________________________________ 7
1.1. OBJETO Y CAMPO DE APLICACIÓN __________________________________ 8
1.2. VOCABULARIO ___________________________________________________ 8
2. FASE PREANALÍTICA ______________________________________________ 26
2.1 CAUSAS DE LA VARIABILIDAD DE LAS MAGNITUDES ___________________ 26
2.1.1 Variabilidad Biológica _____________________________________________ 26
2.1.2 Variabilidad Analítica ______________________________________________ 27
2.1.3 Variables preanalíticas fisiológicas __________________________________ 27
2.1.4 Variables preanalítica en la toma de muestra __________________________ 28
2.1.5 Variables preanalíticas interferentes __________________________________ 30
2.2 PROCESOS DE LA FASE PREANALÍTICA _____________________________ 30
2.2.1 Solicitud Analítica ________________________________________________ 31
2.2.2 Preparación del Paciente __________________________________________ 31
2.2.3 Obtención de las Muestras Primarias _________________________________ 32
2.2.4 Transporte de Muestras ___________________________________________ 36
2.2.5 Conservación y Estabilidad _________________________________________ 37
2.2.6 Interferencias Preanalíticas. ________________________________________ 39
3. FASE ANALITICA __________________________________________________ 42 3.1 HEMATOLOGÍA_______________________________________________________ 42
3.2 CONTROL DE CALIDAD INTERNO________________________________________ 55
3.2.1 Generalidades ___________________________________________________ 55
3.2.2 Construcción de la Gráfica de Control de Calidad _______________________ 58
3.3 CONTROL DE CALIDAD EXTERNO ___________________________________ 67
3.3.1 Interlaboratorios _________________________________________________ 67
3.3.2 Ciclo de funcionamiento ___________________________________________ 69
3.3.2.1 Flujograma de funcionamiento _____________________________________ 69
3.3.3 Manejo de Resultados ____________________________________________ 70
3.3.4 Evaluación del Desempeño_________________________________________ 70
4. FASE POSTANALÍTICA______________________________________________ 73
4.1 TERMINOLOGÍA __________________________________________________ 74
5. ASEGURAMIENTO METROLÓGICO ___________________________________ 78
5.1 IMPORTANCIA DE LA GESTIÓN METROLÓGICA EN LOS LABORATORIOS DE
DIAGNÓSTICO ______________________________________________________ 78

4 | P á g i n a
5.2 INTERVENCIONES METROLÓGICAS EN EQUIPOS DE LABORATORIO: ____ 78
5.3 FRECUENCIA DE LAS INTERVENCIONES METROLÓGICAS EN EQUIPOS DE
LABORATORIO: _____________________________________________________ 81
5.4 INFRAESTRUCTURA NECESARIA PARA LA INSTALACION DE LOS EQUIPOS DE
LABORATORIO ______________________________________________________ 82
6. VIGILANCIA EN SALUD PUBLICA POR LABORATORIO ___________________ 85
6.1 FUNCIONES _____________________________________________________ 85
6.2 COMITÉ DE VIGILANCIA ___________________________________________ 86
6.3 EVENTOS DE INTERÉS EN SALUD PÚBLICA __________________________ 86
6.4 QUE SE VIGILA ___________________________________________________ 87
6.4.1 Grupo de Transmisibles ___________________________________________ 88
6.4.2 Grupo de No Transmisibles_________________________________________ 88
6.4.3 Grupo Factores de riesgo ambiental y sanitario _________________________ 88
6.5 ROL DEL LABORATORIO CLÍNICO ___________________________________ 89
6.6 RECOMENDACIONES _____________________________________________ 90

5 | P á g i n a
CREDITOS AL DOCUMENTO
FERNANDO DE LA HOZ RESTREPO
Director General Instituto Nacional de Salud
MAURICIO BELTRÁN DURÁN
Director de Redes en Salud Pública
LYNDA PATRICIA PRIETO NAVARRERA
Subdirectora de Gestión de Calidad de LSP
ANTONIO JOSÉ BERMUDEZ FERNANDEZ
Coordinador Grupo Genética Crónicas
AUTORES
GLORIA ISABEL BARAJAS BARBOSA
Fase Pre analítica
Fase Pos Analítica
YADIRA PACHECO ESPITIA
Fase Analítica-Control de Calidad Interno
MARIA CLEMENCIA GARNICA BARRIOS
Vocabulario de términos de metrología
Fase Analítica-Hematología
LINA MARCELA QUEVEDO
Vigilancia en Salud pública por Laboratorio
JENNY MARCELA ROJAS
Aseguramiento Metrológico
ANGIE PAOLA RODRIGUEZ GUERRERO
Aseguramiento Metrológico
ANA MATILDE RODRÍGUEZ BECERRA
Fase Analítica-Control de Calidad Externo
EDICIÓN
Lynda Patricia Prieto Navarrera
Antonio José Bermudez Fernández
Ana Matilde Rodríguez Becerra
DISEÑO
Comunicaciones
Instituto Nacional de Salud
ISBN: 978-958-13-0168-3
© Instituto Nacional de Salud, 2014
Avenida calle 26 No. 51-20 Conmutador: (1) 220 77 00 fax 220 7700 Ext. 1200 – 1291
Bogotá, D.C., Colombia

6 | P á g i n a
Presentación
Este Documento, preparado por el Instituto Nacional
de Salud (INS), constituye una herramienta de apoyo
en el trabajo del laboratorio clínico, así como uso de
documento de apoyo con fines educativos.
Está organizado en diferentes aspectos relacionados
con la aplicabilidad al Laboratorio Clínico, se incluye
un vocabulario de términos que se aplica a todos los
aspectos teóricos y prácticos de las medidas y los
dominios de aplicación, de gran utilidad para el
profesional.
Se mantendrá actualizado en la medida que se
cuente con nuevos recursos de información.
La estructuración de cada capítulo de este
documento, son de la exclusiva responsabilidad de
cada autor.

7 | P á g i n a
1. VOCABULARIO DE TÉRMINOS DE METROLOGÍA
PARA EL LABORATORIO CLÍNICO
REVISIÓN (2012)
Maria Clemencia Garnica Barrios; Bacterióloga- Universidad de Colegio Mayor de Cundinamarca;
Instituto Nacional de Salud, Grupo Genética Crónicas. Bogotá, Colombia. [email protected]
En 1984, la Organización Internacional de Normalización (ISO) publicó la primera edición
del Vocabulario internacional de términos fundamentales y generales en metrología (VIM),
elaborada por expertos de la Oficina Internacional de Pesas y Medidas (BIPM), la
Comisión Electrotécnica Internacional (IEC), la Organización Internacional de Metrología
Legal (OIML) y la propia ISO.
En 1993, ISO publicó la segunda edición del vocabulario en cuya elaboración participaron,
además de los organismos implicados en la primera edición, la Federación Internacional
de Química Clínica y Ciencias del Laboratorio Clínico (IFCC), la Unión Internacional de
Química Pura y Aplicada (IUPAC) y la Unión Internacional de Física Pura y Aplicada
(IUPAP).
En 1994, el Centro Español de Metrología (CEM) elaboró una versión en lengua española
basada en la segunda edición del vocabulario.
En 1997 se constituyó el Comité Conjunto para las Guías de Metrología (JCGM), formado
por representantes de los siete organismos internacionales que habían preparado la
segunda edición del VIM.
En el 2005, la Cooperación para la Acreditación Internacional de Laboratorios (ILAC) se
unió oficialmente a las siete organizaciones fundadoras.
El Grupo de Trabajo del Vocabulario Internacional de Metrología del JCGM (JCGM/WG 2)
tiene la misión de revisar y promover el uso del vocabulario, y con este objetivo ha
preparado la tercera edición del mismo. Después de los procesos de consulta, discusión y
votación entre sus miembros, iniciados en 2004, se aprobó y adoptó por unanimidad la
tercera edición en el año 2008, de la cual se ha publicado en 2012 una versión definitiva
que incluye correcciones menores respecto a la de 2008.
Se ha publicado una versión en lengua española de esta tercera edición del VIM a partir
de la colaboración de institutos nacionales de metrología y otras organizaciones
implicadas de diversos países hispanohablantes.
En este documento, la Comisión de Metrología de la Sociedad Española de Bioquímica
Clínica y Patología Molecular (SEQC) recoge las definiciones del VIM que aplican al

8 | P á g i n a
laboratorio clínico. Las definiciones incluidas en este documento sustituyen y actualizan
las contenidas en documentos anteriores publicados por esta Comisión.
Para la consulta de este vocabulario deben tenerse en cuenta las siguientes
consideraciones:
- Las definiciones del VIM son las de la tercera edición. En algunos casos difieren
sustancialmente de las de la segunda edición de 1993. Se han incluido las correcciones
del corrigendum publicado en mayo de 2010.
- Se han incorporado únicamente aquellas notas y ejemplos del VIM que corresponden al
ámbito del laboratorio clínico.
- Las notas y definiciones marcadas con un asterisco han sido elaboradas por la Comisión
de Metrología.
- Se han incluido también definiciones de otros documentos normativos que se detallan en
el apartado de bibliografía.
1.1. OBJETO Y CAMPO DE APLICACIÓN
El presente vocabulario tiene relación con la metrología, “la ciencia de la medida y sus
aplicaciones”, que se aplica a todos los aspectos teóricos y prácticos de las medidas
cualesquiera que sean sus incertidumbres de medida y los dominios de aplicación. El
campo de aplicación es el laboratorio clínico.
1.2 VOCABULARIO
Analito: componente que se indica en el nombre de una magnitud mensurable [ISO
18153:2004, apartado 3.1].
NOTA*: El término “analito” se usa a veces de forma errónea como “mensurando”.
Cadena de trazabilidad metrológica: secuencia de patrones de medida y calibraciones que
es utilizada para relacionar un resultado de medida con una referencia establecida [VIM:
2008, apartado 2.42].
NOTA 1: Una cadena de trazabilidad metrológica se define mediante una jerarquía de
calibración.
NOTA 2: La cadena de trazabilidad metrológica se utiliza para establecer la trazabilidad
metrológica del resultado de medida.
NOTA 3: Una comparación entre dos patrones de medida puede considerarse una
calibración si sirve para comprobar y, si es necesario, corregir el valor y la incertidumbre
de medida atribuidas a uno de los patrones de medida.
Calibración: operación que, en unas condiciones determinadas, establece en un primer
paso una relación entre los valores de una magnitud (con sus incertidumbres de medida)
de unos patrones y las indicaciones correspondientes (con sus incertidumbres de
medida), y que en un segundo paso utiliza esta información para establecer una relación
que permite obtener un resultado de medida a partir de una señal [VIM: 2008, apartado
2.39].
NOTA 1: Una calibración puede expresarse mediante una declaración, una función de
calibración, un diagrama de calibración, una curva de calibración o una tabla de

9 | P á g i n a
calibración. En algunos casos, puede consistir en una corrección aditiva o multiplicativa
de la señal, con una incertidumbre de medida asociada.
NOTA 2: No debe confundirse la calibración con el ajuste de un sistema de medida
(conocido a menudo de forma incorrecta como “auto calibración”), ni con una verificación
de la calibración.
NOTA 3: A menudo, se percibe únicamente el primer paso de la definición como
calibración.
NOTA 4*: El término “indicación” se utiliza a veces en lugar de “señal”. Calibrador: patrón
de medida empleado en calibraciones [VIM: 2008, apartado 5.12].
Capacidad de detección: capacidad de un procedimiento de medida para detectar
pequeñas cantidades de un componente [IUPAC: 1995].
NOTA*: La capacidad de detección se expresa mediante el “valor crítico” y el “límite de
detección”.
Comparabilidad metrológica (de resultados de medida): comparabilidad de resultados
de medida, para magnitudes de un tipo determinado, que son metrológicamente trazables
a la misma referencia [VIM: 2008, apartado 2.46].
NOTA: La comparabilidad metrológica no requiere que los valores medidos y las
incertidumbres de medida asociadas sean del mismo orden de magnitud.
Compatibilidad metrológica (de resultados de medida): propiedad de un conjunto de
resultados de medida correspondiente a un mensurando determinado, tal que, para
cualquier par de valores medidos, el valor absoluto de la diferencia de dos resultados de
medida diferentes es menor que un múltiplo escogido de la incertidumbre de medida
estándar de esta diferencia [VIM:2008, apartado 2.47].
NOTA 1: La compatibilidad metrológica substituye el concepto tradicional de “por debajo
del error”, ya que expresa el criterio para decidir si dos resultados de medida se refieren al
mismo mensurando.
Si en una serie de medidas de un mensurando que se cree constante, un resultado de
medida no es compatible con los otros puede ser que la medida no sea correcta (por
ejemplo, su incertidumbre de medida evaluada es mayor de lo esperado), o bien la
magnitud medida ha cambiado de una medida a otra.
NOTA 2: La correlación entre las medidas ejerce una influencia en la compatibilidad
metrológica. Si las medidas no están correlacionadas, la incertidumbre estándar de su
diferencia es igual a la raíz cuadrada de la suma de los cuadrados de sus incertidumbres
de medida estándar, mientras que es menor para una covariancia positiva o mayor para
una covariancia negativa.
Condición de interés: enfermedad particular, estadio de una enfermedad, estado de salud,
o cualquier otra condición o característica de interés identificable en un individuo, tal como
una enfermedad presente o una condición de salud que puede promover una acción
clínica tal como el inicio, modificación o finalización de un tratamiento [Instituto de
Patrones Clínicos y de Laboratorio, CLSI:2008].

10 | P á g i n a
Condición de precisión intermedia: condición de medida en un conjunto de condiciones
que incluye el mismo procedimiento de medida, el mismo lugar y medidas repetidas del
mismo objeto u objetos similares durante un período de tiempo prolongado, pero que
puede incluir otras condiciones que pueden variar [VIM:2008, apartado 2.22].
NOTA 1: Las “condiciones que pueden variar” pueden ser nuevas calibraciones, patrones,
operadores y sistemas de medida.
NOTA 2: Una especificación relativa a las condiciones debe contener, siempre que sea
posible, las condiciones que varían y las que no.
NOTA 3: En química, a veces se utiliza el término “condición de precisión interserial” para
designar este concepto.
Condición de repetibilidad: condición de medida en un conjunto de condiciones que
incluye el mismo procedimiento de medida, los mismos operadores, el mismo sistema de
medida, las mismas condiciones de funcionamiento y la misma ubicación, así como
también medidas repetidas del mismo objeto u objetos similares durante un corto período
de tiempo [VIM:2008, apartado 2.20].
NOTA 1: Una condición de medida es una condición de repetibilidad si se especifica en un
conjunto determinado de condiciones de repetibilidad.
NOTA 2: En química, a veces se utiliza el término “condición de precisión intraserial” para
designar este concepto.
Condición de reproducibilidad: condición de medida en un conjunto de condiciones que
incluye ubicaciones, operadores y sistemas de medida diferentes, así como también
medidas repetidas del mismo objeto u objetos similares [VIM: 2008, apartado 2.24].
NOTA 1: Los “sistemas de medida diferentes” pueden utilizar procedimientos de medida
diferentes.
NOTA 2: Una especificación relativa a las condiciones debe contener, siempre que sea
posible, las condiciones que varían y las que no.
Conmutabilidad de un material de referencia: característica de un material de
referencia expresada por la concordancia entre los resultados de medida obtenidos para
una magnitud [individual] determinada de este material utilizando dos procedimientos de
medida determinados, por una parte, y la relación entre resultados de medida para otros
materiales determinados, por la otra [VIM:2008, apartado 5.15].
NOTA 1: El “material de referencia” es en general un calibrador, y los “otros materiales
determinados” son generalmente muestras de rutina.
NOTA 2: Los procedimientos de medida son el precedente y el siguiente al material de
referencia usado como calibrador en una jerarquía de calibración (véase la norma ISO
17511:2004).
NOTA 3: La estabilidad de los materiales de referencia conmutables debe comprobarse
regularmente.
Corrección: compensación de un efecto sistemático estimado [VIM: 2008, apartado 2.53].
NOTA: La “compensación” puede tener diversas formas, tales como la adición de un valor
o la multiplicación por un factor, o puede ser deducida de una tabla.

11 | P á g i n a
Curva de calibración: expresión de la relación entre una señal y el valor medido
correspondiente [VIM: 2008, apartado 4.31].
NOTA: Una curva de calibración expresa una relación biunívoca que no proporciona un
resultado de medida porque no contiene ninguna información sobre la incertidumbre de
medida.
Deriva instrumental: variación continua o que se incrementa con el tiempo de una señal,
debida a cambios en las características metrológicas de un instrumento de medida [VIM:
2008, apartado 4.21].
NOTA: La deriva instrumental no está ligada a una variación de la magnitud medida, ni a
una variación de una magnitud influyente identificada.
Desviación de medida: estimación de un error sistemático [VIM: 2008, apartado 2.18].
NOTA*: También suele denominarse “sesgo”.
Efecto matriz: influencia de una propiedad de una muestra, distinta del mensurando, en
la medida del mensurando con un determinado procedimiento de medida y en el valor
medido [ISO 17511:2004, apartado 3.15].
NOTA: Una causa específica de efecto matriz es una magnitud influyente.
Equipo de medida: instrumento de medida, software, patrón de medida, material de
referencia, aparatos auxiliares o una combinación de estos, necesarios para realizar un
proceso de medida [ISO 9000:2005, apartado 3.10.4].
Error aleatorio: componente del error de medida que, en mediciones repetidas, varia de
manera impredecible [VIM: 2008, apartado 2.19].
NOTA 1: El valor de referencia de una magnitud para un error aleatorio es la media que
resultaría de un número infinito de mediciones repetidas del mismo mensurando.
NOTA 2: Los errores aleatorios de medida de un conjunto de mediciones repetidas tienen
una distribución que puede resumirse por su esperanza matemática, que generalmente se
supone es cero, y por su variancia.
NOTA 3: El error aleatorio es igual a la diferencia entre el error de medida y el error
sistemático.
Error de medida: diferencia entre el valor medido y el valor de referencia de una
magnitud [VIM: 2008, apartado 2.16].
NOTA: No debe confundirse el error de medida con un error de producción o una
equivocación humana.
Error de medida máximo permitido: valor extremo del error de medida, con respecto al
valor de referencia de una magnitud conocida, permitido por especificaciones o
regulaciones para una medida, un instrumento de medida o un sistema de medida
determinados [VIM:2008, apartado 4.26].
NOTA 1: Cuando hay dos valores extremos generalmente se utilizan los términos “errores
máximos permitidos” o “límites de error”.

12 | P á g i n a
NOTA 2: No debería usarse el término “tolerancia” para designar el “error de medida
máximo permitido”.
NOTA 3*: También suele denominarse “error total permitido”.
Error sistemático: componente del error de medida que, en mediciones repetidas, se
mantiene constante o varía de forma previsible [VIM: 2008, apartado 2.17].
NOTA 1: El valor de referencia de una magnitud para un error sistemático es un valor
verdadero, un valor medido de un patrón de medida con una incertidumbre de medida
insignificante o un valor convencional.
NOTA 2: El error sistemático y sus causas pueden ser conocidos o desconocidos. Puede
aplicarse una corrección para compensar un error sistemático conocido.
NOTA 3: El error sistemático es igual a la diferencia entre el error de medida y el error
aleatorio.
NOTA 4*: Su estimación es la “desviación de medida” o “sesgo”.
Escala de valores ordinal: escala de valores para las magnitudes ordinales [VIM: 2008,
apartado 1.28].
NOTA 1: Una escala ordinal puede establecerse mediante medidas de acuerdo a un
procedimiento de medida.
NOTA 2*: Se dividen en escalas binarias compuestas de dos valores (por ejemplo: 0/1;
negativo/positivo) y escalas de tres o más valores (por ejemplo: 0/1/2/3;
ausente/poco/moderado/mucho).
Especificidad diagnóstica: porcentaje de individuos sin la condición de interés
(determinada por un criterio diagnóstico preciso) con resultado de la prueba negativo
[CLSI: 2008].
NOTA 1: La “condición de interés” debe ser definida por un criterio independiente de la
prueba a considerar.
NOTA 2: La especificidad se calcula como: 100 x VN / (FP + VN).
Evaluación de tipo A de la incertidumbre de medida: evaluación de un componente de
la incertidumbre de medida mediante un análisis estadístico de los valores medidos
obtenidos en condiciones definidas de medición [VIM: 2008, apartado 2.28].
Evaluación de tipo B de la incertidumbre de medida: evaluación de un componente de
la incertidumbre de medida por otros medios distintos a una evaluación de tipo A de la
incertidumbre de medida [VIM: 2008, apartado 2.29].
EJEMPLOS: Evaluación fundamentada en informaciones:
- asociadas a valores publicados y reconocidos,
- asociadas al valor de un material de referencia certificado,
- obtenidas a partir de un certificado de calibración,
- relacionadas con la deriva,
- obtenidas a partir de la clase de exactitud de un instrumento de medida verificado, y
- obtenidas a partir de límites deducidos de la experiencia personal.

13 | P á g i n a
Exactitud de medida: grado de concordancia entre un valor medido y un valor verdadero
del mensurando [VIM: 2008, apartado 2.13].
NOTA 1: La exactitud de medida no es una magnitud y no se expresa numéricamente. Se
dice que una medida es más exacta si genera un error de medida menor.
NOTA 2: No es conveniente utilizar el término “exactitud de medida” en lugar de
“veracidad de medida”, ni el término “precisión de medida” en lugar de “exactitud de
medida”. Este último está relacionado con la veracidad y con la precisión.
NOTA 3: La exactitud de medida se interpreta, a veces, como la concordancia entre los
valores medidos que son atribuidos a un mensurando.
Factor de cobertura: número superior a uno por el cual se multiplica la incertidumbre de
medida estándar combinada para obtener una incertidumbre de medida expandida [VIM:
2008, apartado 2.38].
NOTA: Un factor de cobertura se simboliza habitualmente mediante el símbolo k
Falso negativo (FN): resultado negativo de una prueba en un individuo que presenta la
condición de interés (determinada por un criterio diagnóstico preciso) [CLSI: 2008].
Falso positivo (FP): resultado positivo de una prueba en un individuo que no presenta la
condición de interés (determinada por un criterio diagnóstico preciso) [CLSI:2008].
Heterocedasticidad*: propiedad de los resultados de un procedimiento de medida por la
cual la desviación estándar depende del valor del mensurando, dentro de un intervalo de
valores particular.
Homocedasticidad*: propiedad de los resultados de un procedimiento de medida por la
cual la desviación estándar es la misma para cualquier valor del mensurando, dentro de
un intervalo de valores particular.
Incertidumbre de medida: parámetro no negativo que caracteriza la dispersión de los
valores atribuidos a un mensurando basado en la información utilizada [VIM: 2008,
apartado 2.26].
NOTA 1: La incertidumbre de medida incluye componentes que provienen de efectos
sistemáticos, tales como los componentes asociados a correcciones y a los valores
asignados a los patrones, así como la incertidumbre de la definición. A veces, los efectos
sistemáticos estimados en lugar de corregirse se incorporan como componentes
asociados a la incertidumbre de medida.
NOTA 2: El parámetro puede ser, por ejemplo, una desviación estándar, denominada
incertidumbre de medida estándar (o uno de sus múltiplos), o la semiamplitud de un
intervalo con una probabilidad de cobertura determinada.
NOTA 3: La incertidumbre de medida incluye en general muchos componentes. Algunos
pueden ser evaluados mediante una evaluación de tipo A de la incertidumbre de medida a
partir de la distribución estadística de los valores que provienen de series de mediciones,
y pueden ser caracterizados por desviaciones estándar.

14 | P á g i n a
Los otros componentes, que pueden ser evaluados mediante una evaluación de tipo B de
la incertidumbre de medida, pueden también ser caracterizados por desviaciones
estándar, evaluadas a partir de funciones de densidad de probabilidad basadas en la
experiencia y otras informaciones.
NOTA 4: En general, para un conjunto dado de información, se entiende que la
incertidumbre de medida se asocia a un valor determinado atribuido al mensurando. Una
modificación de este valor comporta una modificación de la incertidumbre de medida
asociada.
Incertidumbre de la definición: componente de la incertidumbre de medida que resulta
de la cantidad limitada de detalles en la definición de un mensurando [VIM: 2008,
apartado 2.27].
NOTA 1: La incertidumbre de la definición es la incertidumbre mínima que puede
obtenerse en la práctica en cualquier medida de un mensurando determinado.
NOTA 2: Toda modificación de los detalles descriptivos comporta otra incertidumbre de la
definición.
Incertidumbre de medida estándar: incertidumbre de medida expresada en forma de
desviación estándar [VIM: 2008, apartado 2.30].
Incertidumbre de medida estándar combinada: incertidumbre de medida estándar
obtenida utilizando las incertidumbres de medida estándar individuales asociadas a las
magnitudes de entrada en un modelo de medida [VIM: 2008, apartado 2.31].
NOTA: Cuando existen correlaciones entre las magnitudes de entrada en un modelo de
medida, deben también tenerse en cuenta las covariancias en el cálculo de la
incertidumbre estándar combinada.
Incertidumbre de medida estándar relativa: cociente entre la incertidumbre de medida
estándar y el valor absoluto de la magnitud [VIM: 2008, apartado 2.32].
Incertidumbre de medida expandida: producto de una incertidumbre de medida
estándar combinada y de un factor superior a uno [VIM: 2008, apartado 2.35].
NOTA 1: El factor depende del tipo de distribución de probabilidad de la magnitud de
salida en un modelo de medida y de la probabilidad de cobertura escogida.
NOTA 2: El factor que interviene en la definición es un factor de cobertura.
Indicación: valor proporcionado por un instrumento de medida o un sistema de medida
[VIM: 2008, apartado 4.1].
NOTA 1: Una indicación puede presentarse en forma visual o acústica o puede
transferirse a otro dispositivo. A menudo, una indicación viene dada por la posición de una
aguja en una pantalla para las salidas analógicas, por un número visualizado o impreso
para las salidas numéricas, por una configuración codificada para las salidas codificadas,
o por un valor asignado para las mediciones materializadas.
NOTA 2: Una indicación y el valor de la magnitud medida correspondiente no son
necesariamente valores de magnitudes del mismo tipo.
NOTA 3*: El término “indicación” se utiliza a veces en lugar de “señal”.

15 | P á g i n a
Instrumento de medida: dispositivo utilizado para efectuar mediciones, sólo o asociado a
uno o varios dispositivos anexos [VIM: 2008, apartado 3.1].
NOTA 1: Un instrumento de medida que puede ser utilizado sólo, es un sistema de
medida.
NOTA 2: Un instrumento de medida puede ser un instrumento de medida con indicaciones
o una medida materializada.
Intervalo de medida: conjunto de valores de un mismo tipo de magnitud que un
instrumento de medida o un sistema de medida determinado puede medir con una
incertidumbre de medida instrumental dada, en condiciones determinadas [VIM:2008,
apartado 4.7].
NOTA 1: No debe confundirse el “límite inferior de un intervalo de medida” con el “límite
de detección”.
NOTA 2*: En el laboratorio clínico habitualmente abarca desde el “valor crítico” hasta el
“límite de linealidad”.
Intervalo no fiable: zona alrededor del punto de corte que corresponde a un intervalo de
valores donde pueden encontrarse falsos positivos y falsos negativos.
NOTA*: También suele denominarse “zona gris”.
Jerarquía de calibración: secuencia de calibraciones desde una referencia hasta el
sistema de medida final, en la cual el resultado de cada calibración depende del resultado
de la precedente [VIM: 2008, apartado 2.40].
NOTA 1: La incertidumbre de medida aumenta necesariamente durante la secuencia de
calibraciones.
NOTA 2: Los elementos de una jerarquía de calibración son unos patrones de medida y
unos sistemas de medida que se usan siguiendo unos procedimientos de medida.
NOTA 3: La referencia indicada puede ser una definición de una unidad de medida en su
materialización práctica, un procedimiento de medida o un patrón de medida.
NOTA 4: Una comparación entre dos patrones de medida puede considerarse como una
calibración si sirve para comprobar y, en caso necesario, corregir el valor de la magnitud y
la incertidumbre de medida atribuidos a uno de los patrones de medida.
Límite de cuantificación: valor verdadero mínimo de la magnitud que puede estimarse
con una imprecisión aceptable (habitualmente el 10 %) [IUPAC: 1995].
NOTA 1*: También suele denominarse “valor mínimo cuantificable”.
NOTA 2*: No debe utilizarse el término “sensibilidad funcional” en lugar de “límite de
cuantificación”.
Límite de detección: valor medido obtenido mediante un procedimiento de medida
determinado, para el que la probabilidad de declarar falsamente la ausencia de un
componente en un material es β, dada la probabilidad α de declarar falsamente su
presencia [VIM: 2008, apartado 4.18].
NOTA 1: La IUPAC recomienda por defecto valores de α y β iguales a 0,05.
NOTA 2: No debe utilizarse el término “sensibilidad” en lugar de “límite de detección”.

16 | P á g i n a
Magnitud: propiedad de un fenómeno, de un cuerpo o de una sustancia, que se puede
expresar cuantitativamente mediante un número y una referencia [VIM: 2008, apartado
1.1].
NOTA 1: La “referencia” puede ser una unidad de medida, un procedimiento de medida,
un material de referencia o una de sus combinaciones.
NOTA 2: El formato escogido por la IUPAC y la IFCC para la designación de magnitudes
en los laboratorios clínicos es “Sistema−Componente; tipo de magnitud”.
EJEMPLO: Plasma (Sangre)−Ion sodio; concentración de sustancia igual a 143 mmol/L
en una persona determinada en un instante determinado.
Magnitud influyente: magnitud que en una medición directa no afecta a la magnitud
medida realmente, pero que afecta a la relación entre la señal y el resultado de la
medición [VIM: 2008, apartado 2.52].
EJEMPLO: La concentración de sustancia de bilirrubina en una medición directa de la
concentración de sustancia de hemoglobina en el plasma humano.
NOTA 1: Una medida indirecta implica una combinación de medidas directas, sobre cada
una de las cuales pueden tener efecto unas magnitudes influyentes.
NOTA 2*: En el laboratorio clínico se utiliza también el término “interferencia”.
Material de referencia: material suficientemente homogéneo y estable en relación a unas
propiedades determinadas, que se ha establecido como apto para su uso previsto en una
medición o en un examen de propiedades cualitativas [VIM: 2008, apartado 5.13].
NOTA 1: El examen de una propiedad cualitativa comprende la atribución de un valor y
una incertidumbre asociada. Esta incertidumbre no es una incertidumbre de medida.
NOTA 2: Los materiales de referencia con o sin valores asignados pueden servir para
controlar la precisión de medida, mientras que tan solo los materiales de referencia con
valores asignados pueden servir para la calibración o el control de la veracidad de
medida.
NOTA 3: Los materiales de referencia materializan magnitudes y, también, propiedades
cualitativas.
EJEMPLO 1: Ejemplos de materiales de referencia que materializan magnitudes:
a) agua de una pureza determinada, cuya viscosidad dinámica se utiliza para la
calibración de viscosímetros;
b) suero humano sin un valor asignado para la concentración de sustancia de colesterol,
utilizado tan solo para el control de la precisión de medida.
EJEMPLO 2: Ejemplos de materiales de referencia que materializan propiedades
cualitativas:
a) ADN que contiene una secuencia determinada de nucleótidos;
b) orina que contiene 19-androstendiona.
NOTA 4: Algunos materiales de referencia tienen valores asignados que son trazables
metrológicamente a una unidad de medida que no pertenece a un sistema de unidades.
Ejemplos de estos materiales son las vacunas de la Organización Mundial de la Salud
(OMS) a las cuales se han asignado Unidades Internacionales (UI).

17 | P á g i n a
NOTA 5: Un material de referencia determinado puede ser utilizado en una medición
determinada solo para la calibración o para la garantía de la calidad.
NOTA 6: Las especificaciones de un material de referencia deben incluir su trazabilidad,
indicando su origen y procesamiento.
Material de referencia certificado (MRC): material de referencia acompañado de
documentación emitida por un organismo autorizado y que proporciona uno o más valores
de propiedades especificadas, con las incertidumbres y trazabilidades asociadas,
utilizando procedimientos validados [VIM: 2008, apartado 5.14].
EJEMPLO: Suero humano cuyo valor asignado para la concentración de colesterol y la
incertidumbre de medida asociada están indicados en un certificado, y que se utiliza como
calibrador o como material de control de la veracidad de medida.
NOTA 1: La documentación citada es emitida en forma de certificado.
NOTA 2: En la definición, el término “incertidumbre” puede designar tanto una
incertidumbre de medida como la incertidumbre asociada al valor de una propiedad
cualitativa, como la identidad o la secuencia. El término “trazabilidad” designa la
trazabilidad metrológica del valor de una magnitud o la trazabilidad del valor de una
propiedad cualitativa.
NOTA 3: Los valores de magnitudes especificadas de materiales de referencia
certificados han de tener una trazabilidad metrológica conocida y una incertidumbre de
medida asociada.
Medida: proceso para obtener experimentalmente uno o más valores que se pueden
atribuir de forma razonable a una magnitud [VIM: 2008, apartado 2.1].
NOTA 1: La medida no se aplica a las propiedades cualitativas.
NOTA 2: Una medida implica la comparación de magnitudes o el recuento de entidades.
NOTA 3: Una medida presupone una descripción de la magnitud compatible con la
utilización prevista de un resultado de medida, un procedimiento de medida y un sistema
de medida calibrado que funciona según un procedimiento de medida especificado,
incluyendo las condiciones de medida.
Mensurando: magnitud que se quiere medir [VIM: 2008, apartado 2.3].
NOTA 1: La especificación de un mensurando requiere el conocimiento del tipo de
magnitud, la descripción del estado del fenómeno, cuerpo o sustancia que contiene la
magnitud, incluyendo cualquier componente pertinente, y las entidades químicas
involucradas.
NOTA 2: En química, se utiliza a menudo el término “analito” o el nombre de una
sustancia o componente en lugar de “mensurando”.
Esta utilización es errónea porque estos términos no se refieren a magnitudes.
EJEMPLO 1: Concentración de masa de albúmina en plasma.
EJEMPLO 2: Concentración catalítica de alanina-aminotransferasa en plasma medida
mediante la velocidad de conversión del NADH en el procedimiento de medida de
referencia primario de la IFCC.

18 | P á g i n a
Método de medida: descripción genérica de la organización lógica de las operaciones
utilizadas en una medida [VIM: 2008, apartado 2.5].
Patrón de medida: realización de la definición de una magnitud determinada, con un
valor determinado y una incertidumbre de medida asociada, utilizada como referencia
[VIM: 2008, apartado 5.1].
EJEMPLO 1: Patrón de masa de 1 kg con una incertidumbre de medida estándar
asociada de 3 μg.
EJEMPLO 2: Solución tampón patrón con un pH de 7,072 con una incertidumbre de
medida estándar asociada de 0,006.
EJEMPLO 3: Serie de soluciones de referencia de cortisol en suero humano, en la que
cada solución tiene un valor certificado con una incertidumbre de medida.
EJEMPLO 4: Material de referencia que suministra valores con las incertidumbres de
medida asociadas para la concentración de masa de diez proteínas diferentes.
NOTA 1: La “realización de la definición de una magnitud determinada” puede ser
proporcionada por un sistema de medida, una medida materializada o un material de
referencia.
NOTA 2: Un patrón sirve a menudo de referencia en el establecimiento de valores
medidos e incertidumbres de medida asociadas para otras magnitudes del mismo tipo,
estableciendo así una trazabilidad metrológica mediante la calibración de otros patrones,
instrumentos de medida o sistemas de medida.
NOTA 3: El término “realización” se utiliza en su sentido más general, designando tres
procedimientos: el primero, la materialización sensu stricto, es la materialización física de
la unidad a partir de su definición; el segundo, denominado “reproducción” consiste en
establecer un patrón altamente reproducible fundamentado en un fenómeno físico, no en
materializar la unidad a partir de su definición; y el tercer procedimiento consiste en
adoptar una medida materializada como patrón, es el caso del patrón de 1 kg.
NOTA 4: La incertidumbre estándar asociada a un patrón es siempre un componente de
la incertidumbre estándar combinada en un resultado de medida obtenido utilizando el
patrón. A menudo este componente es pequeño en relación con otros componentes de la
incertidumbre estándar combinada.
NOTA 5: El valor de la magnitud y la incertidumbre de medida deben determinarse en el
momento en el que se utiliza el patrón.
NOTA 6: Diversas magnitudes del mismo tipo o de tipo diferente pueden materializarse
con un mismo dispositivo, denominado también patrón.
NOTA 7*: Existe una jerarquía metrológica de patrones de medida (véase la norma ISO
17511:2004).
Patrón de medida de referencia: patrón de medida concebido para la calibración de
otros patrones del mismo tipo de magnitud en una determinada organización o en un
lugar determinados [VIM:2008, apartado 5.6].
Patrón de medida internacional: patrón de medida reconocido por los signatarios de un
acuerdo internacional para una utilización mundial [VIM:2008, apartado 5.2].

19 | P á g i n a
EJEMPLO: Coriogonadotropina, cuarto patrón internacional de la OMS, 1999, 75/589, 650
UI por vial.
Patrón de medida nacional: patrón de medida reconocido por una autoridad nacional
para servir, en un estado o una economía, como base para asignar valores de
magnitudes a otros patrones del mismo tipo de magnitud [VIM:2008, apartado 5.3].
Patrón de medida primario: patrón de medida establecido mediante un procedimiento de
medida primario o creado como objeto escogido por convenio [VIM:2008, apartado 5.4].
EJEMPLO 1: Patrón primario de concentración de sustancia preparado disolviendo una
cantidad de sustancia conocida de un compuesto químico en un volumen conocido de
solución.
EJEMPLO 2: El prototipo internacional del kilogramo en tanto que objeto designado por
convenio.
Patrón de medida secundario: patrón de medida establecido mediante una calibración con
respecto a un patrón de medida primario del mismo tipo de magnitud [VIM: 2008,
apartado 5.5].
NOTA: La relación entre el patrón de medida primario y el secundario se puede obtener
directamente o mediante un sistema de medida intermedio calibrado con el patrón
primario, que asigna un resultado de medida a un patrón secundario.
Precisión de medida: concordancia entre las indicaciones o los valores medidos
obtenidos mediante medidas repetidas del mismo objeto u objetos similares en
condiciones especificadas [VIM:2008, apartado 2.15].
NOTA 1: La precisión de medida se expresa por lo general numéricamente mediante
medidas de imprecisión tales como la desviación estándar, la variancia o el coeficiente de
variación en las condiciones especificadas.
NOTA 2: Las “condiciones especificadas” pueden ser, por ejemplo, condiciones de
repetibilidad, condiciones de precisión intermedia o condiciones de reproducibilidad
(véase la norma ISO 5725-1:1994).
NOTA 3: La precisión de medida se utiliza para definir la repetibilidad de medida, la
precisión intermedia de medida y la reproducibilidad de medida.
NOTA 4: A veces el término “precisión de medida” se utiliza de forma inadecuada para
designar la “exactitud de medida”.
Principio de medida: fenómeno que sirve como base de una medida [VIM:2008,
apartado 2.4].
EJEMPLO 1: Efecto termoeléctrico aplicado a la medición de la temperatura.
EJEMPLO 2: Absorción de energía aplicada a la medición de la concentración de
sustancia.
EJEMPLO 3: Disminución de la concentración de glucosa en sangre en ayunas aplicada a
la medición de la concentración de insulina en una preparación.
NOTA: El “fenómeno” puede ser de tipo físico, químico o biológico.

20 | P á g i n a
Procedimiento de medida: descripción detallada de una medida de acuerdo con uno o
más principios de medida y con un método de medida determinado, basado en un modelo
de medida e incluyendo cualquier cálculo destinado a obtener un resultado de medida
[VIM:2008, apartado 2.6].
NOTA 1: Un procedimiento de medida está habitualmente documentado con suficiente
detalle para permitir que un operario efectúe la medida.
NOTA 2: Un procedimiento de medida puede incluir una declaración referente a un
objetivo de incertidumbre de medida.
NOTA 3: A veces a un procedimiento de medida se le denomina en inglés standard
operating procedure (SOP) y en español “procedimiento normalizado de trabajo” (PNT).
NOTA 4*: Existe una jerarquía metrológica de procedimientos de medida (véase la norma
ISO 17511:2004).
Procedimiento de medida de referencia: procedimiento de medida que se considera
produce resultados de medida apropiados a su uso previsto para la evaluación de la
veracidad de los valores medidos obtenidos mediante otros procedimientos de medida
para magnitudes del mismo tipo, para una calibración o para la caracterización de
materiales de referencia [VIM:2008, apartado 2.7].
Procedimiento de medida de referencia primario: procedimiento de medida de referencia
utilizado para obtener un resultado de medida sin relación con un patrón de medida de
una magnitud del mismo tipo [VIM: 2008, apartado 2.8].
EJEMPLO: El volumen de agua dispensado por una pipeta de 5 mL a 20 °C se mide
pesando el agua dispensada por la pipeta en un recipiente, considerando la diferencia
entre la masa del recipiente que contiene el agua y la masa del recipiente inicialmente
vacío, y corrigiendo después la diferencia de masa por la temperatura real del agua,
utilizando la densidad de masa.
Propiedad cualitativa: propiedad de un fenómeno, un cuerpo o una sustancia que no se
puede expresar cuantitativamente [VIM: 2008, apartado 1.30].
EJEMPLO 1: Sexo de una persona.
EJEMPLO 2: Código ISO de país, de dos letras.
EJEMPLO 3: Secuencia de aminoácidos de un polipéptido.
NOTA: Una propiedad cualitativa tiene un valor que puede expresarse mediante palabras,
códigos alfanuméricos u otros medios.
Punto de corte (de un procedimiento cualitativo): umbral por encima del cual el
resultado es informado como positivo y por debajo del cual es informado como negativo
[CLSI:2008].
NOTA*: También suele denominarse “valor discriminante”.
Repetibilidad de medida: precisión de medida de acuerdo con un conjunto de
condiciones de repetibilidad [VIM:2008, apartado 2.21].
Reproducibilidad de medida: precisión de medida de acuerdo con un conjunto de
condiciones de reproducibilidad [VIM:2008, apartado 2.25].

21 | P á g i n a
Resolución: variación mínima de una magnitud medida que produce una variación
perceptible de la señal correspondiente [VIM:2008, apartado 4.14].
NOTA: La resolución puede depender del valor de la magnitud medida.
Selectividad de un examen cualitativo*: capacidad de un examen cualitativo de
distinguir la señal correspondiente a la propiedad en estudio de otras propiedades
potencialmente influyentes
Selectividad de un sistema de medida: propiedad de un sistema de medida, utilizando
un procedimiento de medida determinado, gracias a la cual el sistema proporciona valores
medidos, para uno o más mensurados, de forma que los valores de cada mensurando
son independientes de otros mensurados u otras magnitudes del fenómeno, cuerpo o
sustancia que se investiga [VIM:2008, apartado 4.13].
EJEMPLO: Capacidad de un sistema de medida para medir la concentración de sustancia
de Creatinina en el plasma mediante el procedimiento de Jaffé, sin ser influido por las
concentraciones de glucosa, urato, cetona y proteína.
NOTA 1: En química, las magnitudes medidas implican a menudo diferentes componentes
en el sistema que se está midiendo y estas magnitudes no son necesariamente del mismo
tipo.
NOTA 2*: En el laboratorio clínico también se usa el término “especificidad analítica” para
indicar la “selectividad de un sistema de medida”.
Sensibilidad de un sistema de medida: cociente entre la variación de una señal de un
sistema de medida y la variación correspondiente del valor de la magnitud medida [VIM:
2008, apartado 4.12].
NOTA 1: La sensibilidad de un sistema de medida puede depender del valor de la
magnitud medida.
NOTA 2: La variación considerada del valor de la magnitud medida ha de ser elevada en
comparación con la resolución.
NOTA 3*: El término “indicación” se utiliza a veces en lugar de “señal”.
Sensibilidad diagnóstica: porcentaje de individuos con la condición de interés
(determinada por un criterio diagnóstico preciso) con resultado de la prueba positivo
[CLSI: 2008].
NOTA 1: La “condición de interés” debe ser definida por un criterio independiente de la
prueba a considerar.
NOTA 2: La sensibilidad se calcula como: 100 x VP / (FN + VP).
Sistema de medida: conjunto de uno o más instrumentos de medida y a menudo otros
dispositivos, incluyendo cualquier reactivo y suministro, ensamblados y adaptados para
proporcionar informaciones destinadas a obtener valores de medida dentro de intervalos
especificados para magnitudes de un tipo determinado [VIM:2008, apartado 3.2].
NOTA: Un sistema de medida puede consistir en un único instrumento de medida.

22 | P á g i n a
Trazabilidad metrológica: propiedad del resultado de una medida mediante la cual este
resultado puede relacionarse con una referencia por medio de una cadena ininterrumpida
y documentada de calibraciones, cada una de las cuales contribuye a la incertidumbre de
medida [VIM: 2008, apartado 2.41].
NOTA 1: La “referencia” puede ser una definición de una unidad de medida materializada
de forma práctica, un procedimiento de medida que incluya la unidad de medida, cuando
se trata de una magnitud que no sea ordinal, o un patrón de medida
NOTA 2: La trazabilidad metrológica requiere la existencia de una jerarquía de calibración.
NOTA 3: La trazabilidad metrológica de un resultado de medida no garantiza que la
incertidumbre de medida sea adecuada para un propósito determinado ni la ausencia de
errores.
NOTA 4: Una comparación entre dos patrones de medida puede considerarse como una
calibración si se usa para verificar y, en caso necesario, corregir el valor y la
incertidumbre de medida atribuidos a uno de los patrones.
NOTA 5: ILAC considera que los elementos necesarios para confirmar la trazabilidad
metrológica son: una cadena de trazabilidad metrológica ininterrumpida hasta un patrón
internacional o un patrón nacional, una incertidumbre de medida documentada, un
procedimiento de medida documentado, una competencia técnica reconocida, la
trazabilidad metrológica al SI de unidades y unos intervalos entre calibraciones.
NOTA 6: El término abreviado “trazabilidad” se utiliza a veces para designar la trazabilidad
metrológica, así como otros conceptos como la trazabilidad de una muestra, de un
documento, de un instrumento o de un material, en el que se hace referencia a la historia
(“rastro”) de la entidad. Es, por tanto, preferible utilizar el término completo “trazabilidad
metrológica” si existe el riesgo de confusión.
Validación: verificación de que los requisitos especificados son adecuados para un uso
determinado [VIM:2008, apartado 2.45].
EJEMPLO: Un procedimiento de medida, habitualmente utilizado para la medición de la
concentración de masa de nitrógeno en agua, también puede ser validado para la
medición de la concentración de masa de nitrógeno en suero humano.
NOTA 1*: La validación de las características metrológicas de un procedimiento de
medida es la confirmación mediante el suministro de pruebas objetivas de que el
procedimiento de medida cumple los requisitos metrológicos del laboratorio clínico para la
medición de una magnitud en las muestras procedentes de pacientes.
NOTA 2*: La validación se realiza cuando se implanta el procedimiento de medida y cada
vez que se modifica de tal forma que puedan verse afectadas sus características
metrológicas.
Valor convencional de una magnitud: valor atribuido a una magnitud mediante acuerdo
para un propósito determinado [VIM:2008, apartado 2.12].
EJEMPLO: Valor convencional de un patrón de masa determinado,
m = 100,00347 g.
NOTA 1: A veces se utiliza el término “valor convencionalmente verdadero” para este
concepto, pero no se aconseja su uso.
NOTA 2: A veces un valor convencional es la estimación de un valor verdadero.

23 | P á g i n a
NOTA 3: Generalmente, un valor co0nvencional se considera asociado a una
incertidumbre de medida convenientemente pequeña, que puede ser cero.
Valor crítico: valor mínimo de la estimación de una magnitud para el cual la probabilidad
de que el valor verdadero de la magnitud sea cero es a (habitualmente 0,05) [IUPAC:
1995].
NOTA 1*: También suele denominarse “nivel crítico”.
NOTA 2*: No deberían usarse los términos “límite de identificación”, “sensibilidad”, “dosis
mínima detectable” y “límite de detección” en lugar de “valor crítico”.
NOTA 3*: Un valor estimado superior o igual al valor crítico corresponde (habitualmente
con el 95 % de probabilidad) a una muestra con analito. Por el contrario, un valor
estimado inferior al valor crítico no demuestra ausencia de analito.
Valor de referencia de una magnitud: valor de una magnitud que sirve de base de
comparación con valores de magnitudes del mismo tipo [VIM: 2008, apartado 5.18].
NOTA 1: El valor de referencia de una magnitud puede ser un valor verdadero de un
mensurando, que es desconocido, o un valor convencional, que es conocido.
NOTA 2: Un valor de referencia de una magnitud asociado a su incertidumbre de medida
se refiere habitualmente a:
a) un material, por ejemplo un material de referencia certificado,
b) un procedimiento de medida de referencia,
c) una comparación de patrones de medida.
NOTA 3*: El término “valor de referencia de una magnitud” corresponde a un concepto
metrológico y no debe confundirse con el término “valor de referencia” o “valor de
referencia biológico” correspondiente al concepto central de la teoría de valores de
referencia propia del laboratorio clínico.
Valor nominal de una magnitud: valor redondeado o aproximado de una magnitud
característica de un instrumento de medida o de un sistema de medida que sirve de guía
para su utilización [VIM: 2008, apartado 4.6].
EJEMPLO 1: El valor 1.000 mL indicado en un matraz aforado.
EJEMPLO 2: El valor 0,1 mol/L de la concentración de sustancia de una solución de ácido
clorhídrico, HCl.
EJEMPLO 3: El valor de 20 ºC como temperatura máxima de almacenamiento.
Valor verdadero de una magnitud: valor de una magnitud compatible con su definición
[VIM: 2008, apartado 2.11].
NOTA 1: En la descripción de la medida en el enfoque “error”, el valor verdadero es
considerado como único y, en la práctica, imposible de conocer. El enfoque
“incertidumbre” consiste en reconocer que, debido a la cantidad intrínsecamente
incompleta de detalles en la definición de una magnitud, no hay un solo valor verdadero
sino un conjunto de valores verdaderos compatibles con la definición. A pesar de esto,
este conjunto de valores es, en principio y en la práctica, imposible de conocer. Otros
enfoques evitan completamente el concepto de valor verdadero y evalúan la validez de
los resultados de medida con ayuda del concepto de compatibilidad metrológica de
resultados de medida.

24 | P á g i n a
NOTA 2: En el caso particular de las constantes fundamentales, se considera que la
magnitud tiene un único valor verdadero.
NOTA 3: Cuando la incertidumbre de la definición asociada al mensurando se considera
insignificante comparada con otros componentes de la incertidumbre de medida, puede
considerarse que el mensurando tiene un valor verdadero esencialmente único.
Este enfoque es el adoptado por la Guía para la Expresión de la Incertidumbre de Medida
(GUM) y otros documentos asociados, en los que se considera redundante el término
“verdadero”.
Veracidad de medida: concordancia entre la media de un número infinito de valores
medidos repetidos y un valor de referencia de una magnitud [VIM: 2008, apartado 2.14].
NOTA 1: La veracidad de medida no es una magnitud y no puede expresarse
numéricamente.
NOTA 2: La veracidad de medida varía en sentido inverso al error sistemático, pero no
está relacionada con el error aleatorio.
NOTA 3: No debería usarse el término “exactitud de medida” en lugar de “veracidad de
medida” ni viceversa.
Verdadero negativo (VN): resultado negativo de una prueba en un individuo que no
presenta la condición de interés (determinada por un criterio diagnóstico preciso) [CLSI:
2008].
Verdadero positivo (VP): resultado positivo de una prueba en un individuo que presenta
la condición de interés (determinada por un criterio diagnóstico preciso) [CLSI:2008].
Verificación: provisión de evidencia objetiva de que una entidad dada satisface unos
requisitos determinados [VIM:2008, apartado 2.44].
EJEMPLO 1: Confirmación de que un material de referencia determinado es, tal como se
declara, homogéneo para el valor y el procedimiento de medida en cuestión para
muestras con un valor de hasta un mínimo de 10 mg de masa.
EJEMPLO 2: Confirmación de que las propiedades relativas a las prestaciones o a los
requisitos legales son satisfechas por un sistema de medida.
EJEMPLO 3: Confirmación de que un objetivo de incertidumbre de medida puede
conseguirse.
NOTA 1: Cuando sea aplicable, debe tenerse en cuenta la incertidumbre de medida.
NOTA 2: La “entidad” puede ser, por ejemplo, un proceso, un procedimiento de medida,
un material, un componente o un sistema de medida.
NOTA 3: Los “requisitos determinados” pueden ser, por ejemplo, que se cumplan las
especificaciones de un fabricante.
NOTA 4: No debe confundirse la verificación con la calibración. No toda verificación es
una validación.

25 | P á g i n a
BIBLIOGRAFÍA
Fuente Primaria
- Sociedad Española de Bioquímica Clínica y Patología Molecular
Comité Científico
Comisión de Metrología
F. Canalias Reverter, N. Alonso Nieva, B. Boned Juliani, FJ. Gella Tomás, S. Izquierdo
Álvarez, R. López Martínez,N. Serrat Orús
Fuentes secundarias
Asociación Española de Normalización (AENOR). Exactitud (veracidad y precisión) de
resultados y métodos de medición. Parte 1: Principios generales y definiciones. UNE
82009-1. Madrid: AENOR; 1998 (equivalencia
ISO 5725-1:1994).
- Asociación Española de Normalización (AENOR). Productos sanitarios para diagnóstico
in vitro. Medición de magnitudes en muestras de origen biológico. Trazabilidad
metrológica de los valores asignados a los calibradores y a los materiales de control.
UNE-EN ISO 17511.
Madrid: AENOR; 2004.
- Asociación Española de Normalización (AENOR). Productos sanitarios para diagnóstico
in vitro. Medición de magnitudes en muestras de origen biológico. Trazabilidad
metrológica de los valores de concentración catalítica de los enzimas asignados a los
calibradores y materiales de control. UNE-EN ISO 18153. Madrid: AENOR; 2004.
- Asociación Española de Normalización (AENOR). Sistemas de gestión de la calidad –
Fundamentos y vocabulario. UNE-EN ISO 9000. Madrid: AENOR; 2005.
- Clinical and Laboratory Standards Institute (CLSI): User protocol forevaluation of
qualitative test performance; approved guideline 2nd edition. EP12-A2. Vol. 28. No. 3.
Wayne: CLSI; 2008.
- International Union of Pure and Applied Chemistry. Nomenclature in evaluation of
analytical methods including detection and quantification capabilities (IUPAC
Recommendations 1995). Pure Appl Chem 1995;67:1699-1723.
- International Vocabulary of Metrology. Basic and general concepts and associated terms
(VIM). ISO/IEC Guide 99-12:2007. Comité Conjunto para las Guías de Metrología
(JCGM); 2012 (www.bipm.org).

26 | P á g i n a
2. FASE PREANALÍTICA
Gloria Isabel Barajas Barbosa- Bacterióloga y Laboratorista Clínica - Universidad Colegio Mayor de
Cundinamarca; Instituto Nacional de Salud. Bogotá, Colombia. [email protected]
Es responsabilidad del laboratorio clínico garantizar la calidad de los exámenes que
proporciona sobre el estado de salud de una persona y para ello se debe controlar todos
los procedimientos desde que el médico solicita el análisis hasta que éste recibe el
informe final.
Para obtener resultados precisos y confiables es necesario obtener muestras ideales
puesto que no podemos olvidar que el propósito de cualquier examen de laboratorio es
proporcionar resultados con un alto nivel de exactitud, de tal manera que se puedan
alcanzar conclusiones para el diagnóstico y el tratamiento del paciente.
La fase preanalítica comienza cronológicamente a partir de la petición del médico clínico e
incluyen la petición de los análisis la preparación del paciente, la recogida de la muestra
primaria y el transporte hasta y dentro del laboratorio, y
que termina cuando comienza el procedimiento analítico.
El objetivo de este capítulo es establecer una serie de recomendaciones para mejorar la
calidad de la fase preanalítica y minimizar en lo posible el efecto de las interferencias y
evitar molestias en los pacientes.
2.1 CAUSAS DE LA VARIABILIDAD DE LAS MAGNITUDES
Las magnitudes biológicas están sometidas a dos tipos de variabilidad; la variabilidad
biológica y la analítica, responsables de que los valores de un determinado analito varíen
entre diferentes individuos y que incluso en una misma persona difieran en el tiempo.
2.1.1 Variabilidad Biológica
La variabilidad biológica consta de dos componentes bien diferenciados, el intraindividual
y el interindividual. La variabilidad intraindividual se define como la fluctuación aleatoria
en la concentración de un determinado analito alrededor de su punto de equilibrio
homeostático. Estos puntos de equilibrio presentes en una población son diferentes para
cada individuo, siendo esta variación lo que se conoce como variabilidad biológica
interindividual. Ambas se expresan como norma general en forma de coeficiente de
variación (CVw y CVg para intra e interindividual respectivamente). La variación puede
suceder a corto o largo plazo y su origen puede ser:
Sistemático: relacionado con los ritmos biológicos o con la edad debido a las
modificaciones que comportan el crecimiento o el envejecimiento.

27 | P á g i n a
Aleatorio; Causado por las variaciones metabólicas relacionadas con la hemostasis. La
variación es tanto menor cuento más estrecho sea el control o la regulación metabólica
del analito. También forma parte del componente aleatorio las variaciones introducidas
por la dieta, clima, estados emocionales, etc.
2.1.2 Variabilidad Analítica
La variabilidad analítica engloba a todos aquellos factores que pueden afectar la muestra
durante todo el proceso analítico. Se entiende como proceso analítico al conjunto de
procedimientos que tienen lugar desde la solicitud del análisis y preparación del paciente
hasta que el informe llega al médico que lo solicito. Está dividido en tres fases:
Preanalítica: comprende la fase desde la preparación del paciente y toma de muestra
hasta la reparación de ésta para su análisis.
Analítica: abarca todos los procedimientos relacionados con la medida de la magnitud que
se estudia.
Postanalítica: Incluye la elaboración del informe analítico y envío al médico solicitante.
Todas estas fases presentan una gran importancia ya que un error en cualquiera de ellas
puede llegar a invalidar el informe final.
Los principales factores que pueden influir en la calidad de la muestra en la fase
preanalítica y que debemos conocer para poder realizar una correcta interpretación del
resultado son:
Variables preanalíticas fisiológicas
Variables preanalíticas en la toma de muestra
Variables preanalíticas interferentes
2.1.3 Variables preanalíticas fisiológicas
Edad es imprescindible la edad del paciente para poder interpretar correctamente un
resultado, dado que algunos mensurandos presentan valores diferentes en niños y
adultos.
Género aparte de las diferencia en los niveles de hormonas sexuales, también existen
diferencias en otros mensurandos como CK, Creatinina, etc.
Etnia hay mensurandos que pueden verse alterados, por ejemplo en la raza negra tienen
los leucocitos más bajos y las lipoproteínas más altas que los de raza blanca, esto debido
a una reducción del número de granulocitos. Los monocitos son más elevados en la raza
blanca.
Tiempo de muestreo los parámetros biológicos sufren cambios frecuentes siguiendo
ritmos biológicos. Los ritmos más comunes son el circadiano y el menstrual que afecta
los resultados. Variaciones circadiana/diurnas: los niveles de muchos componentes

28 | P á g i n a
sanguíneos varían o fluctúan de forma diurna, los factores que influyen en las variaciones
incluyen la postura, actividad, alimentación, luz de día u oscuridad, iniciando al levantarse
o acostarse. Los niveles máximos de renina y TSH son en las horas previas de la
mañana durante el sueño, otros componentes están en los niveles aumentan la
bilirrubina, hemoglobina, insulina, hierro, potasio, testosterona, recuento de rojos,
eosinófilos en sangre, Creatinina, glucosa, triglicéridos. La disminución del fosfato es en
la mañana. Estas variaciones diurnas pueden ser muy grandes, el cortisol, la TSH y el
hierro, hasta de un 50% entre la mañana y la tarde.
Tiempo estacional algunos mensurandos varían por efecto de la estación del año.
Altitud debido a la menor presión de oxigeno, el recuento de glóbulos rojos, la
hemoglobina y el hematocrito están más elevados. Hay también un aumento en la
proteína C reactiva, la adaptación a la altura tarda unos días.
Embarazo se produce una hemodilución fisiológica por un aumento del volumen
plasmático, mas marcado en el tercer trimestre que disminuye el hematocrito y la
hemoglobina, hay cambios hormonales, los niveles séricos de lípidos se incrementan
conforme avanza el embarazo, etc.
Deshidratación la disminución del fluido corporal total, produce hemoconcentración
afectando los glóbulos rojos, enzimas, hierro, calcio, sodio y factores de coagulación por
lo que estos valores no reflejarían el verdadero estado del paciente.
Fiebre afecta el nivel de un número de hormonas. La fiebre inducida por hipoglicemia
aumenta la insulina seguida por un aumento del glucagón. También aumenta el cortisol y
puede interrumpir su variación diurna normal.
2.1.3 Variables preanalítica en la toma de muestra
Ayuno como norma general se recomienda un ayuno previo a la extracción de 8 horas. La
composición de la sangre es alterada significativamente por la ingestión de alimentos.
La glucosa en sangre aumenta dramáticamente con la ingesta y se normaliza a las 2 h, si
su metabolismo es normal.
La ingesta de grasas aumenta los niveles de lípidos en la sangre y produce la lipemia que
puede persistir hasta por 12 h y algunas pruebas químicas no pueden realizarse por la
turbidez del suero o plasma que interfiere en el procedimiento de examen.
La ingesta de carnes y algunos vegetales afectan la prueba de investigación de sangre
oculta en heces.
El consumo excesivo de líquidos puede aumentar los niveles de hemoglobina y alterar el
balance electrolítico.
El consumo de bebidas como cafeína puede afectar los niveles de cortisol.
El consumo crónico de alcohol o grandes cantidades puede causar hipoglicemia y
aumentar los triglicéridos.

29 | P á g i n a
Medicamentos los medicamentos pueden interferir por competencia con los reactivos o
incrementar la reacción y alterar los resultados. Se debe registrar el medicamento y la
dosis que se está administrando.
Fumar previo a la toma ocasiona el aumento del colesterol, cortisol, glucosa, triglicéridos y
recuento de leucocitos. En el fumador crónico hay aumento en el recuento de glóbulos
rojos, disminución de la hemoglobina de la IgA, IgG e IgM, pero aumento de la Ig. E.
Etanol los efectos agudos entre 2-4 h son disminución de la glucosa y el lactato, hay una
acidosis metabólica y aumentan los uratos. En un alcoholismo crónico hay un aumento
de las enzimas hepáticas, por efecto tóxico sobre el hígado y un aumento de triglicéridos.
Drogas adictivas las anfetaminas producen aumento de ácidos grasos; la morfina
aumenta la amilasa, lipasa, bilirrubina, prolactina y disminuye la secreción de insulina. La
heroína aumenta el colesterol, la tiroxina y el potasio. La marihuana aumenta los iones, la
urea, la insulina y por lo tanto disminuye la glucosa. También afecta los uratos.
Postura los cambios posturales pueden alterar la composición del fluido corporal.
Estrés emocional como ansiedad, miedo o trauma puede causar aumento transitorio del
recuento de glóbulos blancos, en los niños el llanto altera los resultados. También puede
ocasionar disminución del hierro y en las hormonas adrenales como el cortisol,
aldosterona, TSH, hormona del crecimiento en los niños. El estrés que causa una toma de
muestra en el paciente en general se subestima.
Temperatura y Humedad pueden causar alteraciones en los fluidos corporales como
concentrar el plasma.
Ejercicio provoca mayor volumen entre los compartimentos intravasales e intersticiales.
Perdida de volumen por el sudor. Afecta las concentraciones hormonales.
Energético Puede ocasionar que los eritrocitos u otras células sanguíneas sean
excretadas por la orina.
Breve Altera significativamente la Creatinina, potasio y proteínas.
Regular Incrementa enzimas con actividad muscular, ácido úrico.
Intensivo Aumenta los anteriores más el potasio, bilirrubinas, pero la glucosa disminuye
significativamente.
Uso del torniquete si se mantiene más de lo recomendado (10 15 segundos) puede
producirse una hemoconcentración, aumentando la concentración de las moléculas
grandes y alteración de algunos parámetros de coagulación.
Anticoagulantes es fundamental que el anticoagulante empleado en cada muestra sea el
adecuado según la prueba de que se trate. Otro aspecto importante es mantener la
proporción adecuada entre la cantidad de sangre y el anticoagulante.

30 | P á g i n a
Orden de extracción de los tubos se recomienda seguir un orden predeterminado en la
extracción de sangre venosa para evitar contaminación microbiológica o contaminación
cruzada de aditivos entre tubos cuando utilizamos sistemas de extracción por vacio que
pueden interferir en la medición de determinados analitos.
2.1.4 Variables preanalíticas interferentes
Hemólisis causada por extracción defectuosa, permanencia prolongada de sangre total
sin centrifugar, contaminación por detergentes, antisépticos, agua o reactivos residuales,
choque térmico (calentamiento o enfriamiento excesivo).
Lipemia puede deberse a no guardar el ayuno recomendado o a problemas metabólicos.
Ictericia originada por una alta concentración de bilirrubina en el suero o plasma.
Autoanticuerpos halterófilos, aglutinas, crioaglobulinas la presencia de cualquiera de estos
anticuerpos circulantes puede afectar bien por interferir sobre el analito mismo o bien en
el proceso de medición (reacción antígeno anticuerpo).
Fármacos Producen interferencias en la medida de un gran número de analitos.
Evaporación reducir el contacto con el aire hasta donde sea posible. Si esto no se hace,
los efectos de evaporación producirán un aumento en la concentración/actividad de todos
los componentes no volátiles. Esto aplica particularmente para el caso cuando el volumen
de la muestra es relativamente pequeño y el área de la superficie es relativamente
grande, como en muestras pediátricas.
Efecto de la luz evitar la exposición directa a la luz solar durante el almacenamiento y
transporte de la muestra, especialmente en el caso de analitos fotosensibles como la
bilirrubina, vitamina C. porfirinas, creatincinasa (CK), y ácido fólico.
2.2 PROCESOS DE LA FASE PREANALÍTICA
Como se ha comentado anteriormente, la fase preanalítica es la secuencia de
acontecimientos que tiene lugar antes de que la muestra sea procesada. Ver figura No. 1.
Actualmente se considera la fase más crítica del proceso ya que en ella es donde se
produce un mayor número de errores y donde se puede perder más tiempo.
Figura 1
Solicitud analítica
Preparación del
paciente
Obtención de las
muestras
primarias Transporte
Conservación
estabilidad
Interferencias

31 | P á g i n a
Las etapas que forman parte de esta fase son:
2.2.1 Solicitud analítica
La hoja de solicitud debe contener toda la información pertinente al paciente y al médico
solicitante así como proporcionar los datos clínicos pertinentes.
Tener en cuenta los siguientes elementos:
Identificación única del paciente: nombres y apellidos completos, número de
identificación, género, edad o fecha de nacimiento, ciclo menstrual, y datos para
la localización.
Información del médico solicitante: nombre completo, teléfono o dirección de
localización.
Tipo de muestra primaria y el lugar anatómico de origen
Nombre claro y completo del examen solicitado (abreviaturas)
Fecha y hora de la toma de la muestra primaria
Fecha y hora de la recepción de las muestras por el laboratorio
Descripción de intervalos de tiempo, prioridad e información clínica suficiente
(Impresión Diagnóstica).
2.2.2 Preparación del paciente
Cada examen requiere de condiciones generales que permiten una óptima obtención de
la muestra; estas indicaciones son entregadas por el laboratorio clínico o por el médico
tratante de acuerdo al Manual de toma de muestras del laboratorio clínico.
Se recomienda un período de 8 hora después de la última comida, es decir 8 horas de
ayuno antes de la extracción de la sangre porque la concentración de diversos
metabólicos de los alimentos pueden aumentar en la sangre venosa o alterarse debido a
efectos hormonales posabsortivos. El ayuno prolongado pude dar lugar a resultados
inesperados.
Se debe Limitar la ingesta de café, grasas, consumo de tabaco y ejercicio en las 24 horas
previas a la extracción.
Hay pruebas específicas que requieren que en los días previos se siga una dieta especial,
están se darán verbalmente y por escrito.
Lo ideal es que el paciente no debe esperar de pie ni cargar objetos pesados mientras
espera su turno de atención.

32 | P á g i n a
2.2.3 Obtención de las muestras primarias
El laboratorio debe asegurar que el material de extracción y el de recogida de muestras,
tienen la calidad adecuada.
Las variables a controlar son múltiples, destacando entre otras, la identificación de la
muestra, el tipo de espécimen, el procedimiento de obtención, el recipiente y/o aditivos
necesarios para cada prueba.
Manejo y atención del paciente Por principio debe tranquilizarse al paciente con palabras
amables y mediante una actitud de confianza y seguridad del personal que toma la
muestra. La serenidad contribuirá a establecer una adecuada relación. Recuerde que el
estrés puede afectar los resultados de laboratorio. La ansiedad del paciente puede
producir cambios en la concentración de catecolaminas y gases en sangre, a través de
efecto hormonal directo e hiperventilación. Por lo tanto, es preciso realizar todos los
esfuerzos para tranquilizar al paciente antes de efectuar la punción.
Consentimiento informado
El laboratorio, al igual que en las otras disciplinas, también se debe tener en cuenta los
principios éticos del consentimiento informado, que está fundamentado en la relación
médico paciente, para lo cual el personal del laboratorio, está obligado a realizar un
proceso personalizado con el paciente, brindándole información clara, suficiente, en
términos que sean de comprensión, verbal y escrita, que le permita participar en la toma
de decisiones respecto al diagnóstico.
Para la mayoría de las pruebas de laboratorio podría plantearse que la información
suministrada al paciente debe contener:
Objetivo del análisis
Explicaciones de los posibles resultados y su significado
Molestias y riesgos derivados de su realización
Beneficios esperados
Garantías de confidencialidad de resultados
Otra información que pueda ser requerida, además de las recomendaciones para
una obtención adecuada de la muestra.
Adicionalmente, se debe informar sobre el derecho de conocer sus resultados y el
deber de recoger el resultado. Cuando un paciente adecuadamente informado
acepta que se realice la obtención de la muestra u otro procedimiento del
laboratorio, se entiende que ha dado el consentimiento informado.
En el caso de pacientes hospitalizados, se mantendrá un registro adecuado de la
información, que debe formar parte de la documentación de la historia clínica. En el
paciente ambulante pude ser útil la información divulgativa como folletos u otros impresos.

33 | P á g i n a
Identificación del paciente La identificación correcta debe registrarse en el recipiente que
contiene la muestra y en la hoja de solicitud. Antes de la toma de la muestra se debe
verificar que se cuenta con el correcto recipiente y anticoagulante apropiado Se lleva a
cabo mediante la comparación de la información contenida en la solicitud analítica del
paciente y la identificación numérica, código de barras, hora de la extracción y nombre del
responsable de que toma la muestra o cualquier otro criterio predeterminado por la
institución.
Posición
El debe encontrarse en una posición estable (sentado o acostado) por lo menos 15
minutos antes de la extracción.
Acostado: Existe un acomodo o distribución hemodinámica y de otros líquidos corporales.
Sentado: Empieza la salida de líquido del espacio intravascular al espacio intersticial y por
lo tanto se produce hemoconcentración.
Preparación el material para la extracción Con anterioridad, el personal debe tener a su
disposición el siguiente material:
Sistema de extracción consta de una aguja multimuestreo, portatubos y tubos de
extracción de sangre
Guantes desechables
Torundas de algodón
Solución desinfectante o alcohol al 70%
Apósitos
Torniquete
Contenedor de desecho
Etiqueta para la muestra con identificación del paciente
Preparación del paciente
Preséntese ante el paciente y pregúntele su nombre y apellidos
Verifique la correspondencia entre el formulario de solicitud de análisis clínico y la
identidad del paciente (es decir, fíjese si los detalles del paciente coinciden con la
información del formulario de solicitud para garantizar la fiel identificación de la persona).
Venopunción
Tener cuidado de no tomar la muestra en el brazo donde esté colocado algún tipo de
venoclisis.
Observe siempre las dos extremidades superiores (brazos), para elegir el mejor sitio de
punción. Inspeccionar la vena que se va a puncionar (preferiblemente que sea visible y
fácilmente palpable). Ver figura No. 2

34 | P á g i n a
Si la vena no es muy visible ni palpable, realice un suave masaje en el antebrazo, con
movimientos desde la muñeca hacia el codo.
Limpiar con solución antiséptica (alcohol 70%) y algodón, haciendo movimientos
circulares de adentro hacia afuera. Deje secar la zona por lo menos 30 segundos.
No toque el lugar desinfectado y sobre todo no ponga el dedo sobre la vena para guiar el
eje de la aguja expuesta, si ha de tocar el lugar repita la desinfección.
El torniquete debe ubicarlo a unos 4 ó 5 dedos de distancia por encima del pliegue de la
fosa antecubital. Nunca colocar sobre lesiones, contusiones, hematomas o equimosis. la
zona de Venopunción y vuelva a examinar la vena, el torniquete puede permanecer de 10
a 15 y ser soltado tan pronto fluya sangre en el primer tubo.
Se debe tener en cuenta que algunas pruebas de laboratorio deben ser tomadas sin la
utilización del torniquete.
Retire la aguja con delicadeza y presione el lugar ligeramente con un algodón limpio o una
torunda de algodón seca, con el brazo extendido y levantado. Dígale que NO doble el
brazo, pues eso puede provocar hematoma.
Para la obtención de varios tubos de sangre, use tubos al vacío con aguja y portatubos.
Este sistema permite el llenado directo de los tubos. Si no se dispone de este sistema,
use una jeringa o un equipo de Venopunción con agujas provistas de aletas.
Coloque los tubos de extracción de sangre en el orden correcto para evitar la
contaminación de aditivos entre tubos.
Invierta los tubos con aditivos el número de veces que sea necesario.
Deseche la aguja en el recipiente para objetos cortopunzantes.
Descarte los elementos utilizados con los desechos de la categoría apropiada.
Lávese las manos nuevamente.
Controle nuevamente las etiquetas de los tubos y los formularios antes de su envío.
Avise al paciente en cuanto acabe el procedimiento.
Pregunte cómo se siente al paciente, inspeccione el lugar de la punción para asegurarse
de que no sangre.
Informe al Coordinador cuando se presente un caso de hematoma o el paciente
manifieste dolor en el momento y/o después de la punción, para hacer el seguimiento
pertinente. EVENTO ADVERSO

35 | P á g i n a
Figura 2
Llenado de los tubos
Los tubos se deben llenar hasta su capacidad establecida nunca se deben llenar menos,
ya que los tubos que contiene aditivos pueden generar resultados erróneos en las
pruebas por alteración en la relación sangre anticoagulante.
Para que no se presenten interferencias en los resultados de los análisis por
contaminación con los diferentes anticoagulantes de los tubos empleados, se debe seguir
un orden de llenado, teniendo en cuenta si los tubos son de plástico o de vidrio. Ver figura
No. 3
El nivel de llenado de cada tubo está indicado por el fabricante y se encuentra registrado
en la marquilla respectiva del tubo. La toma de muestra con el sistema al vacio al ser un
sistema cerrado, facilita la extracción de fluido sanguíneo y llenado adecuado para cada
tubo
Tras la toma de muestras todos los tubos deben invertirse hasta que la burbuja de aire se
desplaza de un extremo a otro del tubo. Ver figura No. 3

36 | P á g i n a
Figura No. 3
2.2.4 Transporte de muestras
El transporte de las muestra debe realizarse con cuidado y evitando derrames, pérdidas o
contaminación de las mismas por otras sustancias, así como las que se pueden presentar
por acciones mecánicas, calentamiento excesivo o exposición a la luz.
Las personas responsables del transporte tienen que seguir las instrucciones de trabajo
correspondientes con el fin de conservar las características originales de las muestras
diagnósticas.
El transporte de las muestras tiene como prioridad su integridad con la finalidad de
mantener la estabilidad de las propiedades biológicas que las componen.
Después de asegurar que las muestras están correctamente identificados (cuando
existan centrifugas en los puntos de extracción) y se envían en gradillas, de forma
ordenada según códigos y tipo de tubo, en posición vertical para evitar interferencias.
Recomendaciones:
Elementos a evaluar con relación al transporte:
HEMOCULTIVO Mezclar de 8-10 veces
COAGULACIÓN Mezclar de 3-4 veces
TUBOS DE SUERO Mezclar 5 veces
TUBOS DE HEPARINA Mezclar 8-10 veces
TUBOS DE EDTA Mezclar de 8-10 veces
TUBO DE GLUCOSA Mezclar de 8-10 veces
OTROS TUBOS

37 | P á g i n a
Materiales
Contenedores de transporte homologados.
Gradillas que permitan mantener tubos en posición vertical.
Refrigerante
Control de la temperatura (termómetro de máximos y mínimos)
Personal
Preparación de las muestras previo al transporte
Conductores y personal que realiza el transporte
Personal que recibe las muestras en el laboratorio
Medios de transporte – preparación de vehículos
Logística – Rutas – control del tiempo
El transporte de las muestras se realizará:
En recipientes herméticos y a prueba de fugas de líquido.
Si el recipiente es un tubo preferiblemente plástico, debe estar herméticamente cerrado
con tapa hermética y colocado de tal forma que mantenga su posición vertical.
Los recipientes con muestras y/o gradillas deben colocarse en una caja resistente de
plástico, y a prueba de fugas de líquido, que contenga una cubierta segura y que cierre
perfectamente señalando el lado superior hacia arriba.
La solicitud del análisis y la identificación de las muestras deben acompañar a cada tubo.
Nunca transportar las muestras de laboratorio en la mano o en los bolsillos de la bata.
En el transporte de la caja plástica que contiene las muestras de laboratorio se deberá
utilizar guantes como medida de Bioseguridad.
No entregar muestras de laboratorio para transporte extra institucional a los pacientes o
familiares, el envío es responsabilidad únicamente del laboratorio clínico, quien deberá
coordinar el transporte de las mismas.
2.2.5 Conservación y estabilidad
En algunos casos, las muestras no son procesadas inmediatamente y por ello necesitan
un periodo de almacenamiento; además, después del análisis de las muestras, éstas
también son conservadas durante un periodo de tiempo determinado.
En general, se establece que la conservación de las muestras tiene las siguientes
condiciones:
4 - 8 horas a temperatura ambiente (preferentemente 1-2 horas).
Hasta 7 días en nevera.
Dos ó tres meses en congelador a – 20 ° C.
Si tenemos que congelar una muestra, se realizará de forma rápida y evitando los ciclos
de congelación-descongelación repetidos, que pueden originar efectos que alteren
algunas estructuras moleculares, como las proteínas.

38 | P á g i n a
Al guardar un espécimen de sangre en la nevera, es preferible centrifugarla primero,
manteniendo separado el suero ó el plasma de las células. Así evitamos interferencias en
algunos parámetros, como el potasio, debido a que la refrigeración inhibe la bomba sodio-
potasio, condicionando posteriormente un mayor nivel de dicho ión si la separación la
efectuamos después de cierto tiempo en la nevera.
Anticoagulantes más usados:
EDTA: Este tipo de anticoagulante es utilizado principalmente para estudios de células
sanguíneas (Hemograma y Citometría de flujo) y estudios de Biología molecular
(extracción de ADN y ARN y FISH).
CITRATO DE SODIO: En concentraciones al 3.8 %se utiliza en estudios de coagulación.
HEPARINA: Se utiliza tanto en algunos estudios de rutina como especializados. Su
presentación puede incluir heparina con concentraciones de sodio o litio. En general, la
heparina con litio es utilizada para estudios de química de urgencias y la heparina sódica
se utiliza para estudios de linfocitos.
OXALATOS: Son anticoagulantes menos comunes, utilizados ocasionalmente en las
determinaciones de glucosa.
FLUORURO DE SODIO: Anticoagulante que se utiliza para la determinación de Glucosa
en sangre, cuando esta no puede ser procesada dentro del plazo prudente.
Los agentes de separación (gel) utilizados en tubos de suero mejoran el rendimiento del
suero y permiten mantenerlos en los tubos primarios. Estos tubos deben mantenerse
siempre verticales para favorecer el proceso de coagulación aproximadamente 30 min
antes de su centrifugación para favorecer la retracción del coágulo.
Variables que afectan la estabilidad se encuentran:
• Condiciones en el transporte de las muestras: temperatura, tiempo y alteraciones
mecánicas, como vibración, a las que son sometidos los especímenes durante su
transporte.
• Condiciones en la conservación de la muestra desde la extracción hasta su
centrifugación o preparación previa al procesamiento: forma de almacenamiento,
evaporación, decantación o separación en alícuotas, temperatura de conservación y
tiempo transcurrido.
Factores que pueden influir en la estabilidad de las muestras biológicas son:
• Condiciones inherentes al sujeto relacionadas con la variabilidad biológica
intraindividual, como factores fisiológicos, patológicos, ingesta de fármacos, y regulación
homeostática.
• Condiciones de obtención de la muestra, tales como la dificultad de la extracción,
tiempo de aplicación del torniquete y tiempo total del procedimiento de extracción.
• Condiciones propias al contenedor de la muestra: componentes intrínsecos del
material, aditivos, conservantes y fases de separación.

39 | P á g i n a
• Metodología analítica empleada y propiedad fisicoquímica que se mide.
También se conoce que todas las variables anteriormente citadas afectan por desigual a
los diferentes componentes de la muestra. Algunas propiedades fisicoquímicas son más
sensibles que otras a la acción de estos factores, no se alteran en la misma cuantía y la
acción de alguna de las variables puede influenciar con signo contrario sobre diferentes
componentes de una misma muestra.
Evitar el efecto de la luz, ésta afectará disminuyendo los valores de bilirrubina, vitamina C.
porfirinas, creatincinasa (CK), y ácido fólico.
Reducir el contacto con el aire hasta donde sea posible. Si esto no se hace, los efectos de
evaporación producirán un aumento en la concentración/actividad de todos los
componentes no volátiles. Esto aplica particularmente para el caso cuando el volumen de
la muestra es relativamente pequeño y el área de la superficie es relativamente grande,
como en muestras pediátricas.
Evitar guardar sangre entera o total sin centrifugar, revisar la información sobre los
analitos sensibles.
Evitar la glucólisis en muestra para determinar glucosa y lactato. La glucólisis puede ser
evitada por la adición de un inhibidor (Fluoruro sódico), junto con un anticoagulante si la
muestra no va a ser procesada dentro del plazo de tiempo.
2.2.6 Interferencias preanalíticas.
El término interferencia analítica se usa, en amplio sentido, para designar el efecto que
ejerce una sustancia, distinta a la que estamos midiendo, en la determinación de la
concentración o actividad del analito, siendo el analito el componente que intentamos
medir en la muestra y la interferencia, un componente de la muestra, distinto del analito,
que altera el resultado final.
Las interferencias pueden presentarse en la muestra debido a fuentes endógenas o
exógenas. Pueden ser producidas in vivo en ciertas condiciones patológicas (p.e.,
bilirrubina, lípidos, proteínas, hemoglobina, etc.), administradas a los pacientes durante el
tratamiento (p.e., drogas, nutrición parenteral, expansores del plasma, anticoagulantes,
etc.), autoadministradas (suplementos nutricionales, alcohol y drogas de abuso, etc.) o
debidas a contaminación de la muestra (anticoagulantes, conservantes, separadores del
suero, etc.)
Suero Ictérico una concentración de bilirrubina superior a 430 µmol/L (25 mg/L) interfiere
en la medida de distintos analitos dando lugar a incrementos en la concentración de los
mismos. Puede interferir en la determinación de albúmina, colesterol, glucosa y proteínas
totales.
Suero Lactescente.
Una alta concentración de triglicéridos en el suero da lugar a una turbidez, que provoca
resultados elevados para aquellas sustancias cuyas determinaciones se basan en la

40 | P á g i n a
absorbancia a las mismas longitudes de onda en que las partículas de lípido también
absorben luz y en que la lectura final de la absorbancia se utiliza como índice del valor de
la concentración de la sustancia que hay que determinar. Se pueden producir
interferencias en la determinación de albúmina, calcio y fosfato inorgánico. Se produce
una inhibición en la actividad de la amilasa, uricasa y ureasa, y una disminución en las
concentraciones de creatincinasa, bilirrubina y proteínas totales.
Suero Hemolítico
La lisis de los elementos formes de la sangre puede contaminar el suero o el plasma
dando lugar a un incremento o disminución en la concentración de diferentes analitos. Se
producen aumentos en las concentraciones de Lactato deshidrogenasa (LDH), Aspartato
aminotransferasa (ASAT o GOT), Alanin aminotransferasa (ALAT o GPT), Fósforo
inorgánico, Potasio, Calcio, Cinc y Magnesio; se produce un incremento en la actividad
sérica de Fosfatasa ácida y en la concentración de Albúmina y Bilirrubina. Una solución al
1% de eritrocitos lisados produce un aumento del 98% de la actividad media de la
Creatincinasa del suero en individuos sanos . La lisis de las plaquetas provoca fuertes
aumentos en la concentración sérica de Potasio y Magnesio,así como en las actividades
séricas de Fosfatasa ácida y Aldolasa. La granulocitosis produce aumentos en las
concentraciones de Lisozima, Arginasa, Glucosa-6-fosfato deshidrogenasa y Glutamato
deshidrogenasa. La hemólisis puede producir disminución en la concentración de Glucosa
y Sodio.La lisis de los elementos formes de la sangre puede producirse durante la toma
de muestras en los tubos de vacío por la fuerte expansión de sangre o por la mezcla con
un anticoagulante de oxalato. También puede producirse durante la centrifugación y
separación del suero o plasma en el procesamiento de la muestra.
Interferencias Químicas
Son numerosas las sustancias que pueden dar lugar a variaciones en la medida de la
concentración de un analito. En las tablas I y II se recogen los fármacos que producen
interferencias químicas sobre las pruebas clínicas más usuales.
Anticoagulantes y Conservantes
Determinados anticoagulantes como el oxalato potásico provocan un aumento de la
presión osmótica del plasma, dando lugar a un transporte de agua desde las células
sanguíneas al plasma y a una dilución del mismo. Los quelantes del calcio producen
inhibición en la actividad de diferentes enzimas para las cuales este ión es fundamental.
Se produce inhibición en la actividad de la Amilasa, LDH y Fosfatasa ácida.

41 | P á g i n a
BIBLIOGRAFÍA
1. Garzón, Alba; Calidad Analítica en el Laboratorio Clínico Gestión y control. Editorial ACG Ltda.
2006
2. COLOMBIA. 2005. Norma Técnica Colombiana NTC-ISO/IEC 17025. Requisitos Generales
para la Competencia de los Laboratorios de Ensayo y Calibración. ICONTEC. 26 Octubre de
2005, 35 pp.
3. Gella, Javier (2012); Comisión de Metrología de la Sociedad Española de Bioquímica Clínica y Patología Molecular. España Trazabilidad e incertidumbre de la medición en el laboratorio clínico; Universidad Autónoma de Barcelona, Centro Cultural Tlatelolco, Cd de México, 26 de junio de 2012.

42 | P á g i n a
3. FASE ANALÍTICA
3.1 HEMATOLOGÍA
Criterios de acción sugeridos después del análisis del hemograma automatizado para
la revisión microscópica del frotis de sangre periférica y el diferencial leucocitario por
el Grupo Internacional de Consenso para Hematología
Maria Clemencia Garnica Barrios; Bacterióloga- Universidad de Colegio Mayor de Cundinamarca;
Instituto Nacional de Salud, Grupo Genética Crónicas. Bogotá, Colombia. [email protected]
Introducción
La valoración e interpretación del frotis de sangre periférica (FSP) como parte del
hemograma representa el método de laboratorio que se utiliza para el estudio de las
características citológicas y del estado de los elementos celulares de la sangre,
determinar las variaciones y anormalidades de estructura forma y tamaño.
Constituye un examen rutinario, que cuando es debidamente interpretado por el
observador, es de gran utilidad para determinar las etiopatogenias, el seguimiento y
control de una serie de enfermedades hematológicas y no hematológicas además de
servir como indicador de la respuesta y los efectos deletéreos de diferentes tratamientos.
Los nuevos contadores hematológicos, que incorporan modernos métodos automatizados
o semiautomatizados, pueden sustituir en gran parte la revisión manual de los frotis;
para la asistencia en el diagnóstico y monitoreo terapéutico de pacientes; los métodos
automatizados han mejorado las capacidades de desempeño proveen cada vez
resultados con mayor exactitud , precisión , confiabilidad y rapidez tanto en lo cuantitativo
como en lo cualitativo , generando una serie de alarmas para la detección de formas
patológicas o inmaduras que direccionan a cambiar o modificar algunos procedimientos
complementarios en el área de hematología (La acción manual de la revisión del frotis de
sangre periférica)
En los últimos años se ha desatado una polémica sobre la necesidad y rentabilidad de la
lectura del frotis de la sangre periférica que involucran: protocolos, control de calidad,
tiempo de respuesta, costos, eficacia y productividad del laboratorio; como ejemplo, el
tiempo empleado en leer e interpretar 100-200 células en un frotis oscila entre 1,9 y 6
minutos, mientras que en el analizador es de un minuto
Resumen
En la actualidad, el laboratorio clínico juega un papel fundamental en la atención a la
salud por sus aportes en el diagnóstico y monitoreo de las enfermedades. De acuerdo a lo
anterior, cada vez es mayor el uso de innovaciones tecnológicas que le confieran
eficiencia.

43 | P á g i n a
Las ventajas al utilizar los equipos automatizados son la rapidez en el análisis, la
sistematización y la calidad analítica. No obstante, es necesario que el usuario realice una
evaluación para asegurar que los resultados producidos sean lo suficientemente
confiables para ser utilizados en el cuidado de los pacientes.
En este contexto, la revisión microscópica del FSP para proporcionar información
adicional o para confirmar los resultados producidos por el analizador, se genera
usualmente por los criterios o protocolos de cada laboratorio para la revisión del frotis
visual aplicados a los resultados del analizador (confirmar las alarmas de sospecha,
detectar alteraciones cuantitativas o cuantitativas, verificar distribución y conteos
plaquetarios, realizar variaciones en el diferencial leucocitario, caracterizar rasgos
morfológicos, determinar la presencia de elementos atípicos (neoplasia), evaluar y
caracterizar anemias , descartar hemoparásitos y en desordenes hereditarios)
Cada laboratorio debe implementar los criterios de acción después del análisis
automatizado de una muestra sanguínea.
El objetivo es reducir en el mayor grado posible el número de muestras que requieren la
acción (revisión del frotis), mientras no se comprometa al paciente al reportar resultados
falsos negativos (FN) o falsos positivos (FP). Sin embargo, los laboratorios pequeños
tienen poca oportunidad para saber si la guía que siguen es efectivamente óptima para
minimizar los (FP y FN).
El intercambio de información reveló que las proporciones de revisión varían de 5 a 95%
entre los diferentes laboratorios; esto se puede explicar por diferencias en la población de
pacientes o en el desempeño del analizador.
El Dr. Berend Houwen reconoció la necesidad de implementar una guía (reglas) con los
criterios de revisión de los resultados del hemograma automatizado
En el 2002 el Dr. Berend Houwen invitó a 20 expertos a discutir y determinar los criterios
más apropiados para la revisión del FSP posterior al análisis instrumental, inicialmente se
desarrollaron 83 reglas por acuerdo consensual del (ICSH) estas reglas fueron aplicadas
a 13.298 muestras de sangre procesadas en 15 laboratorios, finalmente se redefinieron
41 reglas por consenso.
Las reglas hacen referencia a la validación de los mensajes o alarmas que emite el
instrumento al analizar muestras sanguíneas que requieran una revisión microscópica.
Se incluyen reglas para las muestras por primera vez así como reglas para muestras de
monitoreo o control (reglas delta para las muestras que se repiten en un paciente antes
de las 72 horas)
Se espera que estas reglas sean de utilidad a un gran número de laboratorios
hematológicos, se adjunta una sugerencia de protocolo para llevarlo a cabo antes de la
implementación de estas reglas en la operación rutinaria para las muestras de los
pacientes.

44 | P á g i n a
Que debe conocer el profesional de laboratorio para el manejo de las muestras después
del análisis del hemograma automatizado y la revisión microscópica del FSP
Del instrumento: sensibilidad y especificidad analítica y diagnóstica, principios de
cuantificación y clasificación celular, número de células contadas, número de parámetros
proporcionados, subclasificación leucocitaria (diferencial a 3 partes o a 5 partes),
linealidad del instrumento, mensajes o alarmas de sospecha e implementación del control
de calidad y funcionamiento del analizador adecuado.
De las muestras: procedencia de las muestras, si es de primera vez o segunda vez, si
cumple con los criterios de muestra apropiada para el análisis (ictérica, lipemia,
hemolizada, aglutinada, coagulada) y datos demográficos.
Perfiles epidemiológicos de la población que atiende el laboratorio.
Protocolos estandarizados de los procedimientos técnicos de operación (corrida de las
muestras en los analizadores hematológicos y preparación de los frotis sanguíneos)
Intervalos biológicos de referencia (IBR), para algunas magnitudes será necesario tener
en cuenta el grupo etario y el sexo.
Intervalos de alarma: valores que definen fijar los criterios de revisión del FSP, junto con
los IBR permiten tener tres niveles o grados de patología: resultados dentro de los límites
de normalidad, resultados situados entre la normalidad y los valores de alarma y los
resultados situados fuera de los valores de alarma.
Alarmas de sospecha: sea cual sea el sistema de obtención del diferencial leucocitario de
un analizador, se basa en su capacidad de identificar 5 poblaciones normales, la aparición
de células infrecuentes (granulocitos inmaduros, blastos, eritroblastos), el analizador
genera alarmas de sospecha, los casos con alarma de sospecha positiva son candidatos
a realizar la extensión y la valoración microscópica.
El Delta Check: corresponde al porcentaje máximo de variación permitido para una
magnitud determinada desde su última determinación en el mismo paciente, depende del
valor absoluto de la propia magnitud (de difícil aplicación a valores muy bajos), del tipo de
magnitud (mayor estabilidad de los parámetros de serie roja que los de serie blanca), del
tiempo transcurrido entre ambas determinaciones y también del tipo de paciente (paciente
ambulatorio, paciente hematoncológico o ingresado en UCI).
Comentarios al hemograma: constituye un “valor agregado” a los resultados cuantitativos
y pueden orientar el diagnóstico del paciente, es importante que permanezcan en el
registro histórico del paciente
Generales: muestra insuficiente, coagulada, hemolizada, hemograma procesado a 37 C,
parte de los resultados están afectados por aglutinación de la muestra a pesar de una
incubación de la muestra a 37C, resultados comprobados al microscopio y dada la
ausencia de cambios significativos desde el último control, no se realiza revisión al
microscopio.

45 | P á g i n a
Plaquetas: se observan agregados o cúmulos plaquetarios, satelitismo plaquetario,
gigantismo plaquetario, macrodismorfia plaquetar y por extensión la cifra de plaquetas
parece normal.
Hematíes: se observa población dimorfica, aglutinación de los hematíes, fenómeno de
roleaux.
Leucocitos
Granulocitos: neutrófilos con granulación tóxica, degranulados, hipogranulados, con
cuerpos de Döhle, con anomalía de Pelger, hipersegmentación nuclear.
Linfocitos: de aspecto normal, de citoplasma amplio con granulación, de aspecto
linfomonocitario, de aspecto linfomatos, de aspecto atípico, de tipo LLC, sombras de
Gumprecht
Monocitos: de aspecto atípico
Eosinofilos: hípervacuolados, degranulados
Reglas propuestas por el ICSH 1 para la revisión microscópica del frotis de sangre
periférica y el diferencial leucocitario posterior al análisis del hemograma automatizado
Reglas de validación del hemograma

46 | P á g i n a
Reglas direccionadas al diferencial leucocitario

47 | P á g i n a
Reglas orientadas a alarmas de sospecha

48 | P á g i n a
Reglas establecidas a señales de alarma
Pasos para la validación de las reglas
Definir los criterios para un hallazgo positivo en el FSP (ver protocolo de laboratorio).
Determinar el número de muestras para el estudio.
Las muestras para el análisis deben proceder de pacientes con diversas patologías con el
fin de capturar todas las reglas necesarias.
El 80% de las muestras analizadas deben ser muestras de primera vez con el fin de
probar “la observación de primera vez de la regla”.
El 20% de las muestras probadas deben ser muestras “repetidas” con el fin de probar las
reglas delta.
Efectúe el ejercicio en un período de +/- 5 días con la finalidad de eliminar cualquier
variabilidad del analizador así como capturar todos los tipos de muestras de la población
de pacientes.
Procesar las muestras en el instrumento e imprimir los resultados.
Realizar la revisión de los frotis sanguíneos de las muestras analizadas (participación de
dos lectores para mayor seguridad de las lecturas), los diferenciales manuales solo deben
realizarse cuando hay una necesidad específica para hacerlo (ejemplo rechazo del
diferencial, alarma de células anormales, etc.).
Diligencie los datos recolectados en un formato preestablecido (hoja para análisis).
Anote los datos para cada muestra en una fila separada debajo de los enunciados de
cada columna.
Nombre de las columnas: número de identificación de la muestra, identificación del
paciente, número de la regla disparada (uno para cada regla), número total de reglas

49 | P á g i n a
disparadas, alarmas de sospecha del instrumento (una para cada alarma), número total
de alarmas del instrumento, hallazgos positivos de la revisión del frotis (uno para cada
hallazgo) y número total de hallazgos positivos del frotis.
NOTA I: A fin de rastrear las muestras que pueden haber tenido un problema (caso de
investigar una rata de FN que parecen ser muy altos), se sugiere tener columnas
separadas para cada regla, alarma, y hallazgos positivos del frotis.
NOTA 2: Para análisis posteriores, en una columna totalice las reglas, las alarmas, los
hallazgos positivos del frotis, aplicadas por muestras individualizadas
Elaborar tablas de desempeño (tablas de la verdad) comparar las alarmas emitidas en los
hemogramas con los hallazgos de los frotis sanguíneos.
Si una alarma es disparada y el frotis tiene un hallazgo positivo, la muestra se clasifica
como verdadera positiva (VP)
Si una alarma es disparada, pero el frotis no presenta ningún hallazgo, la muestra se
clasifica como falsa positiva (FN)
Si una alarma no es disparada y en el frotis no tiene ningún hallazgo positivo, la muestra
se clasifica como verdadero negativo (VN)
Verifique que la rata de “falsos negativo” es < 5%. El grupo de consenso opina que esta
es la rata máxima aceptable para garantizar la seguridad del paciente. Si el análisis de las
reglas da una rata de falsos negativos superior se sugiere:
Comprobar la hoja y la impresión para cualquier error de transcripción.
Verifique las asignaciones a la tabla de la veracidad.
Revise en la hoja para identificar cual(es) regla(s) son causa específica de los (FN)
Ajuste las reglas como lo necesite.
Si necesita validar nuevas reglas por cambio de tecnología hágalo de la misma forma
Repita los pasos a-e si es necesario.
Si la rata de “falsos positivos” es mayor a la encontrada por el Grupo Consenso, o por
otras razones observa que el valor es demasiado alto, se sugieren las siguientes
acciones:
a. Determine si una alarma del instrumento en particular está sobre alarmando o no
pareciera ser útil para el laboratorio.
b. Solicite al proveedor del instrumento asesoría para identificar si esto es una
característica del instrumento o si la sensibilidad puede ajustarse.
c. Si el laboratorio escoge ignorar una alarma de sospecha, esto debe estar claramente
documentado, ya que operaría el analizador fuera de las recomendaciones del fabricante.
Implemente.
a. Las sesiones de entrenamiento con el personal son cruciales para efectos del cambio.
b. Actualice el manual de procedimientos y cualquier instructivo en el área de trabajo.
c. Actualice cualquier regla computarizada en el LIS.

50 | P á g i n a
Consideraciones finales
Las características actuales de los analizadores hematológicos junto al desarrollo de los
sistemas informáticos, han facilitado el trabajo técnico y el control en los laboratorios de
hematología
La implementación de los “Criterios de acción sugeridos después del análisis del
hemograma automatizado para la revisión microscópica del frotis de sangre” es:
responsabilidad de los profesionales del laboratorio, permitirá mayor interacción entre los
datos proporcionados por el instrumento aplicar correlación clínico patológica, hacer
seguimiento de pacientes y permite dedicar más tiempo a las muestras que realmente
justifican la revisión microscópica.
El grupo consenso ICSH presenta estas reglas a la comunidad hematológica como una
guía para ser implementada en los laboratorios clínicos
Los fabricantes de los analizadores hematológicos encontrarán útiles estas reglas cuando
validen el desempeño de nuevos analizadores hematológicos
El laboratorio que adopte estas reglas para el uso con las muestras pacientes deben
validarlas antes de implementarlas; este procedimiento debe combinarse con cualquier
requerimiento regulatorio bajo los cuales operan los laboratorios.
Criterios para un frotis positivo
Fecha: ID
Muestra:
ID Paciente:
Realizado Por:
N
o. Morfología SP SI NO
La única excepción es malaria, donde cualquier
hallazgo se considerará positivo
1 Morfología de eritrocitos ya sea 2+/moderado o
mayor
2 Morfología plaquetaria ya sea 2+/ moderado o
mayor
3 Acúmulos plaquetarios
4 Cuerpos de Dohle ya sea 2+/ moderado o mayor
5 Granulación tóxica ya sea 2+/ moderado o mayor
6 Vacuolas ya sea 2+/ moderado o mayor
7 Bastos = > 1
8 Metamielocitos > 2

51 | P á g i n a
9 Mielocitos / promielocitos = > 1
10 Linfocitos atípicos > 5
11 Normoblastos > 1
12 Células plasmáticas > 1
Número total de hallazgos positivos en FSP
Tablas guía de las reglas de validación del hemograma
Fecha: ID
Muestra:
ID Paciente:
Realizado Por:
No Reglas SI NO Acciones
PARÁMETROS
1 Neonato Revisar frotis
2
Glóbulos blancos, glóbulos rojos , hemoglobina ,
plaquetas y reticulocitos que excede linealidad
Diluir muestra y reanalizar
3 Glóbulos blancos y plaquetas , menor que linealidad Seguir protocolo del
laboratorio
4 Glóbulos blancos, glóbulos rojos , hemoglobina ,
plaquetas , Rechazo
Verificar integridad de la
muestra, reanalizar
5 Glóbulos blancos <4.0 o > 30.0 mm3 y primera vez Revisar frotis
6 Glóbulos blancos <4.0 o > 30.0 / mm3 Y falla Delta Revisar frotis
7 Plaquetas <100 o > 1000 / mm3 y primera vez Revisar frotis
8 Plaquetas cualquier valor Y falla delta Revisar frotis
9 Hemoglobina < 7g/dL o > 2g/dL sobre rango Revisar frotis e integridad
de la muestra
10 MCV <75 o > 105 fL, 1ra vez, adulto, <24horas Revisar frotis,
11
MCV > 105 fL, adulto, >24h
Revisar frotis
Cambios macrocíticos
asociados
Solicitar nueva muestra
12 MCV cualquier valor y falla Delta
Verificar integridad de la
muestra
13
MCHC >= 2 unidades sobre rango
Verificar aglutinación,
lipemia, esferocitos
14 MCHC <30 y MCV norma o alto
Investigar posible
contaminación
15 RDW >22 y primera vez Revisar FSP
FÓRMULA DIFERENCIAL
16 No diferencial o diferencial incompleto Revisar FSP
17 #Neutrófilos <1.0 o >20.0 mm3 Revisar FSP
18 #Linfocitos >5.0 adulto a >7.0/ mm3 (>12 años) Revisar FSP
19 #Mono >1.5 adulto o >3.0 /mm3 (>12 años) Revisar FSP
20 # Eosinófilos >2.0/ mm3 y primera vez Revisar FSP
21 #Baso >0.5 mm3
y primera vez Revisar FSP

52 | P á g i n a
22 #NRBC cualquier valor y primera vez Revisar FSP
23 #Reticulocitos absoluto >0.100 /mm3 y primera vez Revisar FSP
SEÑAL DE ALARMA
24 Alarma (excepto granulocitos inmaduros /Banda) y
primera vez y adulto Revisar FSP
25 Alarma y primera vez y niño Revisar FSP
26 Leucocitos no confiable, alarma
Verificar integridad de la
muestra
27 Eritrocitos fragmentos Revisar FSP
28 Eritrocitos dimórficos Revisar FSP
29
Eritrocitos resistentes a lisis
Revisar histogramas,
dispersogramas
Revisar FSP
30
Plaquetas acúmulos, alarma, cualquier valor
Chequear muestra
(coágulos)
Revisar FSP
31 Plaquetas alarma PLT&MPV banderas, excepto
cúmulos Revisar FSP
32 Granulocitos inmaduros alarma y primera vez Revisar FSP
33 Granulocitos inmaduros y resultados previo y Delta
positivo Revisar FSP
34 Desviación a la izquierda alarma
Siga protocolo del
laboratorio
35 Linfocitos atípicos/variantes y primera vez Revisar FSP
36 Linfocitos atípicos/variantes y previos y Delta
positivo Revisar FSP
37 Blasto alarma y primera vez Revisar FSP
38 Blasto alarma y previo y Delta pasa o negativo para
WBC Revisar FSP
39 Blasto alarma y previo y Delta falla positivo para
glóbulos blancos Revisar FSP
40 Eritrocitos nucleados (Normoblastos) alarma Corregir blancos si aplica
41
Reticulocitos, patrón anormal Evaluar desempeño del
equipo
Número de reglas aplicadas
Número total de reglas positivas
Alarmas de instrumento aplicadas
Número total de alarmas de instrumentos positivas

53 | P á g i n a
BIBLIOGRAFÍA
FUENTE PRIMARIA
RepreClinLab ( Representaciones clínicas de Laboratorio) Ramón Ayats Aubert Departamento de Hematología Biológica Hospital de la Santa Creu i Sant
Pau (Barcelona)
“Consejo Internacional de Estandarización en Hematología”
FUENTES SECUNDARIAS
Leeuwenhoek A. Microscopical Observations. Philos Trans R Soc Lond. 1674; 9:121-128
Ehrlich P. Beitrag zur Kenntnis der Anilinfarbungen und iher Verwendung in der mikroskopischen
Technik. Arch Mikr Anat. 1877; 13:263-277
Coulter WH. High speed automatic Blood cell counter and cell size analizar. Proc National
Electronics Conf. 1956; 12: 1034-1040.
Mansberg HP, Saunders AM, Groner W. The Hemalog D whitw cell differential system. J
Hidtochem Cytochem. 1974; 22:711-724
MacDonald AJ, Bradshaw AE, Holmes W.A & Lewis SM: The impact of an integrated haematology
screening system on laboratory practice. Clin. Lab. Haem. 1996; 18: 271-276.
Chapman M: Hematology review criteria and its impact on workflow and productivity. Lab Hematol
1997; 3: 48-52. Smith N, Rosenfeld D & Watman R. Hematology autovalidation system. Lab
Hematol 1999; 5: 52-55.
Mc Fadden S: Automating the review process to improve productivity in the hamatology laboratory.
Lab Hematol 2002; 8: 225-229.
Simson E & Villarrubia J: Decision rules for hematology review: An international perspective.
Abstracts of the XVII
International Symposium on Thechnological Innovations in Laboratory Hematology. May 2004.
Barcelona.
Jenny A, Senn F, Wey J, Tschopp M, Wuillemin WA & Linssen J: Comparison of thecnical
validation before and after implementation of the work area manager SIS 2.0 with standard rule
package. Sysmex J Int 2005; 15: 7-12.
Rümke CL. Imprecision of ratio-derived differential leukocyte counts. Blood Cells 1985; 11: 311-4.
Koepke J. Reference Leukocyte (WBC) Differential Count (Proportional) and Evaluation of
Instrumental Methods; Approved , Standard. 2nd ed. (H20-A2). Clinical and Laboratory Standard
Institute (CLSI); 2007.
Davis B. Extended Differential Blood Count. International Council for Standardization in
Haematology (ICSH). Meeting Minutes. General Assembly: Miami; May 2007.
Barnes PW, McFadden SL, Machin SJ, Simson E. Internacional consensus group for haematology.
The International Consensus Group for Haematology review: suggested for action following CBC
and WB differential analysis. Lab Hematol 2005; 11 (2): 83-90.
Cornbleet PJ. Cinical utility of the band count. Clin Lab Med 2002; 22 (1): 101-36.
Davis BH, Bigelow NC. Comparison of neutrophil CD64 expresion, manual myeloid immaturity
counts and automated haematology analyzer flags as indicator of infection and sepsis. Lab
Hematol 2005; 11 (2): 137-47.
Johannessen B, Roemer B, Flatmoen L, Just L, Aarsand AK, Scott S. Implementation of
monoclonal antibody fluorescence on the Abbott CELL-DYN Saphire haematology analyser:
evaluation of lymphoid, myeloid and platelet markers. Clin Lab Haematol 2006; 28 (2): 84-9

54 | P á g i n a
Molero T, Roemer B, Perera MM, Lemes A, De la Iglesia S, Palacios G, Scout CS. Analysis and
enumeration of T cells, B cells and NK cells using the monoclonal antibody fluorescente capability
of a routine haematology analyser. Clin Lab Haem 2005; 27: 224-34.
Novis DA, Walsh M, Wilkinson D, St Louis M, Ben-Ezra J. Laboratory productivity and the rate of
manual peripheral blood smear review. Arch Pathol Lab Med 2006; 130: 596-601.
Pierre RV. Peripheral blood review. The demise of the eyecount leukocyte differential. Clin Lab Med
2002; 22 (1): 279-97.

55 | P á g i n a
3.2 CONTROL DE CALIDAD INTERNO
Yadira Pacheco Espitia: Bacterióloga y Laboratorista Clínico- Universidad Colegio Mayor de Cundinamarca;
Instituto Nacional de Salud, Dirección de Redes en Salud Pública, Bogotá, Colombia. [email protected]
3.2.1 Generalidades
El objetivo fundamental del
control de calidad en el
laboratorio clínico es
garantizar la confiabilidad y la
utilidad médica de los
resultados, por lo cual resulta
indispensable generar
mecanismos para controlar
cada una de las fases del
control de calidad:
preanalítica, analítica y pos
analítica
La fase analítica incluye entre
otros el control de calidad
interno y el control de calidad
externo. Según la norma ISO
15189, numeral 5.6.1 “El
laboratorio debe diseñar un
sistema de control de calidad
interno adecuado para
verificar el logro de la calidad
esperada en los resultados….
“; por esta razón se debe
construir un efectivo sistema de control de calidad interno, para incrementar la
probabilidad de que cada resultado reportado por el laboratorio sea médicamente útil y
permita al médico tomar una decisión diagnóstica o terapéutica acertada.
El material de control se utiliza para la detección de tendencias, cambios y de errores
analíticos, dicho control puede ser de primera opinión (control ofrecido por el mismo
fabricante del equipo ó reactivo) ó de tercera opinión (obedece a un control que no
pertenece a la misma casa comercial del equipo o reactivo).
Al realizar la selección del material de control de calidad el laboratorio, deben tenerse en
cuenta algunos aspectos importantes como:
Matriz: cada laboratorio clínico decide qué tipo de matriz adquirir de acuerdo a sus
necesidades, ésta en lo posible debe asemejarse a las muestras de los pacientes que se
procesan en el laboratorio; puede ser matriz humana, bovina ó sérica.

56 | P á g i n a
Presentación: la matriz puede ser líquida ó liofilizada. En los controles líquidos se reducen
la variabilidad de vial a vial y se elimina la reconstitución, mientras que los controles
liofilizados pueden almacenarse con mayor facilidad y estabilidad, siempre y cuando se
sigan las condiciones de reconstitución del fabricante y se utilice agua grado reactivo.
Analitos: el material control debe incluir los analitos que se procesan en el laboratorio
(controles multi-analito), por lo cual es importante verificar si el volumen del contenido es
adecuado para controlar todos los analitos.
Controles valorados: el material control debe cubrir los intervalos biológicos de referencia
por lo cual debe contener distintas concentraciones del analito para evaluar el
comportamiento del procedimiento de medición a concentraciones bajas, medias y
elevadas.
Caducidad: debe ser prolongada y el número de lote debe ser el mismo, con el fin de
mantener herramientas de validación constante, propias del laboratorio. Una vez
reconstituido el material control debe ser utilizado dentro del tiempo límite establecido por
el fabricante
Almacenamiento: para garantizar la estabilidad del material control, este debe
conservarse siguiendo las recomendaciones dadas por el fabricante.
Manejo del material de control: en cuanto a la reconstitución y alicuotado del material
control, el laboratorio debe revisar y cumplir las recomendaciones del fabricante, de ser
necesario se realizará una guía rápida que describa el procedimiento para el manejo, guía
que debe ser conocida por todos los profesionales del laboratorio. Al momento de
manipular la matriz de control de calidad se deben seguir las normas de bioseguridad,
dado que todas las muestras biológicas deben ser tratadas como potencialmente
infecciosas.
Niveles de control: el laboratorio debe establecer el número de controles, no obstante
entre menor sea el número se minimizará la detección del error; la recomendación
general es utilizar mínimo dos controles: uno de nivel normal y otro de nivel anormal.
Teniendo en cuenta que la Ronda ó corrida analítica “se define como el intervalo de
tiempo en el que se llevan a cabo una serie de mediciones de manera estable en términos
de precisión y exactitud” (CLSI), el laboratorio debe procesar los controles por corrida
analítica.
Requisitos del proveedor:
En cuanto a la producción del material control el proveedor debe contar con un sistema de
calidad para asegurar la confiabilidad del producto.
Debe existir la posibilidad de reservar a largo plazo un mismo número de lote del material
control por parte del fabricante.
El laboratorio debe disponer de un programa de evaluación Interlaboratorios, que permita
revisar información valiosa de grupos pares.
Para el manejo de datos de control de calidad, el laboratorio debe disponer de un software
que facilite la revisión del desempeño de las pruebas, empleando herramientas como
importación automatizada de datos, reglas de control del proceso y opciones de revisión
de control de calidad.

57 | P á g i n a
La evaluación de los controles (nivel normal y patológico) para cada prueba debe
realizarse al menos diariamente para monitorear el proceso analítico. Si la prueba no es
estable por 24 horas u ocurre algún cambio que afecte la estabilidad de la prueba, los
controles deberán analizarse con más frecuencia.
Para cada nivel de control así como para cada prueba se deben calcular los estadísticos y
rangos a partir de los datos obtenidos. Con respecto al software para el manejo de los
datos de control de calidad dado que los cálculos se realizan automáticamente, es
importante entender las matemáticas en las que este software se basa.
Herramientas estadísticas básicas de uso en el laboratorio clínico:
Según la guía CLSI C24:A3 “Control de Calidad estadístico para los procedimientos de
medida cuantitativos: principios y definiciones” se recomienda al menos 20 mediciones
de material control, para cada nivel de control y en días separados. Se recomienda
establecer medias y límites propios para el material de control.
Cálculo de la media
Medida de tendencia central que identifica el “valor objetivo” de un conjunto de datos, es
el resultado de la suma de todas las mediciones dividido entre el número de mediciones.
Para calcular la media para un nivel de control específico, primero sume todos los valores
recopilados para ese control. Después divida la suma de esos valores entre el número
total de valores.
http://www.cybertesis.edu.pe/sisbib/2002/arevalo_le/html/sdx/arevalo_le-TH.4.html
Cálculo de la desviación estándar (S)
Medida de dispersión que cuantifica qué tan cerca están los valores numéricos. La DE
se calcula a partir de los mismos datos usados para obtener la media.
Coeficiente de variación (CV)
Se expresa en porcentaje, es la relación de la desviación estándar con respecto a la
media y se expresa como porcentaje. Este estadístico es comúnmente usado para

58 | P á g i n a
comparar especificaciones del fabricante, resultados del control de calidad interno, para
hacer pruebas de precisión y cuando se compara el desempeño de instrumentos.
Índice de Desviación Estándar SDI
Este estadístico se usa para comparar los resultados del laboratorio cuando es
comparado con un grupo análogo ya sea de un programa de control de calidad externo ó
con un programa Interlaboratorios.
SDI = (media del laboratorio – media del grupo análogo)
Desviación estándar del grupo análogo
Un SDI = 0, indica que el desempeño del laboratorio es idéntico al promedio del grupo
análogo.
Un SDI ≤ 1.25 se considera aceptable.
Un SDI entre 1.25 y 1.49 se considera aceptable para el desempeño marginal. Es posible
que se requiera alguna revisión del sistema de análisis.
Un SDI entre 1.5 y 1.99 se considera como desempeño marginal y se recomienda una
revisión del sistema de análisis.
Un SDI ≥ 2.0 se considera como desempeño inaceptable y usualmente se requieren
acciones correctivas.
3.2.2 Construcción de la gráfica de control de calidad
La gráfica de Levey Jennings es una de las herramientas más utilizadas en control de
calidad en los laboratorios clínicos, proporciona una buena representación de la precisión
y la exactitud; permite observar el comportamiento de los resultados diarios y sucesivos
de las mediciones realizadas al suero control.
Esta gráfica corresponde a una distribución Gaussiana en la cual los datos se distribuyen
alrededor de la media, ± 1DE, ± 2DE y ±3DE:
Tomado de: www.qualitasaagg.wordpress.com
Para cada analito y para cada nivel de control (como mínimo dos para química), se debe
construir la gráfica de Levey Jennings.

59 | P á g i n a
Media 199,8
DE 6,27
Media + 3DE 218,61
Media + 2DE 212,34
Media + 1DE 206,07
Media 199,8
Media - 1DE 193,53
Media - 2DE 187,26
Media - 3DE 180,99
Elaboración de la gráfica:
Solicitar al proveedor que el lote de suero control sea el mismo durante 1 año (mínimo 6
meses); para evitar recalcular los datos estadísticos (media y desviaciones estándar), con
cada cambio de lote.
Cada vez que se inicie un nuevo lote de suero control se recopilan de 20 a 30 datos; la
validación de éstos es la única que se hará con la media y los rangos dados por el inserto.
Ejemplo: Datos de Colesterol, Nivel 1 obtenidos durante el mes de diciembre de 2011
Día 1 Día 2 Día 3 Día 4 Día 5 Día 6 Día 7 Día 8 Día 9 Día 10 Día 11 Día 12 Día 13 Día 14 Día 15 Día 16 Día 17 Día 18 Día 19 Día 20
200 205 195 202 186 207 194 209 200 196 190 204 196 207 200 205 209 197 196 198
Con los datos obtenidos (de 20 a 30), se calculan la media y la desviación estándar (Ver
educación continuada SC-2011). Recuerde que mientras se mantenga el mismo lote de
suero control no es necesario recalcular estos datos estadísticos.
Colesterol, Nivel 1 (diciembre de 2011)
Para el ejemplo se utilizan 20 datos, en caso de haber obtenido mayor número, se hace el
mismo procedimiento
Realizar los cálculos para establecer los límites estadísticos en la gráfica ± 1DE, ± 2DE y
±3DE así:
Colesterol, Nivel 1 (diciembre de 2011)
Con los límites estadísticos se dibuja la gráfica x-y, donde en el eje x se colocan los días y
en el eje y se colocan los valores estadísticos obtenidos en el paso anterior (media, ±
1DE, ± 2DE y ±3DE)

60 | P á g i n a
Ubique los resultados obtenidos diariamente en la gráfica anterior, teniendo en cuenta los
criterios de validación (no es suficiente con registrar los datos, se debe graficar cada
punto y evaluarlo) utilizados por el laboratorio (Ejemplo: Sistema Multireglas de Westgard)
Cuando los laboratorios procesan los controles internos deben:
Seleccionar mínimo dos niveles de control para cada analito.
Recolectar entre 20 y 25 datos de cada nivel de suero control
Eliminar los datos aberrantes aplicando herramientas estadísticas
(Ej. Filtro de Dixon)
Calcular la media (X) y las Desviaciones Estándar (DE)
Construir la gráfica de Levey Jennigs
Recuerde que los pasos 2, 3, 4 y 5 se realizan por analito y para cada nivel de control
Después de procesar se ubican los puntos directamente sobre las gráficas de Levey
Jennigs, antes de correr las muestras de los pacientes.
Para la validación de los datos del suero control se utilizan “Las reglas de Westgard”, las
cuales fueron desarrolladas por el Doctor James Westgard, basado en conceptos
estadísticos que aumentan la posibilidad de detectar errores dentro del laboratorio.
Las 6 reglas básicas para la validación son 12s, 13s, 22s, R4s, 41s y 10x:

61 | P á g i n a
12s
Un punto de cualquiera de los niveles de control se ubica por fuera de
± 2DE; cuando solo sucede en uno de los niveles de control se pueden correr y validar
las muestras de los pacientes.
Es la única regla que permite la validación de resultados, es decir es una regla de alarma.
13s
Un punto de cualquiera de los niveles de control se ubica por fuera de ± 3DE
22s
Dos puntos consecutivos del mismo nivel de control se ubican por fuera de ± 2DE

62 | P á g i n a
Ó cuando dos puntos de los dos niveles de control en la misma corrida se ubican por
fuera de ± 2DE, al mismo lado de la media.
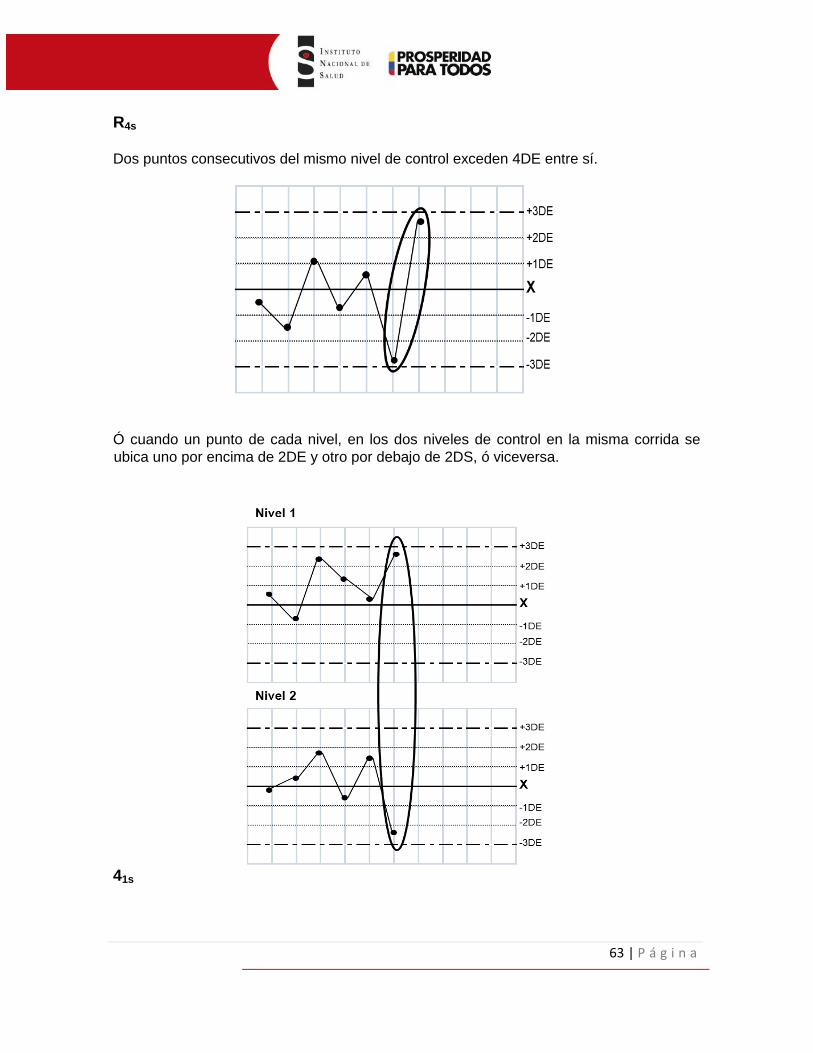
63 | P á g i n a
R4s
Dos puntos consecutivos del mismo nivel de control exceden 4DE entre sí.
Ó cuando un punto de cada nivel, en los dos niveles de control en la misma corrida se
ubica uno por encima de 2DE y otro por debajo de 2DS, ó viceversa.
41s

64 | P á g i n a
Cuatro puntos consecutivos del mismo nivel de control se ubican por fuera de ± 1DE
Ó cuando dos puntos consecutivos de los dos niveles de control en dos días seguidos se
ubican uno por encima ó por debajo de 2DE
10x

65 | P á g i n a
Diez puntos consecutivos del mismo nivel de control se ubican hacia el mismo lado de la
media, sin importar las Desviaciones Estándar.
Ó cuando la sumatoria de los puntos consecutivos de los dos niveles de control que se
ubican al mismo lado de la media dá 10, sin importar las Desviaciones Estándar.
Ej.: En el nivel 1 por encima de la media hay 8 puntos consecutivos y en el nivel 2 por
encima de la media hay 2 puntos consecutivos ó 5 en un nivel y 5 en el otro consecutivos
A excepción de la regla 12s, cuando se cumple alguna de las otras cinco reglas, NO se
permite la validación de los resultados de los pacientes hasta que se detecte el error y se
corrija.

66 | P á g i n a
BIBLIOGRAFÌA
1. CAREY R. Neill, PhD. COOPER W. Gregory CLS, MHA. Sistemas de Control de Calidad
Básico e intermedio para el laboratorio Clínico. División de Control de Calidad Irvine, CA. 2
edición – 2007.
2. COOPER W. Gregory, CLS, MHA. Construcción de un Sistema de Control de Calidad efectivo
para el laboratorio. Take Control IFCC 2007; 3(9): 3 – 7.
3. Roy N. Barnett. Estadística en el laboratorio clínico: Aplicaciones al Control de Calidad y
Valores de Referencia. Editorial Reverté. Barcelona. 2002.
4. E. Boquet Jiménez. Mejoría Continua de La Calidad. Editorial Panamericana. México, 1996.
5. Educación continuada SB 2011 Aspectos Importantes del material de Control de Calidad
Analítico para pruebas cuantitativas del laboratorio clínico. Melania Ríos Parra. Profesional
Especializado – Grupo de Química Clínica, SRNL – INS.
6. Educación continuada SC 2011 Herramientas estadísticas básicas del control de calidad.
Gloria Isabel Barajas B. Profesional Especializado- Grupo Química Clínica, SRNL – INS.
7. Educación continuada SD 2011 Construcción de la gráfica de Control de Calidad Yadira
Pacheco Espitia Profesional Especializado – Sara Milena Núñez Almonacid – profesional
Especializado. Grupo de Química Clínica. SRNL – INS.
8. Sistemas de Control de Calidad Básico e intermedio para el Laboratorio Clínico. División de
Control de Calidad Irvine, CA. 2 edición – 2007.
9. Avances de la Guía CLSI C24:A3 “Control de Calidad estadístico para los procedimientos de
medida cuantitativos: principios y definiciones”.
10. COLOMBIA, NORMA TÉCNICA COLOMBIANA NTC-ISO 15189. Laboratorios Clínicos.
Requisitos Particulares relativos a la Calidad y la Competencia.2009-12-16

67 | P á g i n a
3.3 CONTROL DE CALIDAD EXTERNO
Ana Matilde Rodríguez Becerra; Bacterióloga y Laboratorista Clínica- Universidad de Boyacá;
Instituto Nacional de Salud, Subdirección de Gestión de Calidad de LSP. Bogotá, Colombia.
Figura No.1
El concepto de Control de Calidad Externo abarca la puesta en marcha de diferentes
actividades encaminadas a evaluar la exactitud de los resultados generados por un
laboratorio, mediante la intervención de una entidad externa y ajena al participante.
Encontramos como en los Ensayos de Aptitud, la calidad se ha convertido en un factor
indispensable, ya que no solo se refiere al producto o servicio como tal, sino que es la
mejora permanente y contínua de otros aspectos relacionados tales como organización,
gerencia, manejo y gestión.

68 | P á g i n a
Se observa a lo largo de la historia que desde sus inicios el hombre ha tenido la
necesidad de satisfacer sus obligaciones más elementales, sin tener ningún otro concepto
de calidad más que el de asegurarse que este producto que ha elaborado, cumpla sus
necesidades básicas, la calidad, ha ido evolucionando rápidamente en los últimos años
en función de los cambios requeridos por las necesidades que surgen de los diferentes
actores, no solo a nivel salud sino también a nivel empresarial, con el fin de ser
competitivos , crecer, y que con estos cambios se logre satisfacer necesidades en cuanto
a productos, así como a sus miembros.
En las pruebas de ensayo, la generación de resultados válidos, debe estar ligada en cada
una de sus fases a la implementación de programas de aseguramiento de la calidad, con
el fin de obtener como producto final resultados de análisis con un porcentaje alto de
exactitud y precisión.
Si se busca garantizar la confiabilidad de los resultados emitidos por su laboratorio y a la
vez garantizar que sea comparable con los de otros laboratorios, la mejor estrategia es
participar en Programas de Evaluación Externa del Desempeño. El objetivo de estos
programas es evaluar un método o un grupo de métodos analíticos, y medir la aptitud de
diferentes laboratorios por la comparación de sus resultados contra valores establecidos o
Interlaboratorios con todos los participantes.
En este sentido el asegurar que los resultados sean reproducibles y comparables entre
resultados de otros laboratorios ha sido clave para evaluar su funcionamiento. Stull et al.
Variatión in Proficiency Testing Perormance by Testing Site, (1998), refieren que los
primeros estudios que se realizaron fue en Estados Unidos en 1947, y en sus
conclusiones se revela la variabilidad significativa que se presenta en los resultados entre
los laboratorios participantes.
3.3.1 Interlaboratorios
Dentro de los ensayos de aptitud se encuentran los Programas de Evaluación
Externa del Desempeño (PEED), que funcionan como herramienta para la evaluación
contínua de la competencia de un grupo de laboratorios, mediante el envío continuo y a
determinadas frecuencias de matrices caracterizadas.
Los Programas de Evaluación Externa del Desempeño tienen como objetivo principal
conocer la comparación de los resultados analíticos de diferentes Laboratorios ya que
permite comparar nuestros resultados con respecto a los resultados de otros laboratorios
en una región (país)
La normatividad actual define a la comparación Interlaboratorios como “Organización,
realización y evaluación de mediciones o ensayos sobre el mismo ítem o ítems similares
por dos o más laboratorios, de acuerdo con condiciones predeterminadas.

69 | P á g i n a
Dentro de este esquema, las comparaciones Interlaboratorios, tiene varios propósitos,
entre los cuales están:
Evaluar el desempeño de los laboratorios para llevar a cabo ensayos o mediciones
y hacer seguimiento del desempeño.
Identificar problemas en os laboratorios y de esta manera iniciar acciones para la
mejora
Establecer la eficacia y la comparabilidad de los métodos de ensayo o medida
Aumentar la confianza e identificar las diferencias entre laboratorios. Entre otras.
Si se lleva a cabo una adecuada participación y se asegura el funcionamiento de estos
programas, se estará sumando cada día, gran valor para aportar a la mejora de los
estándares en los laboratorios clínicos.
Pueden constituir una herramienta valiosa ya que gracias a los datos generados se
pueden llevar a cabo comparación de procedimientos analíticos que ya se encuentran
establecidos y a la vez indagar sobre los métodos nuevos que salen al mercado; ya que al
proporcionar un control igual a todos los participantes, se puede luego hacer un contraste
de los resultados mediante un procesamiento estadístico. Además, proporcionar datos
claves que permitan tomar decisiones para aplicar acciones correctivas y proporcionar la
oportunidad de aprovecharlo como un impulso educativo para mejorar el desempeño de
los participantes, teniendo en cuenta que los requerimientos para un aseguramiento de la
calidad son cada vez más exigentes.
La necesidad de confianza constante en el desempeño, no solo es esencial para los
laboratorios y sus clientes, sino también para otras partes interesadas, tales como las
entidades reguladoras, los organismos acreditadores y demás organismos que
especifican y vigilan requisitos para los laboratorios. La norma NTC-ISO/IEC 17011
(ISO/IEC 17011 Numeral (7.15.3)) exige que los organismos de acreditación aseguren la
participación y el desempeño en los ensayos de aptitud de los laboratorios u otros
programas de comparación y que la frecuencia sea la adecuada)
En la actualidad los laboratorios clínicos deben contemplar sistemas para implementar el
control de calidad a fin de garantizar que los resultados generados sean confiables,
adicionalmente cumplir con los requerimientos establecidos por la normatividad actual
NTC-ISO 15189:2009 , que dice “El laboratorio debe desarrollar un mecanismo para
determinar la aceptabilidad de los procedimientos que no se hayan evaluado” (ISO
15189:2009). Así mismo normativamente, los prestadores de servicios de salud deben
cumplir con procedimientos y condiciones para habilitarse y poder prestar los servicios
como se referencia en la Resolución 1441:2013, “Cuenta con: Programa de control de
calidad interno y externo y sus respectivos manuales y Análisis de los reportes del
control de calidad y toma de medidas correctivas documentadas” ((R, de 06 de Mayo de
2013).

70 | P á g i n a
3.3.2 Ciclo de Funcionamiento
El funcionamiento de los Programas Nacionales, es muy similar con respecto a los
Programas Internacionales, en el cual se envía a los laboratorios participantes una o más
matrices caracterizadas, las cuales el laboratorio debe analizar según indicaciones dadas
por cada distribuidor.
El laboratorio debe remitir estos resultados al programa donde estos serán calificados y
estas calificaciones serán procesadas mediante herramientas estadísticas, las cuales
facilitarán la genera ración de un informe para cada laboratorio participante.
3.3.2.1 Flujograma de Funcionamiento
Figura 1. Flujograma de funcionamiento
El participante realiza un proceso de preinscripción y pago (en caso que esto aplique) a
su programa de interés.
El programa realiza el envío de la matriz o matrices a los laboratorios participantes.
El laboratorio participante recibe y analiza la matriz y remite los resultados al Programa.
El Programa recibe y valida los resultados enviados por los participantes.
Se realiza el análisis estadístico de los resultados
Se genera el informe de las evaluaciones con resultados recibidos de los participantes.
La frecuencia y número de materiales a utilizar dependerá de cada programa según sus
necesidades y requerimientos. Así mismo cada programa establece tipo de materiales,

71 | P á g i n a
numero de analítos y suministra la información necesaria en cuanto a material de control,
instrucciones de manejo y condiciones de almacenamiento.
3.3.3 Manejo de Resultados
Los resultados de los Programas de Evaluación Externa se pueden presentar de muchas
maneras y el manejo que se le da, requiere de herramientas estadísticas la cuales deben
ser apropiadas según el tipo de datos que se empleen.
Los datos de los resultados emitidos por parte del laboratorio participante, se pueden
presentar de tipo cuantitativo, cualitativo o semi-cuantitativo, los cuantitativos, a fin de
facilitar su interpretación y permitir la comparación con los objetivos definidos, tienen que
ser trabajados como datos estadísticos de desempeño. El propósito es medir la
desviación con respecto al valor asignado de manera que permita la comparación con los
criterios establecidos de desempeño.
Para los resultados cualitativos o semi-cuantitativos, si se decide usar métodos
estadísticos, estos tienen que ser apropiados de acuerdo a la naturaleza de los datos
utilizados como respuesta. Para los datos cualitativos la técnica apropiada es comparar el
resultado de un participante con el valor asignado. Si son idénticos el desempeño es
aceptable. Si no son idénticos, se necesita un juicio experto para determinar el manejo
que se le dará a este (11).
Determinación del valor asignado
Al comparar el resultado obtenido con un valor asignado, se logra proporcionar una
valoración del error cometido y este valor asignado es un valor convencionalmente
verdadero, que puede estimarse de diversas formas y esto, va a depender de cada
programa (11), entre los procedimientos más comunes para establecerlo tenemos:
Valores conocidos: Por ejemplo resultados determinados por el fabricante
Valores de referencia certificados: Por métodos de ensayo o de medida absolutos.
Valores de referencia: Como los determinados por análisis o comparación del ítem, con
un material de referencia o patrón
Valores consensuados por expertos: Calculando la media de resultados de un grupo de
laboratorios seleccionados por el elevado nivel de calidad de sus resultados.
Valores consensuados por los participantes: Calculando la media de resultados de todos
los laboratorios participantes.
3.3.4 Evaluación del desempeño
Los Programas de Control Externo ofrecen una manera de hacer seguimiento al
desempeño de cada participante, mediante la elaboración de informes de desempeño ya
sea periódico (después de cada medición) o global (al completar o finalizar el periodo
establecido) sobre los resultaos obtenidos con respecto a los métodos utilizados y se
brinda información sobre el laboratorio, el conjunto de laboratorios participantes y sobre
los métodos de medida.

72 | P á g i n a
s importante que el participante tenga claridad a la hora de interpretar los datos que
proporciona un programa de evaluación externo, y que por errores de interpretación, no
se interfiera en el cumplimiento de su objetivo, ya que lo que se busca con estos es,
estimar el error sistemático del procedimiento de medida y estimular la mejora continua de
un sistema de calidad ante la posibilidad de detectar errores que no se han identificado
con el control interno, evitando así la toma de decisiones incorrectas para iniciar acciones
oportunas.
BIBLIOGRAFIA
1. Historia de la calidad, Cronología del Siglo XX;
http://www.tecnologiaycalidad.galeon.com/calidad/6.htm.
2. Historia del Control de Calidad Total;http://www.calidad.ugto.mx/archivos/historia.pdf.
3. SOLANO Nereida1, FLORES, Daniel; GONZÁLEZ Rafael,UZCÁTEGUI Luz, VERDE
Zuliamar, MEZA Aliria, RODRÍGUEZ Carmen, APONTE Dayabel: EVALUACIÓN
EXTERNA DE LA CALIDAD EN LABORATORIOS CLÍNICOS PÚBLICOS Y PRIVADOS
EN EL ÁREA DE BIOQUÍMICA CLÍNICA DE CIUDAD BOLÍVAR; Saber, Universidad de
Oriente, Venezuela.Vol. 20. Nº 2: 155-162. 2008.
4. Stull, Thomas L. Hearm,PhD;John S. Hancock; James H. HAndsfield, MPH; Carlyn L.
Collins, MD,MPH; Variatión in Proficiency Testing Perormance by Testing Site, Tina M
Stull,MD.
5. Control de Calidad Externo; LIC INTERLAB;
http://www.interlab.com.ec/htm/aseguramiento_externo.htm
6. Cromidion; Abastecimiento Integral Hospitalario, RIQAS Control de Calidad Externo;
http://www.cromoion.com/content.php?id=361&cc=36 .
7. Vargas,Luz Mila; Guevara, Milena; Angel, Blanca Celmira; Cano, Luis Gerardo; Guía de
Control de Calidad Interno y Externo de Laboratorio Clínico. Versión 01-2007
8. Rodríguez Socarrás, Isis, Lic; Carbajales León, Ana Isabel, Dra.; Jiménez Marrero,Annia;
Dra; Sistema integral de evaluación externa de la calidad para los laboratorios clínicos
(versión 2); Revista Archivo Médico de Camagüey versión ISSN 1025-0255 AMC v.14 n.2
Camagüey mar.-abr. 2010.
9. Gella, F. Javier; Control de la Calidad en el Laboratorio Clínico, BioSystems S.A. 2
Edición,Barcelona (España), Julio 2005.
10. COLOMBIA NTC-ISO/IEC 17043:2010 Requisitos Generales para los Ensayos de
Aptitud.ICONTEC.

73 | P á g i n a
4. FASE POSTANALÍTICA
Gloria Isabel Barajas Barbosa- Bacterióloga y Laboratorista Clínica - Universidad Colegio Mayor de
Cundinamarca; Instituto Nacional de Salud. Bogotá, Colombia. [email protected]
Independientemente del cuidado y la atención que se hayan dedicado a las fases
preanalítica y analítica, se deben realizar varios pasos importantes durante la fase
postanalítica para asegurar la calidad y utilidad de los resultados del laboratorio clínico.
La fase postanalítica incluye:
Confirmación de los resultados
Intervalos Biológicos de Referencia
Puntualidad
Reporte de resultados
Confidencialidad
Cada uno de estos pasos requiere de procedimientos y decisiones cuidadosos para
incrementar la calidad de los resultados.
Confirmación de resultados
La práctica clínica requiere cada vez más información para poder tomar decisiones
correctas que afectan al cuidado de los pacientes. Uno de los principales proveedores de
información es el laboratorio clínico. Está publicado que entre el 60-70% de las decisiones
que implican ingresos, altas y elección terapéutica están apoyadas en los resultados de
laboratorio. Puede afirmarse, que un laboratorio genera valor para su institución, cuando
verdaderamente actúa como un eje central en el proceso de decisión clínica.
Para realizar esta misión, los laboratorios no sólo deben proporcionar resultados fiables y
en tiempos de respuesta adecuados, sino también intervenir en la elección racional de las
pruebas solicitadas y participar activamente en la interpretación de los resultados para
convertirlos en información y conocimiento.
La validación es un proceso postanalítica, mediante el cual los resultados obtenidos, son
revisados y quedan disponibles para su uso clínico. Su propósito principal es prevenir y
evitar la emisión de información de laboratorio con errores o incongruencias que puedan
traducirse en decisiones médicas incorrectas.

74 | P á g i n a
4.1 TERMINOLOGÍA
Son diversos los términos utilizados para referirse al proceso de validación. En ocasiones
se habla de liberación, aprobación, aceptación, verificación, etc.
• Verificación. Confirmación del cumplimiento de los requisitos especificados por
medio del examen y la aportación de evidencia objetiva.
• Validación. Confirmación del cumplimiento de los requisitos particulares para un
uso concreto específico por medio del examen y la aportación de evidencia objetiva.
Proceso de validación / autovalidación
El objetivo principal de la validación de resultados es asegurar que no hay razones obvias
que comprometan su validez antes de que estén disponibles para su uso clínico.
Con la paulatina introducción de la informatización en los laboratorios clínicos, la
validación pasa a realizarse asistida por ordenadores atendiendo fundamentalmente a:
Los avisos, alarmas, mensajes de los equipos automatizados.
Resultados fuera de los intervalos biológicos de referencia.
Concordancia con respecto a resultados previos o de otras pruebas relacionadas
fisiopatológicamente.
Cualquier otra fuente de información clínica relevante.
La autovalidación tiene dos posibles consecuencias:
• La validación de los resultados que cumplen los criterios o reglas
establecidas.
• La retención para ser validados de forma manual de aquellos resultados que
precisan una revisión personal por los profesionales del laboratorio y si es
preciso generar una acción (repetición de la prueba, adición de nuevos test, aviso al
clínico, etc.).
Los criterios, algoritmos o reglas de validación deben ser previamente definidos por cada
laboratorio. Debe hacerse con solidez científica y consistencia. El proceso de
autovalidación, siempre utilizará los mismos criterios, revisará toda la información
disponible y su trabajo no será interrumpido por asuntos menores. De esta forma se
elimina o minimiza la variabilidad interindividual y la posibilidad de cometer errores
asociados a la fatiga de los procesos repetitivos.
La guía “Guidelines for autoverification (Auto 10-A)” del Clinical and
Laboratory Stan- dards Institute (CLSI, anteriormente NCCLS) es quizás la publicación
más exhaustiva sobre autovalidación. La última versión es de 2006, y recomienda que los
laboratorios clínicos tienen que establecer políticas y procedimientos de

75 | P á g i n a
autoverificación previamente a la aplicación de estos procesos sobre los resultados de
pacientes.
Intervalos Biológicos de Referencia
Para poder interpretar con fines diagnósticos un valor medido de una magnitud biológica
de un paciente es imprescindible conocer uno o más valores de esa magnitud medidos en
individuos similares y compararlos. Debido a esta necesidad surge el concepto de
Intervalos Biológicos de Referencia.
Un Intervalo Biológico de Referencia es un valor medido de una magnitud particular
obtenido con fines comparativos en un individuo —llamado individuo de referencia— que
cumple unos requisitos preestablecidos. Estos requisitos pueden ser muy diversos,
dependiendo de la finalidad del Intervalo, si son de individuos sanos o afectados de una
enfermedad concreta, lo imprescindible es que la descripción de dichos individuos no sea
ambigua. No obstante, los más usados son los correspondientes a individuos
presuntamente sanos; tanto es así que habitualmente, si no se indica lo contrario, al
hablar de Intervalo Biológico de Referencia se supone que se trata de una población de
individuos sanos.
Queda claro, pues, que en el proceso diagnóstico un valor observado en un paciente sólo
debería compararse con Intervalos Biológicos de Referencia suministrados por el mismo
laboratorio que ha efectuado la medición de la magnitud biológica en cuestión. En todo
laboratorio clínico se debería incluir en los informes los intervalos de referencia biológicos
correspondientes a las magnitudes biológicas medidas en cada paciente. Los intervalos
de referencia pueden estimarse a partir de valores de referencia producidos por el propio
laboratorio, de forma individual, o formando parte de un equipo de laboratorio. Si no
dispone de Intervalos Biológicos de Referencia propios debe adoptar unos de ajenos. Los
intervalos ajenos sólo debería adoptarse después de validarlos en el propio laboratorio,
pero, desgraciadamente, en la mayor parte de los casos esto no es así.
La Federación Internacional de Química Clínica (IFCC) ha elaborado recomendaciones
para la producción de Intervalos Biológicos de Referencia. En el texto “Intervalos de
referencia biológicos Xavier Fuentes Arderiu Laboratori Clínic Hospital Universitario de
Bellvitge L’Hospitalet de Llobregat (Barcelona) Cataluña, España” y en el documento CLSI
C28-P3
Puntualidad, urgencias e informes
El tiempo global de obtención de un resultado (tiempo de respuesta es el tiempo que
transcurre desde el momento en que el análisis se solicita hasta el momento en que se
entrega el resultado al médico o paciente. Durante la fase postanalítica el tiempo global
de conclusión puede variar dependiendo de los cálculos y la elaboración del informe.

76 | P á g i n a
El tiempo de respuesta de todos los resultados del laboratorio clínico debe ser tan corto
como sea posible sin sacrificar calidad, sin embargo, los esfuerzos por acortar
excesivamente el tiempo de respuesta pueden conducir a errores importantes.
Una prueba de laboratorio se solicita y se procesa como urgente sólo cuando una
decisión médica crítica depende directamente de la puntualidad del resultado, por
ejemplo, cuando se miden marcadores del infarto al miocardio en la sangre de un
paciente antes de iniciar terapia trombolítica. En esta situación un sacrificio parcial de la
calidad metrológica del resultado, por ejemplo por medio de un análisis descentralizado
con un procedimiento menos exacto, se justifica puesto que un resultado puede llegar
demasiado tarde si la muestra debe transportarse a un laboratorio clínico distante. Sin
embargo, no existe justificación para sacrificar la calidad de ningún resultado de
laboratorio. Por ejemplo iniciar a un paciente en terapia de esteroides por lupus
eritematoso sistémico es una decisión clínica crítica, sin embargo la puntualidad no
justifica que se utilice un método de aglutinación de látex para detectar anticuerpos
nucleares “rápidamente”.
Informe
Las cantidades y las unidades son cruciales en los informes. Las mediciones de
laboratorio utilizan principalmente cinco cantidades que no son derivadas y que son el
tiempo, la longitud, la masa, la cantidad de sustancia y el número de entidades. El
Sistema Internacional de Unidades (Systeme International d´Unite, abreviado SI) definido
por la Conferencia Internacional de Pesos y Medidas en el sistema de unidades adoptado
por todos los asignatarios de la Convención Diplomática del Metro.
Cuando proceda, la descripción de los análisis realizados y sus resultados deberían
seguir vocabulario y la sintaxis recomendada por una o más de las siguientes
organizaciones (3):
Consejo Internacional de Normalización en Hematología (ICSH)
Sociedad Internacional de Hematología (ISH)
Federación Internacional de Química Clínica (IFCC)
Unión Internacional de Química Pura y Aplicada (IUPAC)
Sociedad Internacional de Trombosis y Hemostasia (ISTH)
Comité Europeo de Normalización (CEN).
Cuando proceda, la descripción y los resultados deberían seguir la nomenclatura
recomendada por una o más de las siguientes organizaciones:
Unión Internacional de Bioquímica y Biología Molecular (UBMB).
Unión Internacional de Sociedades de Microbiología (IUMS).
Unión Internacional de Sociedades de Inmunología (IUIS).
SNOMED Internacional, (Colegio Americano de Patólogos).
Organización Mundial de la salud (OMS).
La dirección del laboratorio debe ser responsable del formato dado a los informes de
resultados. Los resultados deben ser legibles, sin errores de transcripción y comunicados
a personas autorizadas para recibirlos y utilizar la información clínica. El informe de
resultados debe incluir pero no estar limitado a lo siguiente:

77 | P á g i n a
Identificación clara y no ambigua del análisis, incluyendo cuando sea apropiado, el
procedimiento de medición.
Identificación del laboratorio que emite el informe.
La identificación única y la dirección del paciente, cuando sea posible y destino del
informe.
El nombre u otro identificador único del solicitante y la dirección del mismo.
Fecha y hora de la obtención de la muestra primaria, cuando tal información esté
disponible y sea pertinente para la asistencia al paciente y la hora de recepción por el
laboratorio.
Fecha y hora de emisión del informe la cual, si no aparece en el mismo debe ser
fácilmente accesible cuando se necesite.
El origen y el sistema (o el tipo de muestra primaria).
Nombre del médico solicitante.
Resultados expresados en Unidades del SI o unidades trazables a unidades del SI,
cuando proceda.
Los Intervalo Biológico de Referencia, si procede.
Otros comentarios por ejemplo calidad o idoneidad de la muestra primaria que puede
haber comprometido el resultado.
La identificación de la persona que autoriza la entrega del informe de resultados.
Los resultados originales y los corregidos si procede.
La firma o autorización de la persona que verifica o entrega el informe de resultados,
cuando sea posible.
BIBLIOGRAFÍA
1. M.L. E. Boquet Jiménez; M.L. Castillo Sánchez; Mejoría de la Calidad Guía para los
laboratorios clínicos de América Latina –Editorial médica Panamericana. Pág. 89
2. Cava Valenciano, Fernando; Autovalidación de resultados en el laboratorio clínico área de
Laboratorio Hospital Universitario Fundación Alcorcón. Ed Cont Lab Clín; 13: 104-135 2009-
2010.
3. COLOMBIA, NTC-ISO 15189, Laboratorios Clínicos. Requisitos particulares relativos a la
calidad y la competencia. ICONTEC 21 de Mayo de 2014. 48 pp.

78 | P á g i n a
5. ASEGURAMIENTO METROLÓGICO
Jenny Marcela Rojas Morales, Ingeniera Biomédica- Universidad Manuela Beltrán; Subdirección
Gestión de Calidad de LSP-INS. Bogotá, Colombia. [email protected]
Angie Paola Rodríguez Guerrero- Ingeniera Biomédica- Universidad Manuela Beltrán
Subdirección de Gestión de Calidad de LSP-INS; Bogotá, Colombia. arodrí[email protected]
5.1 IMPORTANCIA DE LA GESTIÓN METROLÓGICA EN LOS
LABORATORIOS DE DIAGNÓSTICO.
La Gestión Metrológica es un proceso fundamental que proporciona herramientas
necesarias para lograr la confirmacion metrologica y el control continuo de los procesos
de medicion que aportan de manera significativa en la toma de decisiones en el contexto
del aseguramiento de la calidad de los mètodos de ensayo, con el fin de proporcionar
fiabilidad en los resultados de un Laboratorio.
En este capitulo se busca proporcionar información a los profesionales (usuarios de los
equipos de laboratorio) que les permita contextualizar y aclarar, inquietudes con respecto
a los términos comúnmente empleados en Metrología, fortaleciendo la gestion de
laboratorio con el fin de minimizar las probabilidades de error al momento de reportar
resultados de los ensayos realizados por deficiencias en el funcionamiento de los
equipos.
5.2 INTERVENCIONES METROLÓGICAS EN EQUIPOS DE LABORATORIO
Figura 1. Intervenciones Metrológicas

79 | P á g i n a
¿Qué es un mantenimiento?
Es la revisión y/o reparación del equipo para disponerlo dentro de las especificaciones
requeridas.
¿Qué tipo de mantenimientos hay?
Predictivo: Facilita tomar decisiones antes de que se presente cualquier mal
funcionamiento en el equipo.
Preventivo: Como objetivo principal, reduce las fallas y tiempos muertos lo que
incrementa la disponibilidad de cada equipo.
Correctivo: Son las acciones de reparación de daños causados por deterioros normales ó
uso por acciones extrañas o imprevistas.
¿Qué es una verificación?
Es la aportación de evidencia objetiva de que un equipo valorado satisface los requisitos
especificados, ya sea según norma o fabricante.
¿Qué tipo de verificaciones hay?
Tabla 1: Tipos de verificaciones en equipos de Laboratorio.
TIPO DE
VERIFICACIÓN
EJECUTOR DE LA
ACTIVIDAD
FUNCIÓN
Intermedia
interna
Entidad
responsable del
equipo.
Realizar la verificación con patrones físicos o
Materiales de Referencia Certificados
calibrados, propios de la entidad propietaria del
equipo, con el fin de garantizar entre períodos
de calibración y/o verificación (según aplique)
la conservación de las caracterísitcas
metrológicas de dicho equipo.
Intermedia
externa
Proveedor
contratado
Realizar la verificación con patrones físicos o
Materiales de Referencia Certificados
calibrados, suministrados por el proveedor,
con el fin de garantizar entre períodos de
calibración y/o verificación (según aplique) la
conservación de las características
metrológicas de dicho equipo.
Externa Proveedor
contratado
Realizar la verificación con patrones físicos o
Materiales de Referencia Certificados
calibrados, suministrados por el proveedor,
con el fin de garantizar posterior a cada
mantenimientos realizado la conservación de
las características metrológicas de dicho
equipo.

80 | P á g i n a
¿Qué es una calibración?
Es medir contra un patrón o Material de Referencia Certificado y registrar los resultados
para saber con certeza qué tan cercanos son los resultados que proporciona el
instrumento de medición a un valor nominalmente verdadero. Para más claridad remitirse
a Figura 2.
¿Qué es una calificación?
Sirve para proveer información acerca del diseño, instalación, operación y desempeño de
cualquier sistema. Es un procedimiento donde se detalla el funcionamiento del equipo
bajo especificaciones establecidas por fabricante y necesidades del usuario.
¿Qué tipo de calificaciones hay?
Calificación de Instalación:
Verificación documentada de que las necesidades claves para la instalación corresponden
a las recomendaciones del fabricante para los cuales fue diseñado el equipo.
Calificación Operacional:
Verificación de que el equipo funciona en la forma esperada y son capaces de operar
satisfactoriamente en el rango de los parámetros para los que ha sido diseñado.
Calificación de Desempeño:
Se demuestra la efectividad del proceso, bajo dos tipos de condiciones: la 1° son las
normales de operación y la 2° bajo límites de operación
Figura 2. Calibración

81 | P á g i n a
Figura 3. Frecuencia de las Intervenciones
5.3 FRECUENCIA DE LAS INTERVENCIONES METROLÓGICAS EN EQUIPOS
DE LABORATORIO:
¿Cómo determinar las frecuencias de las intervenciones Metrológicas?
Es necesario tener en cuenta cuatro factores importantes a nivel general:
Para cualquier tipo de mantenimiento:
La frecuencia de uso, las recomendaciones de fabricante indicadas en el manual de
operación, y el comportamiento del equipo.
Para las verificaciones:
Teniendo en cuenta la tabla n° 1, estas deben ser realizadas de forma inmediata a cada
intervención de mantenimiento.
Para las verificaciones intermedias:
Teniendo claros los tiempos de calibración, garantizando la conservación de las
características metrológicas e identificando el comprotamiento del mismo a través de los
resultados reportados en los certificados de estas intervenciones, se puede determinar
entre períodos de calibración tiempos medios.

82 | P á g i n a
Por ejemplo:
CALIBRACIÓN
1
VERIFICACIÓN
INTERMEDIA 1
CALIBRACIÓN
2
VERIFICACIÓN
INTERMEDIA 2
2013-12-06 2014-06-06 2014-12-06 2015-06-06
Los prinicipales factores que influyen los intervalos son:
Tipo de equipo y uso.
Tendencias de datos obtenidos de registros de calibraciones previas.
Comportamiento del equipo evidenciado en registros o informes.
Condiciones ambientales en las que se encuentra el equipo.
Para calibraciones:
Inicialmente es posible determinar períodos anuales, no obstante, teniendo en cuenta el
comportamiento de los equipos, este puede incrementarse o disminuirse evidenciando los
resultados entre informes de calibración de períodos consecutivos. En caso que el equipo
presente variaciones de un período a otro muy altas debe evaluarse la posibilidad de
recambio tecnológico.
Es posible también determinar estos períodos si el fabricante lo indica.
Para calificaciones:
Para Calificación de Instalación, sólo se realiza una vez siempre y cuando el equipo no
sea trasladado.
Para Calificación Operacional y de Desempeño su vigencia es anual, siempre y cuando el
equipo no requiera ajustes.
5.4 INFRAESTRUCTURA NECESARIA PARA LA INSTALACIÓN DE LOS
EQUIPOS DE LABORATOTIO:
Es necesario realizar el aseguramiento de las condiciones ambientales del área donde se
encuentra instalado cada uno de los equipos, minimizando las probabilidades de daños
provocados por las condiciones a las cuales es expuesto, logrando mantener las
características metrológicas establecidas por el fabricante y garantizando que el ensayo
se desarrolle según lo requerido a nivel general.
Para tal efecto se proponen por secciones las condiciones:
Ambientales.
Eléctricas.

83 | P á g i n a
CONDICIONES AMBIENTALES
TEMPERATURA PRESION HUMEDAD
Debe verificarse según catálogo del equipo a comprar las condiciones ambientales en las
cuales el equipo opera satisfactoriamente, teniendo en cuenta Temperatura, Humedad y la
Altura de la ciudad en la que se encuentra. Esta información usted la encuentra en el catálogo
del producto sección "especificaciones técnicas" o en los manuales de uso proporcionados
por el proveedor autorizado por casa matriz.
CONTROL Y MONITOREO DE CONDICIONES AMBIENTALES
Control:
Debe garantizarse, mediante Aire Acondicionado, u otros equipos
disponibles para que el área no eleve o baje su temperatura de
modo tal que altere el correcto funcionamiento del equipo. El
propósito con estos equipos es regular de manera automática las
condiciones climáticas dentro del laboratorio, teniendo en cuenta
cada Departamento.
Monitoreo:
Contar con sistemas de monitoreo de condiciones ambientales
(temperatura y humedad), tales como termohigrómetros o
sensores de temperatura y humedad calibrados en los puntos
típicos de registro de las condiciones ambientales que se
presenten en el área, abarcando todo el rango, de tal manera que
se puedan realizar las correcciones que den a lugar según
certificado de calibración.
Registrando la temperatura y humedad en los debidos formatos el
comportamiento del área (teniendo en cuenta los criterios del
laboratorio -se recomienda mínimo dos veces por día am/pm-),
con esto evidenciando en caso de que se presenten variaciones
o evidenciar por el contrario homogeneidad en el área donde se
desarrolla el método de ensayo.

84 | P á g i n a
CONDICIONES ELÉCTRICAS
ÍTEM DESCRIPCIÓN
Consumo de corriente Verificar el área donde va a conectar el equipo que la toma
eléctrica en primer lugar se encuentre identificada para poder
determinar en la caja de principal (taco) a qué taco está
asignada y se verifica la capacidad de suministro en corriente del
taco, este debe ser superior al que tiene el equipo por
especificación.
Tipo de toma disponible Verificar que la clavija del equipo a comprar sea la disponible en
las áreas y a su vez que le proporcione el voltaje necesario para
que el equipo encienda, para Colombia hay posibilidad de tomas
de 110v a 120v, y de 220v a 240v.
BIBLIOGRAFÍA.
1. Taller Nacional de Actualización para el Fortalecimiento de la Calidad en la Red Nacional de
Laboratorios, Presentación Power Point. Gestión Metrológica en Laboratorios de Salud Pública.
Dirección Redes en Salud Pública – Subdirección Gestión de Calidad de Laboratorios de salud
Pública. 2013-10-04.

85 | P á g i n a
6. VIGILANCIA EN SALUD PUBLICA POR
LABORATORIO CLÍNICO
Lina Marcela Quevedo Gutiérrez; Bacterióloga y Laboratorista Clínica- Universidad de Boyacá;
Instituto Nacional de Salud, Grupo Genética y Crónicas. Bogotá, Colombia. [email protected]
La vigilancia en general, cualquiera que sea el campo o disciplina que la use, corresponde
a un proceso sistemático, ordenado y planificado de observación y medición de ciertas
variables definidas, para luego describir, analizar, evaluar e interpretar tales
observaciones y mediciones con propósitos definidos.
La vigilancia de la salud es la observación sistemática, el análisis y la interpretación de
datos cuyos resultados se utilizarán en la planeación, implementación y evaluación de la
práctica en salud pública. Ayuda a tomar decisiones oportunas; a establecer prioridades
de salud pública; generar hipótesis de investigación, a identificar y tomar decisiones de
intervención mediante el seguimiento de aquellos eventos o factores determinantes o
condicionantes que puedan modificar el riesgo de ocurrencia, a fin de iniciar y completar
oportunamente las medidas de control necesarias, evaluar programas y a hacer uso
racional de los recursos disponibles en los diferentes niveles de atención.
Actualmente la Vigilancia de la Salud, rompe con una serie de paradigmas, pues
incorpora no solamente las enfermedades infecciosas, e incluye todos aquellos eventos
no transmisibles, factores de riesgo y otras condiciones de importancia en salud pública.
De esta manera, presenta una visión integrada de la práctica de la epidemiología en los
servicios de salud, con el modelo readecuado de atención.
6.1 FUNCIONES
El objetivo fundamental de todo Sistema de Vigilancia de la Salud es actualizar
permanentemente el conocimiento de todos los aspectos relacionados al comportamiento
de los problemas de salud de una población de determinada zona geográfica (país,
región, localidad) con el fin último de controlar y prevenir estos problemas de salud
Para que la vigilancia epidemiológica sea útil y efectiva debe cumplir una serie de
atributos:
1. La información debe recogerse de forma sistemática. La vigilancia de los problemas prioritarios debe ser permanente a lo largo del tiempo. 2. La información ha de ser específica y selectiva. Tan solo debe recogerse aquella que sea útil. El exceso de información puede ser tan perjudicial como la carencia de la misma. 3. La información recogida debe estar vinculada a actuaciones de prevención y control, e s lo que se conoce como información para la acción.

86 | P á g i n a
4. La información recogida debe tener como referencia una población. El objeto es conocer lo que está ocurriendo en la población bajo vigilancia. 5. La vigilancia es una función de Estado generalmente respaldada por Leyes u otro tipo de normativa.
6.2 COMITÉ DE VIGILANCIA
Los comités de vigilancia en salud tienen como objetivo desarrollar los análisis pertinentes
de la información generada por el sistema, establecer criterios para la programación de
actividades y facilitar la toma adecuada de decisiones, que deben traducirse en acciones
concretas, las cuales posteriormente deben ser socializadas y tener seguimiento a través
de indicadores de gestión e impacto en el control de los eventos, brotes o epidemias.
Actuarán como comités de vigilancia en salud pública: los comités de vigilancia
epidemiológica (Cove), los comités de infecciones intrahospitalarias, los comités de
estadísticas vitales, los comités de vigilancia epidemiológica comunitaria (Covecom), y
otros comités afines que se hayan conformado para efectos de análisis e interpretación de
la información de vigilancia en salud pública.
Los comités de vigilancia epidemiológica deberán reunirse ordinariamente una vez al mes
y serán presididos por el director territorial de salud. La secretaría técnica estará a cargo
del responsable de vigilancia epidemiológica de la dirección territorial de salud.
6.3 EVENTOS DE INTERÉS EN SALUD PÚBLICA
Aquellos eventos considerados como importantes o trascendentes para la salud colectiva
por parte del Ministerio de la Protección Social, teniendo en cuenta criterios de frecuencia,
gravedad, comportamiento epidemiológico, posibilidades de prevención, costo-efectividad
de las intervenciones, e interés público; que además, requieren ser enfrentados con
medidas de salud pública.
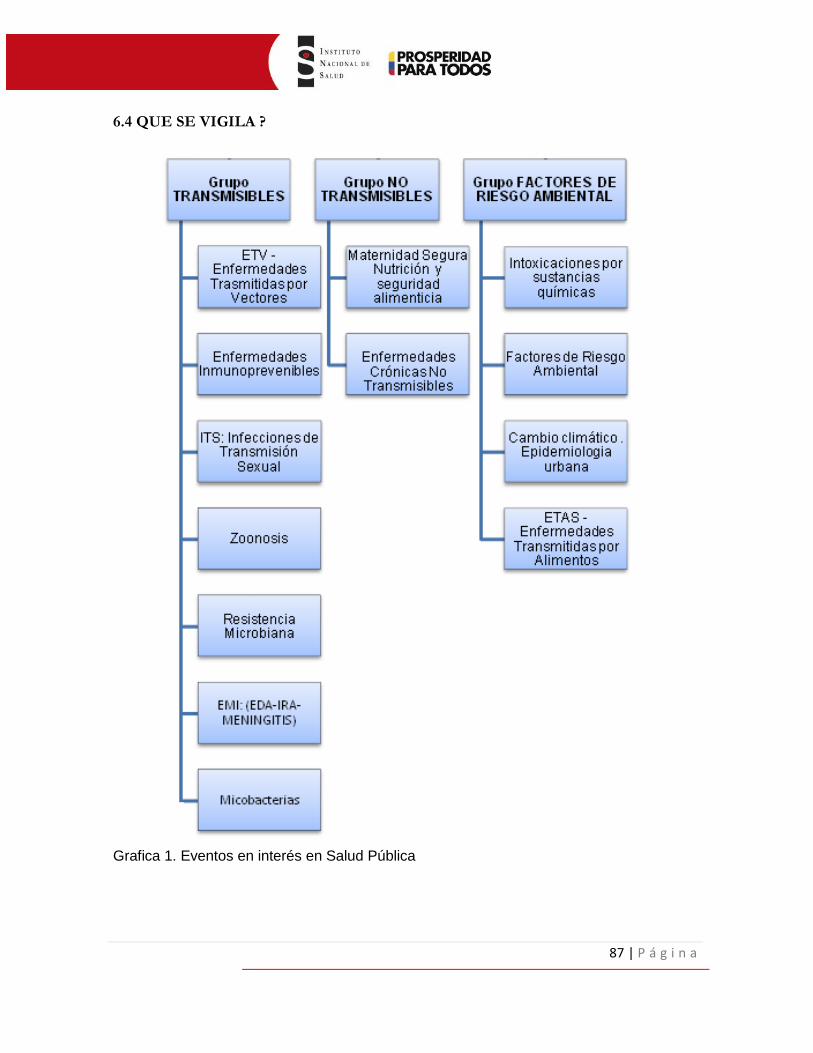
87 | P á g i n a
6.4 QUE SE VIGILA ?
Grafica 1. Eventos en interés en Salud Pública

88 | P á g i n a
6.4.1 Grupo de Transmisibles
Enfermedades transmisores de vectores: Chagas, Dengue, Leishmania, Malaria
Inmunoprevenibles: Parálisis flácida aguda, Sarampión/rubéola y síndrome de rubéola
congénita, Tétanos neonatal, Tétanos accidental, Tos ferina y difteria, Varicela, Eventos
supuestamente atribuidos a la inmunización (Esavi).
Infecciones de transmisión sexual: VIH/SIDA, Sífilis gestacional y congénita, Hepatitis B
Zoonosis: Rabia y agresiones por animales potencialmente transmisores de rabia,
Accidente ofídico, Leptospirosis, Encefalitis equinas, La peste, rickettsiosis, enfermedad
de Creutzfeldt-Jakob, carbunco y cualquier otra zoonosis con alto riesgo de transmisión
(animal-humano o humano-humano)
Resistencia Microbiana
(EDA-IRA-MENINGITIS): Infección Respiratoria Aguda, Enfermedad Similar a Influenza e
Infección Respiratoria Aguda Grave (ESI-IRAG), IRAG Inusitada, mortalidad por IRA en
menores de 5 años y morbilidad por IRA, Morbilidad en todos los grupos de edad y
mortalidad por enfermedad diarreica aguda (EDA) en menores de cinco años, Meningitis
bacteriana aguda.
Micobacterias: Tuberculosis, Tuberculosis farmacorresistente, Lepra,
6.4.2 Grupo de No Transmisibles
En la actualidad, las enfermedades crónicas No trasmisibles son la principal causa de
morbi-mortalidad del país. La importancia adquirida por estos eventos y las opciones
concretas para su prevención, las definen como prioritarias, e indican que se deben
adelantar acciones para enfrentarlas.
Maternidad Segura: mortalidad materna perinatal, neonatal tardía y morbilidad materna
extrema (MME).
Enfermedades crónicas: Cáncer infantil, Defectos congénitos, diabetes.
6.4.3 Grupo Factores de riesgo ambiental y sanitario
Los seres humanos demandan de los recursos disponibles en el medio ambiente para su
supervivencia, salud y bienestar, en el proceso de satisfacer sus necesidades y mejorar
sus condiciones económicas y de vida ha creado también riesgos para la salud y la
supervivencia, esta relación dinámica, abierta y de mutua afectación entre las personas y

89 | P á g i n a
el ambiente, es el contexto de referencia para comprender las relaciones entre el medio
ambiente y la salud. (OMS)
Intoxicaciones por sustancias químicas: Intoxicaciones agudas por Plaguicidas,
Medicamentos, Metanol, Metales pesados, Solventes, Monóxido de Carbono y otros
gases, Sustancias Psicoactivas, Otras sustancias químicas.
Factores de Riesgo Ambiental: Enfermedades transmitidas por alimentos – ETA, Hepatitis
A, Fiebre Tifoidea y Paratifoidea, Cólera
6.5 ROL DEL LABORATORIO CLINICO
El laboratorio cumple un rol fundamental en la vigilancia epidemiológica, pues muchos de
los eventos que se vigilan requieren del diagnóstico de laboratorio. El apoyo de este
resalta ante la diversidad de enfermedades que se presentan manifestaciones en forma
sindrómica, por lo que el médico requiere del soporte del laboratorio para descartar o
confirmar el caso.
Para el desarrollo de sistemas de vigilancia efectivos, ante la aparición de enfermedades
emergentes, el laboratorio debe ser capaz de detectar enfermedades infecciosas nuevas
o amenazas como el resurgimiento de problemas que se creían controlados. La vigilancia
basada en el laboratorio puede proveer datos de la localización y frecuencia de
aislamiento de patógenos específicos, que se presentan en conglomerados o con un
incremento mayor al esperado.
Así, generalmente, los datos de vigilancia se complementan con estudios epidemiológicos
adicionales para determinar en forma más precisa las causas, la historia natural, los
factores de predisposición y los modos de transmisión relacionados con el problema de
salud.
Por ejemplo, cuando los datos de vigilancia son mapeados a un determinado nivel, como
puede ser a nivel de municipio, se pueden detectar una serie de características
geográficas sobre las que pueden sustentarse distintas hipótesis en cuanto a etiología y
riesgo. El reconocimiento de tales conglomerados puede conducir a una investigación
epidemiológica adicional; en ocasiones las personas identificadas mediante la vigilancia
se emplean como sujetos de estudios epidemiológicos.
Para la vigilancia de enfermedades en erradicación, como el sarampión, la carencia de
toma de muestra en casos sospechosos se considera una falla del sistema de vigilancia.
El Laboratorio juega un papel esencial en prácticamente todos los procesos relacionados
con la Vigilancia Epidemiológica, actuando como un principalísimo brazo ejecutor de la
misma:

90 | P á g i n a
Agrega evidencia para la sospecha de casos
Confirma o descarta casos sospechosos
Colabora en la definición y caracterización de brotes y epidemias
Desarrolla o participa en la investigación epidemiológica
Integra sistemas centinelas
Desarrolla la vigilancia de agentes etiológicos y de la resistencia a antimicrobianos
Participa en la vigilancia de enfermedades transmitidas por alimentos, ocupacionales o
crónicas
Participa en el control de vectores, control de alimentos, vigilancia de epizootias, etc.
Realiza el monitoreo de riesgo del ambiente
En todo este conjunto de actividades participan laboratorios con distintas funciones y
diferentes niveles de complejidad. Estos deben estar funcionalmente integrados en una
red con alcance nacional, con el fin de desarrollar adecuadamente las actividades de
Vigilancia bajo la dirección de un laboratorio central de referencia (Instituto Nacional de
Salud)
6.6 RECOMENDACIONES
Uno de los grades inconveniente que a menudo se presenta en la implementación de los
sistemas de vigilancia es la derivación de la información, el análisis y devolución de los
resultados. En tal sentido lo que proponemos es que cada nivel pueda analizar la
información correspondiente al mismo, para ello el nivel local no solo debe recolectar la
información sino que tiene que disponer de herramientas que le permitan efectuar una
tabulación y análisis de la misma a fin de seleccionar prioridades y cumplir con el
verdadero postulado de la vigilancia “INFORMACIÓN PARA LA ACCIÓN”.
La emergencia de nuevos desafíos profesionales para la acción en Salud Pública,
resultante de la mayor complejidad actual de los sistemas y las organizaciones, expande
el espectro de competencias requeridas para un funcionamiento adecuado de los
sistemas de salud, dando lugar a una fuente de trabajo más y más diversificada. Esto
implica nuevos aprendizajes y el desarrollo de capacidades apropiadas en los diferentes
niveles del sistema de salud.

91 | P á g i n a
BIBLIOGRAFÍA
1. SINAVE/Ministerio de Salud y Acción Social – Argentina. “Manual de Normas y
Procedimientos del Sistema Nacional de Vigilancia Epidemiológica” Revisión Internacional
2000.
2. Resolución 1481 de 2013. Por el cual se adopta el Plan Decenal de Salud Pública 2012-
2021, Disponible en
http://www.minsalud.gov.co/Documentos%20y%20Publicaciones/Plan%20Decenal%20-
%20Documento%20en%20consulta%20para%20aprobaci%C3%B3n.pdf.
Consultado el 08 de Agosto de 2014.
3. Decreto 3518 de 2006, disponible en http://www.ins.gov.co/lineas-de-accion/Subdireccion-
Vigilancia/sivigila/Documentos%20SIVIGILA/Decreto%203518%2006%20Crea%20y%20regla
menta%20el%20SIVIGILA.pdf. Consultado el 08 de Agosto de 2014.
4. Vigilancia de la salud en la Argentina. Rol del laboratorio clinico 2006, Disponible en
http://www.fbioyf.unr.edu.ar/evirtual/pluginfile.php/2804/mod_resource/content/0/13_Vigilancia
_de_la_Salud_en_la_Argentina_protegido.pdf. Consultado el 08 de Agosto de 2014.
5. El Laboratorio en la Vigilancia Epidemiológica, Perspectivas y situación actual en Uruguay
2004, Disponible en http://www.bvsops.org.uy/pdf/epidemiol.pdf. Consultado el 08 de Agosto
de 2014.
6. OPS/DPC/CD (Unidad de Enfermedades Transmisibles) – Washington. “Lineamientos para la
elaboración de estrategias y acciones para el fortalecimiento de la capacidad nacional para la
prevención y el control de epidemias”. Documento de trabajo, marzo 2004


















