
INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA - Ferran...
116
MONOGRAFÍAS DR. ANTONIO ESTEVE GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA J.E. Baños • C. Brotons • M. Farré 23 3
Transcript of INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA - Ferran...

puesta
MONOGRAFÍAS DR. ANTONIO ESTEVE
GLOSARIO DEINVESTIGACIÓN
CLÍNICA YEPIDEMIOLÓGICAJ.E. Baños • C. Brotons • M. Farré
23
23
GLO
SARIO
DE
INVES
TIG
ACIÓ
N C
LÍN
ICA
Y EP
IDEM
IOLÓ
GIC
AJ.E
. Bañ
os •
C. B
roto
ns •
M. F
arré

Hace unos días, la señora que limpia nuestroCentro nos sorprendió doblemente. Mujer ro-tunda en el hablar, nos dijo: “No me han deja-do Vds. el timing colgado en la cartelera”. Sihaber olvidado este detalle de organización in-terna ya era causa de sorpresa, no lo era me-nos el uso de este huésped lingüístico super-fluo en boca de una señora más castiza quelas Yemas de Santa Teresa. ¿Por qué tenía ellaesta palabra en la boca?Puede resultar extraño lo que vamos a afirmar:a veces desconfiar, es un talento. Para un ex-tranjero es tan espontáneo decir vinió comopara un camarada español acercarle un vino.Usar palabras poco frecuentes en cualquiercontexto requiere un mínimo de suspicacia -desarrollada como un hábito- para evitar echarmano de palabras en un uso que, a más degratuito, sea erróneo. Cuando nominan a al-guien para un Óscar, ¿a quién le han cambia-do el nombre? ¿Quién se llama desde ahora“Para un Óscar”?, porque “nominar” es “po-ner nombre, denominar”, salvo que lo que sequería decir era “proponer” a Fulanito para re-cibir el premio. El idioma es una víscera que a veces nos aler-ta y no la oímos por falta de entrenamiento pa-ra saber desde dónde echa su solitaria botellaal mar. Esta espontaneidad sospechable flota -y lamentablemente tiñe- diversos ámbitos dela lengua que tiene verdadero beneficio pre-servar. Uno de los trastornos del lenguaje dehoy es el supuesto prestigio que parece otor-gar el uso vacilante de palabras extranjeras enun aparente cientificismo a todo trance. No setrata aquí de arremeter contra un enemigosiempre cambiante y multiforme que adopta laforma de los tan extendidos a nivel de, contac-tar, en base a o incluso jugar un papel. No esuna cruzada contra el inglés ni contra ningúnneologismo de cualquier origen si aporta unanueva y saludable certitud con su uso, ni nosanima un sentido heroico de blandir una espa-da –dicho “blandir” del germánico brand, es-pada, y no del blandire latino, adular- con no
sé qué fantasmas nacionales, sino bien pues-tos en la nueva lógica del provecho, con elidioma como uno de sus instrumentos. Lejísi-mos de nosotros el arresto de hacer un llama-miento de otra especie que no sea eminente-mente práctico. Los ámbitos terminológicosentre hispanohablantes se debaten entre unacierta esperanza y nada a cambio. Por eso,muchos términos en lenguas extranjeras –fun-damentalmente el inglés, lingua franca- vienena llenar este vacío por un mecanismo sencillo:porque no es necesario un acuerdo entre losdos hablantes en su idioma original común, si-no que al disponer de la palabra precisa en in-glés, cada uno recurre a la suya propia al ha-cer la traducción. Y la palabra común en cas-tellano, sigue brillando por su ausencia.Las lenguas minoritarias -a veces habladas porminorías de 300 o 1000 millones –, es decir ypara entendernos, lenguas que no son el in-glés, tienen varias tareas pendientes. Una deellas es disponer de un glosario terminológicoespecífico para cada ámbito, empeño quepuede reportarle pingües ganancias científi-cas, industriales y comerciales si se evita quepatrimonio nuestro pase a terceros. Sin duda, una de las áreas huérfanas de refe-rencias terminológicas es la investigación clíni-ca y epidemiológica, lo cual es especialmentegrave cuando con ellas pretendemos conocermás o mejor alguna población sana o enferma.Para conseguir estos objetivos, necesitamosuna serie de métodos, herramientas y termino-logía que nos permitan no solamente hacer si-no también compartir el resultado de nuestrotrabajo, aprender del de los demás, contrastary discutir; avanzar, en suma. En la medida enque este esfuerzo colectivo por ser menos ig-norantes se hace operativo y se alcanzan todoslos planos de la investigación científica, necesi-tamos consensuar cómo debemos proceder ycómo divulgarlo. Incluso diríamos que es másimportante convenir cómo lo decimos: pode-mos innovar, disgregar o transgredir cualquiermétodo de investigación, siempre y cuando lo
Prólogo
7

expliquemos convenientemente, para permitirsu corroboración o refutación. Pero si no so-mos capaces de describir razonablemente lasecuencia de nuestro proceder o cualquier ma-tiz de nuestro trabajo, estamos dando pábulo atodo tipo de elucubraciones o manipulacionesacerca de lo que hemos hecho.El lenguaje científico, por lo tanto, es una pie-za fundamental y no un mero accesorio parael trabajo científico: permite decir las cosas talcomo las observamos (ser objetivos), para en-tendernos (ser inteligibles) y para expresar só-lo lo suficiente (ser sintéticos). Si ello es así,¿cómo nos explicamos el caótico mundo determinología científica, de acrónimos y deabreviaturas en que nos movemos? ¿Cómo en-tender el poco aprecio por el consenso termi-nológico que podemos observar en nuestrasrevistas y libros de texto, conferencias y semi-narios? Por ejemplo, cuando alguien consultaMedline acerca de todos los ensayos clínicosque contiene sobre un determinado tema, subúsqueda tiene una precisión menor del 35%, independientemente de su experiencia do-cumental. Quiere decir que alguien (los auto-res del estudio, los miembros de los respecti-vos comités editoriales de las revistas, los do-cumentalistas, o todos a la vez), han llamadoensayo clínico controlado a muchos estudiosque no asignaron al azar los pacientes inclui-dos con el fin de comparar dos o más inter-venciones, cuando en cambio no se le dio estaconsideración a estudios que en realidad sí lohicieron y por tanto, lo eran. Si nos sucede es-to ante un tema con el grado de popularidadque tiene el ensayo clínico, ¿qué podemos es-perar cuando manejamos términos que repre-sentan significados tan complejos como la vali-dez o el sesgo protopático?Los investigadores, seres brillantes por defini-ción, pueden tener la tentación de inventarcontinuamente términos nuevos. Incluso algu-nos se han dedicado durante un tiempo a con-quistar términos, especialmente sesgos, quehan quedado bautizados con el nombre propiodel investigador correspondiente, como si deuna vieja colonia se tratara. Pero el sentido co-
mún, la plétora de información científica quenos invade, la necesidad de ser selectivos yeficientes en nuestras actividades de comuni-cación científica, entre otras razones, aconsejaque nos dotemos de algunas herramientas deapoyo, como por ejemplo, glosarios de termi-nología científica como el que tenemos ennuestras manos.Sus autores conocen bien las necesidades quetratan de solventar, siquiera parcial o tempo-ralmente. Han lidiado muchas corridas en dis-tintos hospitales, facultades, centros y comitésde investigación donde el uso del lenguajecientífico ha sido confuso, deficiente o contra-dictorio. Por ello, es encomiable su esfuerzode ampliar y adaptar al castellano obras de re-ferencia que ya existían en otras lenguas paraproducir un glosario que por sí mismo va a serya un referente desde ahora. Hubiera sido ide-al que este trabajo fuera el producto de unconsenso más amplio entre sociedades cientí-ficas, organismos universitarios, etc. peromientras tanto, bienvenido sea para llenar losvacíos existentes. Además, este glosario puedeservir de base, de punto de partida, para lamovilización necesaria de las personas e insti-tuciones que tienen algo que decir al respectoen España e Iberoamérica. En la medida enque se consiga que este proceso sea transpa-rente, reproducible y riguroso (es decir, condetalle explícito de los métodos de búsqueda yconsulta de obras ya existentes, criterios de in-clusión y exclusión de términos y vocablos,mecanismos para resolver las contradicciones,etc.) estaremos aplicando el método científicopara la imprescindible puesta a punto denuestro vehículo colectivo de comunicación eninvestigación clínica y epidemiológica. El repa-so no sólo va a darle brillo y esplendor, sinoque va a permitirnos continuar en mejorescondiciones las futuras batallas para la pene-tración y el derribo de las antiguas y renovadasmurallas de ignorancia e incomunicación.
Xavier Bonfill* y Claudio Etcheverry***Director del Centro Cochrane Español
**Traductor del Centro Cochrane Español
8
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Siendo o habiendo sido miembros de Comitésde Investigación y de Comités Éticos de Inves-tigación Clínica percibimos la necesidad hacetres años de elaborar un glosario de amplio es-pectro- desde la farmacología, a la estadísticapasando por la ética y la epidemiología- no só-lo por la gran variabilidad de proyectos a revi-sar que llegan a estos comités, sino tambiénpara disponer de un ‘diccionario de metodolo-gía de la investigación’ encima de la mesa,por la diferente procedencia de los miembrosque constituyen estos comités lo que daba lu-gar a interpretaciones diversas de un mismoconcepto.La elaboración de este glosario ha sido un tra-bajo arduo en la búsqueda y recopilación, pe-ro muy enriquecedor por las discusiones quemanteníamos los autores y que surgían en ca-da definición. Obviamente no pretendíamoshacer un libro original, ya que existen otrosglosarios publicados, y que consultamos repe-tidamente. Pero sí que estamos satisfechos,con toda modestia, de varios aspectos que ha-cen que esta obra sea, a nuestro entender,singular. Se trata de un amplio glosario dondehay contabilizadas más de 700 entradas, te-niendo en cuenta tanto las propias definicio-nes, las referidas a otras definiciones y las si-glas y acrónimos. Muchas definiciones son ori-ginales (resultantes de un consenso entre losautores), otras son adaptadas de otros textos,y otras son la transcripción literal del Real De-creto 561/1993 o del Diccionario de la LenguaEspañola (aunque además casi siempre se ad-junta una definición propia o adaptada en és-tas); ofrece una traducción al inglés de cadauno de los términos y un índice inglés-castella-
no para facilitar la búsqueda, aspecto éste ne-cesario por el aluvión cada vez mayor de litera-tura anglosajona; ofrece una lista de 85 siglaso acrónimos utilizados frecuentemente en la li-teratura de investigación biomédica y que tam-bién se han incluido en las definiciones; sehan aportado figuras, tablas, y múltiples ejem-plos, fórmulas y representaciones numéricaspara facilitar la comprensión de algunos térmi-nos complejos; y finalmente, se han anexadoel Real Decreto 561/1993 (ensayos clínicoscon medicamentos) y la última versión de laDeclaración de Helsinki. Todos estos aspectos cumplen el objetivo quenos habíamos marcado. Creemos que el pre-sente glosario puede ser útil no sólo para loscomités, sino para cualquier profesional de lasalud que trabaje en el ámbito universitario,hospitalario o de la atención primaria, mínima-mente interesado en la investigación, e inclusopara estudiantes de pre y posgrado de cien-cias médicas.Estamos seguros de haber olvidado conceptosque debieran estar presentes en un glosariode estas características. Probablemente unamayor iconografía habría sido deseable. Y so-mos conscientes de que existen definicionesque se prestan a discusión e incluso a crítica.Todas estas limitaciones las asumimos desdeel primer día que empezamos a preparar esteglosario. En cualquier caso, es mejor la críticaque debatirse en el vacío. Las críticas, cuandojustas, nos ayudaran a mejorar la próxima edi-ción y, por ello, no podemos más que solicitar-las y agradecerlas.
Los autores
Presentación
9

AABAcrónimo de Aumento absoluto del benefi-cio.
AARAcrónimo de Aumento absoluto del riesgo.
Abandono (Drop-out)Sujeto o paciente que no es capaz de conti-nuar, o no quiere hacerlo, con las visitas deseguimiento de un estudio y que decide vo-luntariamente no seguir en la investigación.También se aplica a los pacientes que no fi-nalizan un estudio por razones no relaciona-das claramente con éste (por ejemplo, revo-cación del consentimiento, cambio de domi-cilio). Véase Retirada.
Acontecimiento adverso (Adverse event, AE)Cualquier experiencia no deseable que ocu-rra a un sujeto durante un ensayo clínico, seconsidere o no relacionada con los productosen investigación (RD 561/1993 art. 19.1).Este término se utiliza durante la fase de in-vestigación de un fármaco, mientras quecuando se ha comercializado se emplea re-acción adversa. Véase Reacción adversa.
Acontecimiento adverso grave (Serious adverseevent)
Aquel que produce la muerte, amenaza la vi-da, produce incapacidad permanente o dalugar a hospitalización o prolongación de lamisma. Además, se considerarán siempregraves las anomalías congénitas y los proce-sos malignos (RD 561/1993 art. 19.1).
Acontecimiento adverso inesperado (Unexpec-ted adverse event)
Experiencia no descrita (en naturaleza, gra-vedad o frecuencia) en el manual del investi-gador (RD 561/1993, Art. 19.1).
AdherenciaVéase Cumplimiento.
Agencia de investigación por contratoVéase Organización de investigación por con-trato.
Agencia Española del MedicamentoOrganismo público adscrito al Ministerio deSanidad y Consumo creado por la Ley
66/1997 de 30 de Diciembre (BOE 31-12-1997). En el momento de cerrar la redacciónde este glosario está pendiente su organiza-ción y puesta en funcionamiento (junio de1998). Sus funciones según la mencionadaLey son: a). conceder la autorización de comercializa-ción de especialidades farmacéuticas y otrosmedicamentos de uso humano, así como larevisión y adecuación de medicamentos yacomercializados;b). participar en la planificación y evaluaciónde medicamentos de uso humano que seautoricen en la Unión Europea a través de laAgencia Europea de Evaluación de Medica-mentos;c). evaluar y autorizar los ensayos clínicos ylos productos en fase de investigación clíni-ca;d). autorizar los laboratorios farmacéuticosde medicamentos de uso humano;e). planificar, evaluar y desarrollar el sistemaespañol de farmacovigilancia;f). desarrollar la actividad inspectora y decontrol de medicamentos de competenciaestatal;g). gestión de la Real Farmacopea Española;h). instrucción de procedimientos derivadosde infracciones cuando corresponda al esta-do;i). competencias relativas a estupefacientes ypsicotropos;j). otras que le sean atribuidas por normaslegales o reglamentarias.
Agencia Europea de Evaluación de Medicamen-tos (European Agency for the Evaluation ofMedicinal Products, EMEA)
Agencia de la Unión Europea encargada dela coordinación de la evaluación científica yde la calidad, seguridad y eficacia de los me-dicamentos cuya comercialización esté suje-ta a procedimientos comunitarios de autori-zación. Esta agencia es la encargada de eva-luar los medicamentos que siguen eldenominado procedimiento centralizado; su
Glosario
11

autorización será válida para toda Europa,tramitándose en cada país únicamente elprecio y las condiciones de financiación.El procedimiento centralizado de autoriza-ción es obligatorio para los productos obteni-dos por procedimientos biotecnológicos in-cluidos en la lista A del reglamento 2309/93de la UE (ADN recombinante, expresióncontrolada de genes, hibridomas y anticuer-pos monoclonales). También pueden incluir-se otros tipos de fármacos a criterio de laAgencia.La Agencia cuenta con grupos de trabajo(eficacia, calidad, humano, seguridad, far-macovigilancia y biotecnología) y de expertosde los estados miembros que refuerzan alComité de Especialidades Farmacéuticas(Committee for Proprietary Medicinal Pro-ducts, CPMP) y al Comité de Productos Vete-rinarios (Comittee for Proprietary VeterinaryProducts, CPVP). Se encuentra localizada enLondres.
Ajuste (Standardization)Proceso estadístico utilizado para obteneruna ponderación de tasas específicas por ca-tegorías (por ejemplo, edad o sexo). Existendos métodos de ajuste: ajuste directo e indi-recto. Algunos autores utilizan el término deestandarización. Véanse Ajuste directo yAjuste indirecto.
Ajuste directo (Direct standardization)Proceso estadístico derivado de la aplicaciónde las tasas específicas por categorías (porejemplo, edad o sexo) de una población deestudio a una población estándar, obtenien-do una única tasa sumatoria que refleja elnúmero de acontecimientos que se esperarí-an si la población de estudio tuviera la mis-ma distribución de la característica de inte-rés que la población estándar. Por ejemplo,la tasa de mortalidad por cáncer ajustada poredad en España en 1990 se calcularía apli-cando las tasas específicas de cáncer poredad observadas en 1990 en España a unapoblación estándar como pudiera ser la po-blación mundial; así se obtendría una tasasumatoria que representaría una tasa hipoté-tica que hubiera sido observada en la pobla-ción española de 1990 si ésta tuviera la mis-ma distribución por edad que la poblaciónmundial.
Ajuste indirecto (Indirect standardization)Proceso estadístico derivado de la aplica-ción de las tasas específicas por categorías(por ejemplo, edad o sexo) de una pobla-ción estándar a una población de estudio.
Este tipo de ajuste se utiliza habitualmentecuando el número de individuos observa-dos con la enfermedad en la población deestudio es pequeño y por azar las tasaspueden estar sujetas a oscilaciones impor-tantes. Por ejemplo, en una población detrabajadores expuestos al asbesto, se en-cuentra que a los 20 años de seguimientoel número de muertes observadas por cán-cer de pulmón es de 60. Para calcular elnúmero de muertes por cáncer de pulmónque se esperarían en esta población en elsupuesto de que se hubieran muerto porcáncer a unas tasas iguales a las de unapoblación estándar, se aplicarían las tasasespecíficas por cáncer de pulmón de la po-blación estándar a la población de estudio(trabajadores expuestos al asbesto), obte-niéndose los casos esperados en esa pobla-ción. Éstos se suman y se calcula el índicede mortalidad estandarizado (IME) o razónde mortalidad estandarizada, dividiendo elnúmero de casos observados por el númerode casos esperados (IME = observados/es-perados), y se multiplica por 100. Por ejem-plo, un IME de 140 significaría que estegrupo de trabajadores del asbesto tiene unriesgo de mortalidad por cáncer un 40%superior al de los hombres de esa mismaedad en la población general.
Aleatorio (Random)Característica de una secuencia de observa-ciones, actividades o asignaciones que es elresultado de un proceso regido por el azar,en el que la probabilidad de una secuenciadeterminada es conocida o puede ser cono-cida. Algunos autores también le denominanEstocástico. Véanse Número aleatorio y Va-riable aleatoria.
AleatorizaciónVéase Asignación aleatoria.
Alfa de Cronbach (Cronbach’s alpha)Estadístico que cuantifica la consistencia in-terna y mide la intercorrelación entre un nú-mero diferente de preguntas o ítems que su-puestamente reflejan el mismo concepto.Puede tomar los valores de -1 a +1. VéaseConsistencia interna.
Análisis bayesianoVéase Análisis de Bayes.
Análisis de Bayes (Bayesian analysis)Análisis que provee una distribución de pro-babilidad posterior de algún parámetro quees función de los datos observados y de unadistribución de probabilidad anterior. VéaseTeorema de Bayes.
12
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Análisis de coste-beneficio (Cost-benefit analy-sis)
Evaluación de los resultados del beneficio deuna intervención terapéutica, expresado entérminos monetarios, comparado con loscostes propios de la intervención. El resulta-do se expresa como la razón entre el costede la intervención y el coste del beneficio ob-tenido, y debe medirse con las mismas uni-dades monetarias (por ejemplo, pesetas).También se le conoce como análisis de be-neficio en función del coste. Véase Razón decoste-beneficio.
Análisis de coste-efectividad (Cost-effective-ness analysis)
Evaluación de los resultados obtenidos entérminos de incremento del beneficio tera-péutico que se deriva de los costes extraor-dinarios. Este análisis valora si los benefi-cios aportados compensan el coste añadi-do. El resultado se expresa como la razónentre el coste y la efectividad, midiéndoselos costes en unidades monetarias y los be-neficios en términos de unidades de efecti-vidad, como años de vida ganados. Tam-bién se le conoce como análisis de efectivi-dad en función del coste. Véase Razón decoste-efectividad.
Análisis de coste-utilidad (Cost-utility analysis)Variante del análisis de coste-efectividad enel que la medida de la efectividad se expresaen forma de año de vida ajustado por calidad(AVAC) u otras. También se le conoce comoanálisis de la utilidad en función del coste.Véase Análisis de coste-efectividad.
Análisis de la covarianza (Analysis of covarian-ce, ANCOVA)
Prueba estadística que consiste en la combi-nación de un análisis de regresión y un ANO-VA. Compara una variable dependiente con-tinua con variables independientes que pue-den ser categóricas o continuas. Porejemplo, se puede utilizar este tipo de análi-sis para determinar la relación entre las con-centraciones de hemoglobina (variable de-pendiente) y la ingesta de hierro controladapor el sexo (variables independientes).
Análisis de la varianza (Analysis of variance,ANOVA)
Prueba estadística paramétrica que permitela comparación de una variable cuantitativa(variable dependiente) en más de dos gruposmuestrales. Por ejemplo, permite compararla media de presión arterial sistólica en cincogrupos de pacientes sometidos a tratamien-tos distintos.
Análisis de la varianza multivariado (Multivaria-te analysis of variance, MANOVA)
Prueba estadística paramétrica que permitecomparar más de una variable cuantitativa(variable dependiente) en dos o más gruposmuestrales. Por ejemplo, permite compararla media de la presión arterial sistólica y lamedia del peso en cinco grupos de pacientessometidos a diferentes tratamientos.
Análisis de minimización de costes (Cost mini-misation analysis)
Evaluación económica en la que se suponeque no existen diferencias entre los efectosde las opciones comparadas; es, por tanto,suficiente comparar sus costes para elegir lamás barata.
Análisis de regresión (Regression analysis)Prueba estadística que permite correlacionaruna variable cuantitativa (variable depen-diente) con una o más variables cuantitativas(variables independientes). Permite estudiarla variación de una variable (variable depen-diente) según diferentes valores de otra va-riable (variable independiente). Por ejemplo,permitiría relacionar los cambios de la pre-sión arterial sistólica con las variaciones defrecuencia cardíaca o de peso corporal. Vé-anse Regresión lineal, Regresión logística yRegresión múltiple.
Análisis de sensibilidad (Sensitivity analysis)Procedimiento en el que los resultados de unestudio son recalculados utilizando valoresalternativos para algunas de las variables delestudio, con objeto de ver si se alteran lasconclusiones del mismo.
Análisis de series temporales (seculares)(Analysis of temporal trends, Analysis of secu-lar trends)
Análisis que examina los acontecimientos clí-nicos en el tiempo o entre diferentes áreasgeográficas y los correlaciona con las ten-dencias en la exposición. Las unidades deobservación son habitualmente poblaciones.
Análisis de subgrupos (Subgroup analysis)Análisis practicado en una subpoblación de-terminada de los pacientes participantes enun estudio. Es lícito realizarlo cuando se ha-yan expuesto explícitamente unos criteriosconcretos basados en unas hipótesis plante-adas previamente en el diseño, cuando lamagnitud del efecto observado en la pobla-ción total sea grande (>25%) y el efecto deltratamiento sea altamente significativo, ycuando los resultados sean consistentes conlos de otros estudios o coherentes con la evi-dencia biológica.
13
GLOSARIO

Análisis de supervivencia (Survival analysis)Análisis basado en la cuantificación del nú-mero de acontecimientos observados (muer-te u otros) y el momento en el tiempo en queéstos ocurren en relación a un momento ce-ro. En los ensayos clínicos, el tiempo de unacontecimiento para un individuo se midedesde el momento de su inclusión en el es-tudio. Los efectos del tratamiento se determi-nan comparando las tasas de los aconteci-mientos en los diferentes grupos interveni-dos.
Análisis explicativo (Explanatory analysis)Análisis de los resultados de un ensayo clíni-co según el tratamiento que hayan recibidofinalmente los pacientes independientemen-te del grupo al que habían sido asignadosaleatoriamente. Véase Ensayo clínico explica-tivo.
Análisis intermedio (Interim analysis)Análisis practicado en el curso de un estudiocon el objetivo de monitorizar y evaluar losefectos del tratamiento en un momento de-terminado del mismo. Su realización debeestablecerse previamente en el diseño delestudio. También se le conoce por análisisprovisional.
Análisis por intención de tratar (Intention totreat analysis)
Análisis de los resultados de todos los pa-cientes incluidos en el estudio, manteniendointacta la asignación aleatoria. De esta mane-ra se evita el sesgo que se produce al excluirdel análisis a todos aquellos pacientes quehan tenido un seguimiento incompleto, o elcausado por el cambio del grupo asignadoinicialmente. Véase Ensayo clínico pragmáti-co.
Análisis provisionalVéase Análisis intermedio.
Análisis secuencial (Sequential analysis)Análisis de los resultados después de la in-clusión de un paciente, una pareja o un gru-po de pacientes en el estudio. Permite deter-minar si la diferencia acumulada entre un ti-po de tratamiento y otro se mantiene dentrode unos límites aceptables. Si no se excedenéstos, se puede seguir incluyendo pacientesy, en caso contrario, se puede dar por finali-zado el estudio. Véase Diseño secuencial.
Analogía (Analogy)Relación de semejanza entre dos o más co-sas. Véase Razonamiento por analogía e In-ferencia causal.
ANCOVAAcrónimo de Análisis de la covarianza.
ANOVAAcrónimo de Análisis de la varianza.
Año/s de vida ajustado/s por calidad (AVAC)(Quality-Adjusted Life Year/s, QALY/s)
Años de supervivencia que aporta una deter-minada intervención ponderados en funciónde una escala de calidad de vida. VéaseAnálisis de coste-utilidad.
ApareamientoVéase Emparejamiento.
ApuntamientoVéase Curtosis
Área bajo la curva de características funciona-les (Area under the ROC curve)
Probabilidad de clasificar correctamente pa-res de individuos sanos y enfermos, selec-cionados de la población al azar, mediantelos resultados obtenidos al aplicarles laprueba diagnóstica. Por ejemplo, en el casodel diagnóstico de cáncer de próstata me-diante la utilización del antígeno próstaticoespecífico (PSA), una área de 0,75 significaque un individuo seleccionado aleatoriamen-te del grupo de enfermos tendrá el 75% delas veces un valor de PSA mayor que un in-dividuo elegido al azar del grupo sano. Pue-de tomar los valores de 0,5 a 1. Cuando noexisten diferencias en la distribución de re-sultados de la prueba entre los subgruposenfermo y sano, toma el valor de 0,5, mien-tras que cuando existe una separación per-fecta entre las dos distribuciones toma el va-lor de 1. Valores entre 0,5 y 0,7 indican unaprueba diagnóstica de baja exactitud; entre0,7 y 0,9 pueden ser útiles para algunospropósitos; y un valor de 0,9 o superior indi-ca exactitud alta (Fig. 1). Véase Curva de ca-racterísticas funcionales.
Asignación (Assignment)Proceso que permite la distribución de los in-dividuos en cada uno de los grupos de estu-dio.
Asignación aleatoria (Randomized assignment,Randomization)
Técnica que consiste en distribuir cada suje-to a uno de los grupos de estudio siguiendoun método de azar que asegure que cadauno tenga exactamente las mismas probabili-dades de formar parte de uno u otro grupo.Véanse Aleatorio, Asignación aleatoria estrati-ficada, Asignación aleatoria por bloques yAsignación aleatoria simple.
Asignación aleatoria estratificada (Stratifiedrandomization)
Clasificación de los sujetos, previa a la asig-nación, en categorías o estratos según deter-
14
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

minadas características o criterios pronósti-cos. A continuación se procede a su asigna-ción de forma independiente en cada cate-goría mediante un proceso de aleatorizaciónpropio. De esta manera se consigue que losgrupos contengan aproximadamente el mis-mo número de sujetos en cada categoría oestrato.
Asignación aleatoria por bloques (Blocking,blocked or block randomization)
Distribución de los sujetos a cada uno de losgrupos en forma de bloques, teniendo cadabloque un número fijo o variable de sujetos,de tal manera que el número total de partici-pantes en el estudio debe correspondersesiempre con un múltiplo del bloque o biencon la suma de ellos si son de tamaño varia-ble. Este tipo de asignación se utiliza paragarantizar la homogeneidad entre los grupos.
Asignación aleatoria simple (Simple randomi-zation)
Método elemental de aleatorización que sepuede conseguir tirando una moneda a carao cruz, utilizando una tabla de números alea-
torios, o bien, actualmente, mediante progra-mas informáticos. Cualquiera que sea el pro-cedimiento, la probabilidad de ser asignadoa un grupo u otro está determinada de ante-mano y permanece constante a lo largo delestudio.
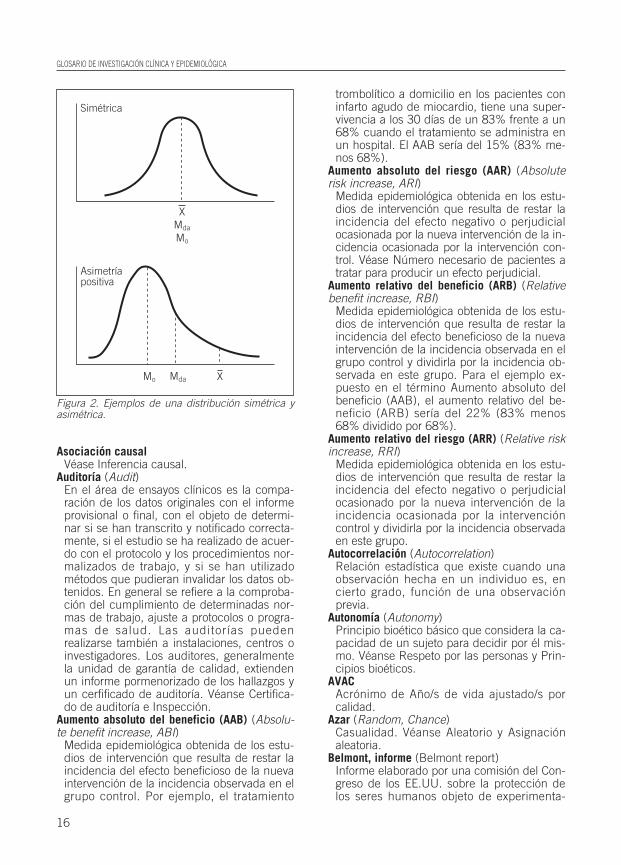
Asimetría (Skewness)Distribución en la que los valores de media(X–), mediana (Mda) y moda (Mo) no coinci-
den y en la que una de las colas es más lar-ga que la otra (Fig. 2).
Asistente de investigación clínica (Clinical Re-search Assistant, CRA)
Profesional que, bajo las órdenes del moni-tor, colabora en diferentes aspectos del dise-ño, búsqueda de investigadores, inicio, mo-nitorización, cierre y evaluación de los ensa-yos clínicos. Sus responsabilidades varíandependiendo de cada compañía farmacéuti-ca.
Asociación (Association)Grado en que dos acontecimientos se rela-cionan con más frecuencia de la que cabeesperar por el azar.
15
GLOSARIO
1,00
0,80
0,60
0,40
0,20
0,000,00 0,20 0,40 0,60 0,80 1,00
Curva ROC
Prueba inexacta
Proporción de falsos positivos(1–especificidad)
Pro
porc
ión
de v
erda
dero
s po
sitiv
os(S
ensi
bilid
ad)
Figura 1. Área bajo la curva de características funcionales.
Asociación causalVéase Inferencia causal.
Auditoría (Audit)En el área de ensayos clínicos es la compa-ración de los datos originales con el informeprovisional o final, con el objeto de determi-nar si se han transcrito y notificado correcta-mente, si el estudio se ha realizado de acuer-do con el protocolo y los procedimientos nor-malizados de trabajo, y si se han utilizadométodos que pudieran invalidar los datos ob-tenidos. En general se refiere a la comproba-ción del cumplimiento de determinadas nor-mas de trabajo, ajuste a protocolos o progra-mas de salud. Las auditorías puedenrealizarse también a instalaciones, centros oinvestigadores. Los auditores, generalmentela unidad de garantía de calidad, extiendenun informe pormenorizado de los hallazgos yun cerfificado de auditoría. Véanse Certifica-do de auditoría e Inspección.
Aumento absoluto del beneficio (AAB) (Absolu-te benefit increase, ABI)
Medida epidemiológica obtenida de los estu-dios de intervención que resulta de restar laincidencia del efecto beneficioso de la nuevaintervención de la incidencia observada en elgrupo control. Por ejemplo, el tratamiento
trombolítico a domicilio en los pacientes coninfarto agudo de miocardio, tiene una super-vivencia a los 30 días de un 83% frente a un68% cuando el tratamiento se administra enun hospital. El AAB sería del 15% (83% me-nos 68%).
Aumento absoluto del riesgo (AAR) (Absoluterisk increase, ARI)
Medida epidemiológica obtenida en los estu-dios de intervención que resulta de restar laincidencia del efecto negativo o perjudicialocasionada por la nueva intervención de la in-cidencia ocasionada por la intervención con-trol. Véase Número necesario de pacientes atratar para producir un efecto perjudicial.
Aumento relativo del beneficio (ARB) (Relativebenefit increase, RBI)
Medida epidemiológica obtenida de los estu-dios de intervención que resulta de restar laincidencia del efecto beneficioso de la nuevaintervención de la incidencia observada en elgrupo control y dividirla por la incidencia ob-servada en este grupo. Para el ejemplo ex-puesto en el término Aumento absoluto delbeneficio (AAB), el aumento relativo del be-neficio (ARB) sería del 22% (83% menos68% dividido por 68%).
Aumento relativo del riesgo (ARR) (Relative riskincrease, RRI)
Medida epidemiológica obtenida en los estu-dios de intervención que resulta de restar laincidencia del efecto negativo o perjudicialocasionado por la nueva intervención de laincidencia ocasionada por la intervencióncontrol y dividirla por la incidencia observadaen este grupo.
Autocorrelación (Autocorrelation)Relación estadística que existe cuando unaobservación hecha en un individuo es, encierto grado, función de una observaciónprevia.
Autonomía (Autonomy)Principio bioético básico que considera la ca-pacidad de un sujeto para decidir por él mis-mo. Véanse Respeto por las personas y Prin-cipios bioéticos.
AVACAcrónimo de Año/s de vida ajustado/s porcalidad.
Azar (Random, Chance)Casualidad. Véanse Aleatorio y Asignaciónaleatoria.
Belmont, informe (Belmont report)Informe elaborado por una comisión del Con-greso de los EE.UU. sobre la protección delos seres humanos objeto de experimenta-
16
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA
Simétrica
XMda
Mo
Asimetríapositiva
MdaMo X
Figura 2. Ejemplos de una distribución simétrica yasimétrica.

ción biomédica y conductual (The NationalCommission for the Protection of humansubjects of biomedical and behavioral rese-arch. The Belmont report, Ethical principlesand guidelines for the protection of humansubjects of research). Se enuncian en esteinforme tres principios básicos de la bioética:respeto por las personas, beneficencia y jus-ticia. Véanse Autonomía, Beneficencia, Justi-cia, No maleficencia, Principios bioéticos yRespeto por las personas.
Beneficencia (Beneficence)Principio bioético según el cual se debe pro-curar favorecer a los sujetos de la investiga-ción, no exponiéndolos a posibles daños, yque la relación beneficio/riesgo, derivada desu participación en el ensayo, sea favorable.Ello implica maximizar el beneficio y minimi-zar el riesgo. Véase Principios bioéticos.
Biodisponibilidad (Bioavailability)Velocidad y magnitud con la que un principioactivo es absorbido a partir de una formula-ción farmacéutica y se hace disponible en ellugar de acción. En la práctica se refiere a lafracción de la dosis administrada que alcan-za la circulación sistémica y a la velocidadcon que lo hace. Por lo general, los estudiosde biodisponibilidad se llevan a cabo en vo-luntarios sanos y las concentraciones del fár-maco se determinan en la sangre (plasma osuero). Véanse Biodisponibilidad absoluta yBiodisponibilidad relativa.
Biodisponiblidad absoluta (Absolute bioavaila-bility)
Comparación de las concentraciones de unfármaco obtenidas por vía intravenosa conlas de otra vía de administración, por ejem-plo la oral. Véase Biodisponibilidad.
Biodisponibilidad relativa (Relative bioavailabi-lity)
Comparación de las concentraciones de unfármaco obtenidas tras su administración pordos vías distintas, ninguna de las cuales es laintravenosa. También se emplea para com-parar dos formulaciones galénicas distintasadministradas por la misma vía, por ejemplo,comprimidos orales y solución oral. VéaseBiodisponibilidad.
Bioequivalencia (Bioequivalence)Comparación entre la biodisponibilidad demedicamentos genéricos y de nuevas mar-cas de fármacos ya comercializados con labiodisponibilidad de la especialidad farma-céutica considerada como referencia. Vé-anse Biodisponibilidad y Medicamento ge-nérico.
Bioética (Biomedical ethics)Parte de la ética que trata de la moral y delas obligaciones de los hombres en el campode la medicina, la investigación clínica y lainvestigación médico-biológica. Véase Princi-pios bioéticos.
BloquesVéase Asignación aleatoria por bloques.
Bonferroni, corrección de, método deVéase Comparaciones estadísticas múltiples.
BPCAcrónimo de Buena Práctica Clínica.
BPFAcrónimo de Buena Práctica de Fabricación.
BPLAcrónimo de Buena Práctica de Laboratorio.
BPMAcrónimo de Buena Práctica de Manufactu-ra.
Buena Práctica ClínicaVéase Normas de buena práctica clínica.
Buena Práctica de FabricaciónVéase Normas de buena práctica de fabrica-ción.
Buena Práctica de LaboratorioVéase Normas de buena práctica de labora-torio.
Buena Práctica de ManufacturaVéase Normas de buena práctica de manu-factura.
Búsqueda de casos (Case-finding)Cribado que realiza de forma activa el médi-co aprovechando las consultas por cualquierotro motivo. En este tipo de cribado -a dife-rencia del cribado masivo- el médico tiene laresponsabilidad de hacer un seguimiento delos resultados positivos. También se le deno-mina búsqueda o localización activa de ca-sos. Véase Cribado.
Calibración (Calibration)Según el Diccionario de la Lengua Española,calibrar es Establecer, con la mayor exactitudposible, la correspondencia entre las indica-ciones de un instrumento de medida y losvalores de magnitud que se mide con él.También puede definirse como comparaciónde los resultados de una nueva técnica conlos obtenidos con un método exacto de refe-rencia.
CalidadVéanse Control de calidad y Garantía de cali-dad.
Calidad de vida (Quality of life)Grado de la interacción del individuo con sumedio en sus diferentes facetas social, física,emocional e intelectual. También se define
17
GLOSARIO

como grado de apreciación totalmente subje-tivo que engloba el sentido general de satis-facción del individuo y la percepción de supropia salud. En esta definición se incluyenfactores como las circunstancias familiares,la economía y la satisfacción en el trabajo.
Calidad de vida relacionada con la salud (He-alth related quality of life)
Aspectos de la vida dominados o influidosúnicamente por la salud personal (salud físi-ca o mental y estado de bienestar). En estadefinición no se tienen en cuenta aspectosde la vida como, por ejemplo, dónde se viveo cómo se vive.
CANDAAcrónimo de Computer Assisted New DrugApplication. Véase Solicitud de autorizaciónde especialidad farmacéutica.
Características basales (Baseline characteris-tics)
Conjunto de variables de un sujeto recogidasantes de la asignación aleatoria y del iniciode la intervención. Véase Variable basal.
Casos y controlesVéase Estudio de casos y controles.
Causalidad (Causality, Causation)Relación de las causas con los efectos queproducen. Véase Inferencia causal.
CECAcrónimo de Comité de Ensayos Clínicos
CEICAcrónimo de Comité Ético de InvestigaciónClínica
Certificado de auditoría (Audit certificate, auditstatement)
Documento que cerfifica que se ha realizadouna auditoría. Puede ser a las instalacionesde un centro, a un investigador, a una orga-nización o a un informe. Véase Auditoría.
Ciego (Blind or blinded, mask or masked)Condición impuesta en un individuo o grupode individuos con el propósito de que no co-nozcan o aprendan algún hecho u observa-ción, como puede ser la asignación del trata-miento. También se utiliza la palabra enmas-caramiento. Véanse Evaluación ciega porterceros, Simple ciego, Doble ciego y Tripleciego.
CocienteVéase Razón.
Cociente de probabilidades (Likelihood ratio)Razón utilizada en pruebas diagnósticascuando los resultados se pueden expresar enmás de dos categorías (como por ejemplo lasconcentraciones plasmáticas de CK para eldiagnóstico de infarto agudo de miocardio).
Resulta de dividir la sensibilidad (a / a + c)por 1 - especificidad (b / b + d) para cadauna de las categorías. Véanse Sensibilidad yEspecificidad.
Código de aleatorización (Randomizationcode)
Documento en el que se especifican los có-digos correspondientes a cada tratamiento osujeto participante en un estudio. Número,cifra, letra o combinación que identifica a unpaciente y el tratamiento que le correspondepor asignación aleatoria. En estudios a dobleciego se guardan en un sobre cerrado, yasea de forma global para todos los partici-pantes, o mejor en sobres individuales paracada paciente. En general existen varias co-pias, al menos una en manos del investiga-dor o de la farmacia que dispensa los fárma-cos, y otra custodiada por el promotor. Encaso de necesidad, como pudiera ser la pre-sencia de un acontecimiento adverso grave,el sobre puede ser abierto.
Código deontológicoVéase Deontología médica, código.
Coeficiente de correlaciónVéase Correlación.
Coeficiente de variación (CV) (Coefficient of va-riation, CV)
Medida utilizada para describir la variaciónen una población que permite comparar ladispersión relativa de los datos. Es la desvia-ción estándar (DE), expresada como porcen-taje de la media (%): CV = (DE/X
–) ( 100.
Coherencia (Coherence)Interpretación de una asociación causal queno es discordante con lo ya conocido de lahistoria natural y biológica de la enfermedad.Véase Inferencia causal.
Colaboración Cochrane (Cochrane Collabora-tion)
Grupo de colaboración internacional creadoen 1992 que tiene como objetivos preparar,mantener y difundir revisiones sistemáticas yactualizadas de ensayos clínicos controladossobre la atención sanitaria. Está formado porgrupos colaboradores de revisión, constitui-dos por individuos que comparten un interésespecífico, que analizan críticamente la evi-dencia existente, y coordinados a su vez porun equipo editorial, que es responsable deensamblar y editar un módulo con las revi-siones preparadas para difundirlo a través deuna base de datos. El nombre de la colabo-ración está dedicado a Archie Cochrane, per-sona interesada en conocer la eficacia realde una gran parte de las prácticas e inter-
18
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

venciones que constituyen la actividad de lossistemas sanitarios. Autor del libro Effective-ness and efficiency: random reflections onhealth services. En España existe un centrode la Colaboración Cochrane.
Comisión Nacional de FarmacovigilanciaÓrgano consultivo del Ministerio de Sanidady Consumo en materia de efectos adversos otóxicos de los medicamentos (OM 25 de ju-nio de 1989).
Comité de Ensayos Clínicos (CEC)Terminología en desuso, ya que fue la anti-gua denominación de los comités que en ca-da centro evaluaban los protocolos de los en-sayos clínicos según la OM del 3 de agostode 1982, que desarrollaba el RD 944/1978.Desde la publicación del RD 581/1993 sehan sustituido por el Comité Ético de Investi-gación Clínica (CEIC). Véase Comité Ético deInvestigación Clínica.
Comité de Especialidades Farmacéuticas (Com-mittee for Proprietary Medicinal Products,CPMP)
Organismo de la Unión Europea encargadode la regulación y control de los medicamen-tos. Véase Agencia Europea de Evaluaciónde Medicamentos.
Comité de Ética (Ethics Committee, Indepen-dent Ethics Committee, Ethical Committee)
Comité encargado de la evaluación de los as-pectos éticos de la investigación con huma-nos. En algunos países los comités de éticatienen una doble vertiente, de manera queevalúan aspectos asistenciales y de investi-gación. En otros se han separado los aspec-tos asistenciales para constituirse en ComitésÉticos de Investigación (Research EthicsCommittee o simplemente Ethics Commit-tee), que se encargan de la revisión de losprotocolos de investigación en humanos, in-cluyendo los ensayos clínicos. En los EE.UU.los protocolos de investigación en humanosdeben ser autorizados por un Consejo Insti-tucional de Revisión (Institutional Review Bo-ard, IRB), que está constituido en la mayoríade hospitales y centros de investigación. Ve-lan específicamente por los derechos y la sa-lud de los individuos, los métodos utilizadospara obtener el consentimiento informado,los riesgos y los potenciales beneficios de lainvestigación. En España estas funciones lasrealizan los Comités Éticos de InvestigaciónClínica (CEIC). También se les denomina Co-mités Éticos Independientes (IndependentEthics Committee). Véase Comité Etico de In-vestigación Clínica.
Comité de Ética Asistencial (Ethics Committee,Ethical Committee)
Comité encargado de revisar los aspectos éti-cos de la asistencia a los pacientes en uncentro hospitalario. En algunas autonomías,un miembro del Comité de Ética Asistenciales miembro obligatorio del Comité Ético deInvestigación Clínica.
Comité de Expertos (Steering Committee)Grupo de especialistas en un área que revisanla marcha de un ensayo clínico. En ocasionesconfirman los diagnósticos, revisan los datosde seguridad y, en caso de que existan análi-sis intermedios, determinan si el estudio debecompletarse o terminar prematuramente.
Comité de Investigación (Research Committee)Comité encargado de evaluar los proyectosde investigación en todos sus aspectos. Enocasiones los CEIC asumen sus funciones,pero la participación de éstos es obligatoriaen los ensayos clínicos con medicamentos yproductos sanitarios. Véanse Comité de Éti-ca, Comité Ético de Investigación Clínica yConsejo Institucional de Revisión.
Comité ÉticoVéase Comité de Ética
Comité Ético de Investigación Clínica (CEIC)Comité encargado de evaluar la idoneidad delos protocolos de ensayos clínicos con medi-camentos y del equipo investigador propues-to, así como de la información escrita que sedará a los posibles sujetos de la investiga-ción, la previsión de compensación en casode lesiones y el tipo de consentimiento queotorgan los participantes. Aunque su crea-ción fue motivada para la supervisión de losensayos clínicos con medicamentos, en algu-nos centros desempeñan las funciones delComité de Investigación del centro. Además,debe realizar el seguimiento del ensayo clíni-co desde su inicio a su finalización. Su com-posición y funciones están reguladas en el tí-tulo III del RD 561/1993. Los CEIC asumenalgunas de las funciones que en algunos paí-ses realizan los Comités de Etica, los ComitésËticos de Investigación o los Consejos Institu-cionales de Revisión. Véanse Comité de Éticay Consejo Institucional de Revisión.
Comparaciones estadísticas múltiples (Multiplecomparison test)
Cuando en una investigación existen tres omás grupos de estudio, es obligatoria unacomparación global utilizando un análisis dela varianza. Si éste resulta significativo, pue-den aplicarse comparaciones entre los distin-tos pares de grupos para encontrar las posi-
19
GLOSARIO

bles diferencias entre ellos. Se recomienda elajuste del valor de p dependiendo del númerode comparaciones realizadas. Por ejemplo, sicomparamos tres grupos (A, B y C), las posi-bles comparaciones serán de A frente a B, deA frente a C y de B frente a C. Se recomiendautilizar algún método para ajustar el valor dep a las tres comparaciones realizadas, porejemplo, utilizando el de Bonferroni. Se divideel valor habitual de p (0,05) por 3 compara-ciones, considerándose entonces significati-vas las diferencias entre pares si p<0,0167.Otros métodos menos conservadores para va-riables paramétricas son la prueba de Tukeyo la de Scheffé que utilizan los valores de lavarianza residual para calcular la diferenciamínima que resultará significativa.
Concordancia (Agreement)Grado de correspondencia entre dos méto-dos u observadores que miden una mismavariable o bien el grado de correspondenciaal medir repetidamente una misma variablecon un mismo método o un mismo observa-dor. Véase Índice de Kappa.
Consejo (Counselling)Intervención consistente en la provisión deinformación y recomendaciones individualesque se relacionan con las conductas perso-nales que pueden reducir el riesgo de enfer-medad. Ejemplos de consejo son el dietéticoo el antitabaco.
Consejo Institucional de Revisión (CIR) (Institu-tional Review Board, IRB)
Comité que revisa y aprueba estudios que serealizan en humanos en las instituciones delos EE.UU. La revisión se focaliza en los as-pectos éticos de la investigación propuesta yen la adecuación del procedimiento para ob-tener el consentimiento informado de los pa-cientes. En España, algunas de estas funcio-nes las ejercen los CEIC. En otros países sedenominan Comités de Ética (Ethical Com-mittees). Véanse Comité de Ética y ComitéÉtico de Investigación Clínica.
Consentimiento informado (Informed consent)Procedimiento formal solicitado a todos lossujetos que intervienen en una investigaciónclínica que debe reunir cuatro característi-cas: informado, comprendido, legalmentecompetente y voluntario. La información de-be darse de tal manera que pueda ser com-prendida por el sujeto de la investigación. Lacompetencia legal plantea la necesidad deobtener el consentimiento a través de un re-presentante legal en los casos de sujetos me-nores de edad e incapaces. La voluntariedad
debe ser en ausencia de control externo (sincoerción, manipulación ni persuasión) y au-téntica (respetando el principio de autono-mía). Según el RD 561/1993 sobre ensayosclínicos: El sujeto expresará su consenti-miento preferiblemente por escrito o, en sudefecto, de forma oral ante testigos indepen-dientes del equipo investigador que lo decla-rarán por escrito bajo su responsabilidad. Enlos casos de sujetos menores de edad e in-capaces, el consentimiento lo otorgará siem-pre por escrito su representante legal. Cuan-do el menor tenga doce o más años, deberáprestar además su consentimiento para par-ticipar en el ensayo, después de haberle da-do toda la información pertinente adaptada asu nivel de entendimiento. El consentimientodel representante legal y del menor, en sucaso, será puesto en conocimiento del Minis-terio Fiscal, previamente a la realización delensayo. En casos excepcionales puede in-cluirse a pacientes en un estudio sin su con-sentimiento, para solicitárselo en cuanto seaposible (consentimiento informado diferido).El RD 561/1993 especifica el formato quedebe reunir la hoja de información del sujeto,el consentimiento por escrito, el consenti-miento oral ante testigo y el consentimientodel representante. Véase Consentimiento in-formado diferido.
Consentimiento informado diferido (Deferredconsent)
Consentimiento que se otorga una vez ya seha incluido al sujeto en el estudio. Se con-templa en situaciones excepcionales, de talurgencia que no es posible disponer del con-sentimiento del propio sujeto ni de su repre-sentante legal. En estos casos la situación de-be estar prevista en el protocolo del ensayoclínico, que será aprobado por el Comité Eticode Investigación Clínica y únicamente proce-derá cuando el estudio tenga un interés tera-péutico particular para el sujeto. El investiga-dor debe informar al Comité Ético de Investi-gación Clínica y al promotor de las razonesque han dado lugar a la situación. El sujeto osu representante legal deberán ser informa-dos en cuanto sea posible y otorgarán su con-sentimiento para continuar en el ensayo si asílo desean (RD 561/1993, art. 12.6).
Consentimiento informado por representanteVéase Consentimiento informado.
Consistencia (Consistency)Observación repetida de una asociación endistintas poblaciones en diferentes circuns-tancias. Véase Inferencia causal.
20
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Consistencia interna (Internal consistency)Propiedad de un cuestionario que valorahasta qué punto cada ítem o cada seccióndel mismo está midiendo la misma cosa. Vé-ase Alfa de Cronbach.
ControlVéase Grupo control.
Control concurrente (Concurrent control)Grupo control basado en datos registradosdurante el mismo período de tiempo que elresto de individuos participantes en el estu-dio.
Control de calidad (Quality assessment, Qualitycontrol)
Grupo de técnicas y actividades realizadasdentro del sistema de garantía de calidad,encaminadas a verificar, generalmente en losestudios de medicamentos, si se han cumpli-do los requisitos de calidad de las activida-des relacionadas con el ensayo (normas deBuena Práctica Clínica, Buena Práctica deLaboratorio y Buena Práctica de Fabrica-ción). Véase Garantía de calidad.
Control histórico (Historical control)Grupo control basado en datos registradosanteriormente a los del estudio actual, y conla finalidad de comparar los datos presentescon los resultados obtenidos entonces. Parala utilización de un grupo control histórico serequiere que exista un buen registro de lostratamientos utilizados anteriormente y desus resultados. Véase Estudio de controleshistóricos.
Correlación (Correlation)Grado de relación entre dos variables. Lamedida utilizada es el coeficiente de correla-ción (r) que cuantifica la relación lineal entrela exposición y la enfermedad (Fig. 3).
Correlación de Pearson (Pearson’s correlation)Medida de asociación que expresa el gradode relación lineal entre dos variables conti-
nuas que siguen una distribución normal yque toma valores entre -1 y +1. Por tanto, 0sería la ausencia total de relación, +1 cuandohay una relación lineal creciente perfecta, y -1 cuando hay una relación lineal decrecienteperfecta.
Correlación de Spearman (Spearman’s correla-tion)
Medida de asociación que expresa el gradode concordancia entre dos variables que nosiguen una distribución normal, comparán-dose los rangos de las mismas. También to-ma valores entre -1 y +1.
Correlación intraclase (Intraclass correlation)Grado de asociación y de acuerdo entre di-ferentes observadores o métodos que midenvariables continuas. Puede tomar los valoresde 0 a 1. Representa la proporción de la va-rianza total (varianza de valores reales másvarianza de los errores de medida) explicadapor la varianza intersujetos de los valoresverdaderos. Se considera que el valor míni-mo de la correlación que indica acuerdo es0,75.
Muymala Muyo nula Mediocre Moderada Buena buena
Valor de r <0,3 0,31-0,5 0,51-0,7 0,71-0,9 ≥0,91
CPMPAcrónimo de Committee for Proprietary Me-dicinal Products. Véase Comité de Especiali-dades Farmacéuticas.
CRAAcrónimo de Clinical Research Assistant. Vé-ase Asistente de investigación clínica.
CRDAcrónimo de Cuaderno de recogida de datos.
CRFAcrónimo de Case report form. Véase Cua-derno de recogida de datos.
21
GLOSARIO
(r cercana a 1)
(a)
Asociación muy positiva
X
Y
(r cercana a 0)
(c)
No asociación
X
Y
(r cercana a –1)
(b)
Asociación muy negativa
X
Y
Figura 3. Ejemplos de correlación.

Cribado (Screening)Identificación de una enfermedad en su esta-dio presintomático mediante la aplicación deuna prueba diagnóstica. Cuando el cribado seaplica a grandes poblaciones no selecciona-das, el proceso se llama cribado masivo o po-blacional (mass screening). En estos casos elcribado no pretende ser diagnóstico, ya quelas personas con un resultado positivo o pato-lógico se deben derivar a su médico para laconfirmación. Un ejemplo de cribado masivosería la toma de la presión arterial o la realiza-ción de una mamografía a las mujeres mayo-res de 50 años en una determinada población.Cuando el cribado se aplica a los pacientesque acuden a la consulta se habla de búsque-da de casos. Véase Búsqueda de casos.
Criterio estándarVéase Patrón de oro.
Criterio de referenciaVéase Patrón de oro.
CROAcrónimo de Contract Research Organiza-tion. Véase Organización de investigación porcontrato.
CruzadoVéase Diseño cruzado.
CTXAcrónimo de Clinical Trial Exemption Certifi-cate. Véase Producto en fase de investiga-ción clínica.
Cuaderno de recogida de datos (CRD) (Case re-port form, CRF)
Formulario para recoger los datos concretosdel estudio y que refleja la logística y el dise-ño del mismo, de acuerdo con lo especifica-do en el protocolo. Puede presentarse en di-ferentes formatos (papel, óptico, electrónico).
Cuadrado latino (Latin square)Diseño utilizado para aquellos ensayos clíni-cos cruzados en los que se utilizan más dedos fármacos, usándose las letras del alfabetoorientadas en sentidos horizontal y vertical,cada una de ellas correspondiendo a una in-tervención. Este tipo de diseño asegura unbuen equilibrio para controlar algunos facto-res pronósticos, individuales o temporales.Un posible ejemplo es un ensayo clínico cru-zado en el que cada paciente debe recibircuatro fármacos distintos (A, B, C, D). Algu-nas de las posibles secuencias a las que de-berían asignarse cuatro pacientes (un pa-ciente por secuencia) se muestran en el si-guiente cuadrado latino 4 x 4. Es importantemencionar que cada paciente recibe los cua-tro tratamientos en distintas secuencias, y
que durante cada período hay un pacienteque está recibiendo cada uno de los posiblestratamientos. Véanse Diseño balanceado yDiseño cruzado.
Ejemplo de cuadrado latino 4 × 4 balancea-do en el que cada fármaco sólo precede o si-gue a otro en una ocasión (secuencia)
Período 1 Período 2 Período 3 Período 4Secuencia 1 A B C DSecuencia 2 B D A CSecuencia 3 C A D BSecuencia 4 D C B A
Cuartil (Quartile)Valores (tres) que dividen la distribución nor-mal en cuatro partes de igual tamaño. El se-gundo cuartil se corresponde con la media-na.
Cuestionario (Questionnaire)Documento estructurado de recogida de da-tos que se utiliza como instrumento paraevaluar el estado de salud, diagnosticar o re-alizar el seguimiento de una situación clínicaconcreta.
Cumplimiento (Compliance, adherence)Grado en el que un paciente sigue el trata-miento prescrito, ya sea farmacológico o no.Se han descrito métodos directos (determi-nación de un fármaco, de sus metabolitos omarcadores) e indirectos (evaluación del cur-so clínico, recuento de comprimidos, entre-vista estructurada) para determinarlo. Tam-bién se utiliza el término de observancia yadherencia. Véase Observancia.
Curriculum vitae (CV) (Curriculum vitae, CV)Según el Diccionario de la Lengua Españolaes la relación de los títulos, honores, cargos,trabajos realizados, datos biográficos, etc.,que califican a una persona. Es obligatoria lapresencia de una copia del CV (completo oreducido) de los investigadores en el archivodel investigador y en el del promotor.
Curtosis (Kurtosis)Grado de estrechez de una distribución uni-modal. Una distribución relativamente planay con colas cortas es de curtosis baja, y unadistribución picuda con largas colas es decurtosis alta. La distribución normal se utilizacomo referente y se considera de curtosis in-termedia. También se la denomina apunta-miento (Fig. 4).
Curva de características funcionales (ReceiverOperating Characteristic Curve, ROC)
Gráfico que muestra todos los pares de sen-sibilidad/especificidad resultantes de la varia-
22
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

ción continua de los puntos de corte en todoel rango de resultados observados. Tambiénse la denomina curva ROC. Véase Área bajola curva de características funcionales.
DatoVéase Variable.
Datos, entrada de (Data entry)Proceso por el que los datos primarios de unestudio se introducen en un programa de or-denador para su análisis estadístico. Se reco-mienda que la entrada se realice por dupli-cado, por dos personas distintas y compro-bando después las diferencias.
Datos, entrada a distancia de (Remote dataentry)
Proceso por el que los datos primarios delestudio se introducen en un ordenador a dis-tancia del central y deben transmitirse hastael mismo. Por ejemplo, en algunos estudioslos investigadores entran los datos en progra-mas específicos de ordenador que despuésenvían al promotor mediante sistemas elec-trónicos de transmisión (modem o correoelectrónico).
DCIAcrónimo de Denominación común interna-cional.
Decil (Decile)Valores (nueve) que dividen la distribuciónnormal en diez partes del mismo número deelementos. El quinto decil se correspondecon la mediana.
Denominación común internacional (DCI) (Inter-national Nonproprietary Name, INN)
Nombre de un fármaco aprobado por la Or-ganización Mundial de la Salud a propuestade su inventor. Se basa en una serie de re-glas con el fin de encontrar raíces o termina-ciones similares para los fármacos de unmismo grupo terapéutico. También se le lla-ma nombre o denominación genérica. En
nuestro país existe la llamada denominaciónoficial española (DOE) que, propuesta por elMinisterio de Sanidad y Consumo, es la ver-sión en castellano de la DCI. Es obligatorio suuso junto a la marca comercial en las espe-cialidades farmacéuticas. Los medicamentosgenéricos se identifican por la denominacióncomún internacional. Véase Medicamentogenérico.
Denominación oficial española (DOE)Véanse Denominación común internacional yMedicamento genérico.
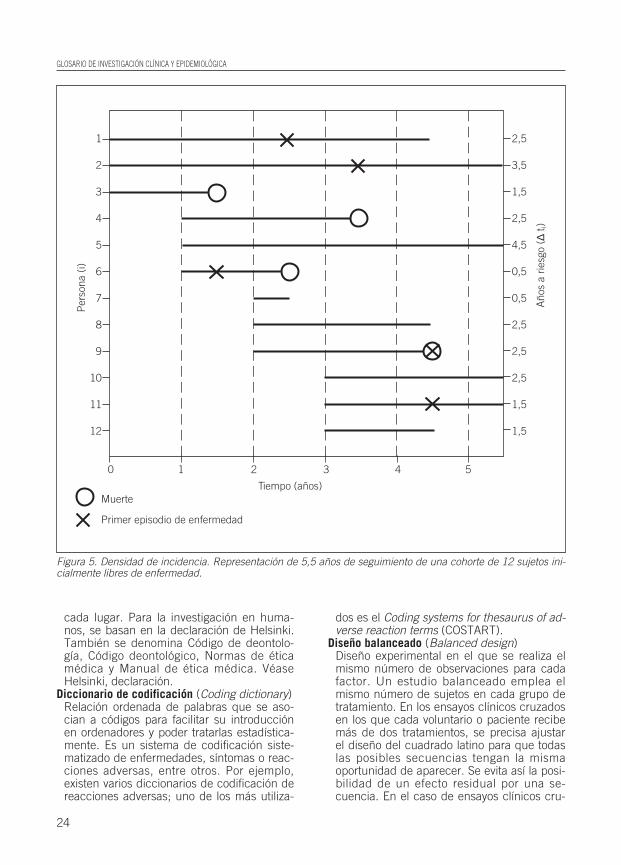
Densidad de incidencia (DI) (Incidence density)Tipo de incidencia en el que se restringe elcálculo a un período de tiempo durante elcual el total de la población provee informa-ción. Ésta se considera una medida de tasainstantánea de desarrollo de una enferme-dad en una población y se define como:
Nuevos casos de una enfermedad durante un período de tiempo
DI = total personas-tiempo de observación
El denominador es la suma del tiempo de ca-da individuo a riesgo o la suma del tiempoque cada persona permanece bajo observa-ción y libre de enfermedad (Fig. 5).
Desigualdad relativaVéase Razón de posibilidades.
Desviación estándar (DE) (Standard deviation,SD)
Parámetro estadístico de dispersión que re-presenta como promedio cuánto los valoresde un individuo se desvían de la media. Seutiliza desviación típica como sinónimo. Véa-se Varianza.Se calcula mediante la fórmula siguiente:
n∑i=1 (xi - x-)2
DE = √ S = √n-1
donde: xi = cada valorx- = median = número de individuos (grados libertad)S = varianza
Desviación típicaVéase Desviación estándar.
Deontología médica, código de (Ethics manual)Relación de principios básicos de actuaciónen la práctica médica. Incluye, entre otros,capítulos referentes a la relación médico-en-fermo, la reproducción humana, la muerte yla experimentación médica sobre las perso-nas. Generalmente son normas aprobadaspor los respectivos colegios de médicos de
23
GLOSARIO
A
B
C
Figura 4. Ejemplos de distribuciones con distintascurtosis. (A: exceso de curtosis; B: normal; C: faltade curtosis).
cada lugar. Para la investigación en huma-nos, se basan en la declaración de Helsinki.También se denomina Código de deontolo-gía, Código deontológico, Normas de éticamédica y Manual de ética médica. VéaseHelsinki, declaración.
Diccionario de codificación (Coding dictionary)Relación ordenada de palabras que se aso-cian a códigos para facilitar su introducciónen ordenadores y poder tratarlas estadística-mente. Es un sistema de codificación siste-matizado de enfermedades, síntomas o reac-ciones adversas, entre otros. Por ejemplo,existen varios diccionarios de codificación dereacciones adversas; uno de los más utiliza-
dos es el Coding systems for thesaurus of ad-verse reaction terms (COSTART).
Diseño balanceado (Balanced design)Diseño experimental en el que se realiza elmismo número de observaciones para cadafactor. Un estudio balanceado emplea elmismo número de sujetos en cada grupo detratamiento. En los ensayos clínicos cruzadosen los que cada voluntario o paciente recibemás de dos tratamientos, se precisa ajustarel diseño del cuadrado latino para que todaslas posibles secuencias tengan la mismaoportunidad de aparecer. Se evita así la posi-bilidad de un efecto residual por una se-cuencia. En el caso de ensayos clínicos cru-
24
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA
2,5
3,5
1,5
2,5
4,5
0,5
0,5
2,5
2,5
2,5
1,5
1,5
1
2
3
4
5
6
7
8
9
10
11
12
0 1 2 3 4 5
Tiempo (años)
Per
sona
(i)
Año
s a
riesg
o (∆
t i)
Muerte
Primer episodio de enfermedad
Figura 5. Densidad de incidencia. Representación de 5,5 años de seguimiento de una cohorte de 12 sujetos ini-cialmente libres de enfermedad.

zados con más grupos de tratamiento queperíodos, para conseguir que al final exista elmismo número de sujetos tratados en cadagrupo deberán balancearse los bloques deasignación aleatoria (diseño de bloques in-completos y balanceados).En el siguiente cuadrado latino no balancea-do para las secuencias, la secuencia A de-lante de B se repite en varias ocasiones,mientras que B nunca precede a A. Si existeun efecto residual de A, siempre nos podráafectar la respuesta de B.
A B C DB C A DC D A BD A B C
En el siguiente cuadrado latino balanceadopara las secuencias, cada fármaco precedeuna sola vez a otro. Existen las dos posiblessecuencias: A precede a B y B precede a A.Se controlan por tanto los posibles efectosresiduales.
A B C DB D A CC A D BD C B A
Véase Cuadrado latino.
Diseño cruzado (Cross-over design)Tipo de ensayo clínico aleatorizado en el quelos sujetos reciben dos o más tratamientosen períodos sucesivos que han sido determi-nados al azar, lo que permite que cada suje-to sea su propio control. Al reducir la variabi-
lidad, estos ensayos son más eficientes y supotencia estadística es mayor. Para evitarque los efectos del primer tratamiento de lasecuencia se manifiesten en el segundo perí-odo, se suele incluir entre los tratamientosperíodos de lavado para evitar los efectos re-siduales. Además, debe tenerse en cuenta elposible efecto del tiempo, ya que su dura-ción es mayor. En general, este tipo de dise-ño sólo puede utilizarse en enfermedades osíntomas estables, ya que se deben adminis-trar varios tratamientos de forma consecuti-va. Véanse Cuadrado latino y Período de la-vado.
Pacientes1, 3, 5, … (n/2) A Período B
de 2, 4, 6, … (n/2) B lavado A
↑ Aleatorización
Diseño factorial (Factorial design)Tipo de ensayo clínico aleatorizado en elque el tratamiento de estudio se utiliza encombinación con al menos otro tratamientode estudio, o en el que múltiples dosis deun determinado tratamiento se están utili-zando en el mismo ensayo. Este tipo de di-seño se utiliza para contrastar hipótesis di-ferentes en un mismo ensayo, o para eva-luar la interacción entre di ferentesfármacos. Un ejemplo de un diseño factorialsería la utilización del ácido acetilsalicílico(AAS) para la prevención cardiovascular y ladel beta-caroteno para la prevención delcáncer (Fig. 6).
25
GLOSARIO
22.071 aleatorizados
11.037 AAS 11.034 placebo AAS
5.517 Beta-caroteno 5.520 Placebobeta-caroteno
5.520 Beta-caroteno 5.514 Placebobeta-caroteno
Figura 6. Diseño factorial (AAS= ácido acetilsalicílico).

Diseño paralelo (Parallel design)Tipo de ensayo clínico aleatorizado en el seasigna a unos pacientes a recibir el trata-miento control, mientras que a otros pacien-tes se les asigna el tratamiento experimental.Así, cada paciente sólo recibe uno de los tra-tamientos de estudio. Es el diseño más utili-zado para evaluar la eficacia de los medica-mentos. Pueden existir estudios de dos, tres,cuatro o más grupos paralelos en caso decomparar dos, tres, cuatro o más fármacos odosis de los mismos.
Pacientes1, 5, 9, … (n/3) A2, 4, 7, … (n/3) B3, 6, 8, … (n/3) C
↑ Aleatorización
Diseño Play-the-winner (“regla de jugar al ga-nador”, Play-the-winner design or trial)
Estudio en el que se sigue una asignaciónadaptativa de los participantes. Así, los resul-tados observados en un paciente determinanel tratamiento que recibirá el siguiente. Por
26
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA
35
30
25
20
15
10
5
0
0 5 10 15 20 25 30 35
A mejor
No diferencia
B mejor
Figura 7. Diseño secuencial. Gráfico estadístico para evaluar la respuesta. Se compara el efecto del producto Acon el del producto B. Si en la primera pareja de pacientes A resulta mejor que B, se traza una cruz en el cuadrosuperior inmediato al negro de la gráfica. Si B resulta mejor que A se traza una cruz en el cuadro contiguo a laderecha del negro. Si no existen diferencias entre ambos productos no se registra nada el las gráficas. Se van re-clutando parejas de pacientes y apuntando los resultados hasta que las cruces toquen el límite de las tres áreas(mejor; B mejor, No diferencia). En este momento termina el estudio.
ejemplo, si el primer paciente recibe el fárma-co A y es eficaz, el siguiente recibirá A. Cuan-do el tratamiento A resulte ineficaz se asignaráal siguiente paciente al fármaco B y viceversa.Es un tipo de diseño muy raramente utilizado.
Número de Tratamientopaciente asignado Resultado
1 A Fracaso2 B Fracaso3 A Éxito4 A Éxito5 A Éxito6 A Fracaso7 B Fracaso8 A Éxito
Diseño secuencial (Sequential design)Tipo de ensayo clínico en el que los indivi-duos del grupo experimental y del grupocontrol se disponen por pares o bloques -unos recibiendo el tratamiento a experimen-tar y otros recibiendo el tratamiento control- yla decisión de incorporar nuevos pacientesviene determinada por el hecho de que la di-ferencia acumulada entre los dos tratamien-tos esté dentro de unos límites específicos.En caso de excederse estos límites, se da porfinalizado el ensayo. Este tipo de diseño sereserva para aquellos ensayos en los que laevaluación de los resultados se puede hacercon cierta rapidez, y cuando los períodos deseguimiento no son excesivamente largos. Eltamaño de la muestra dependerá de las dife-rencias que se van hallando a lo largo del es-tudio (Fig. 7). Véase Análisis secuencial.
Distribución (Distribution)Comportamiento teórico que describe ade-cuadamente cómo se distribuyen las varia-bles.
Distribución binomial (Binomial distribution)Distribución de una variable dicotómica querefleja la probabilidad de que ocurra un nú-mero de sucesos determinados. Por ejemplo,la probabilidad de remisión de la enferme-dad cancerígena después del tratamientoagudo en un grupo de pacientes con cáncer.
Distribución de Bernouilli (Bernouilli distribu-tion)
Distribución de variables con dos resultadosposibles, con probabilidades que pueden serdesiguales. Por ejemplo, la probabilidad deéxito o fracaso de una intervención quirúrgi-ca es diferente según se trate de una apendi-citis o de una operación de derivación aorto-coronaria. Cuando se repite N veces el expe-rimento o intervención, se habla de unadistribución binomial.
Distribución de frecuencias (Frequency distri-bution)
Distribución que refleja cómo se reparten lossujetos de una muestra según una determi-nada característica, y nos da la frecuencia desu aparición. También se denomina distribu-ción de probabilidad o diagrama de frecuen-cias. Gráficamente, en el eje de las abscisasse representan los valores de la variable y enel de las ordenadas su frecuencia relativa(Fig. 8).
Distribución de GaussVéase Distribución normal.
27
GLOSARIO
30
20
10
0Teatro
Por
cent
aje
de a
lum
nos
Bailes Cines Deportes Toros Cafés Otros
Figura 8. Distribución de frecuencias de las preferencias de un grupo de alumnos por diferentes actividades lúdicas.

Distribución de χ2 (Chi-square distribution)Distribución definida únicamente sobre losvalores reales positivos, con un único pará-metro al que se denomina grados de liber-tad. También se le denomina distribución deji al cuadrado, es un anglicismo llamarla dis-tribución de chi-cuadrado.
Distribución de Poisson (Poisson distribution)Distribución que refleja las situaciones en lasque la probabilidad de aparición de un suce-so sea muy pequeña. Por ejemplo, la proba-bilidad de aparición de partos triples o cuá-druples.
Distribución normal (Normal distribution)Distribución cuyas medidas se agrupan alre-dedor de un valor central y que presentanuna frecuencia cada vez menor a medidaque se alejan de dicho valor medio. Muchasvariables biológicas siguen la distribuciónnormal. Este patrón tiene una distribuciónunimodal, de forma acampanada, en la queambas ramas son simétricas. La media po-blacional ±1,96 desviaciones estándar inclu-ye al 95% de la población. También se la co-noce como distribución de Gauss.
Doble ciego (Double blind or blinded, doublemask or masked)
Procedimiento de enmascaramiento, emple-ado habitualmente en los ensayos clínicos,en el que se utilizan unos códigos de tal ma-nera que ni los pacientes ni el investigador opersonal clínico en contacto con el pacienteconocen la asignación a los grupos de trata-miento. Para el caso de medicamentos se re-quiere que su forma de presentación (tama-ño, color) sea idéntica. Véase Ciego.
Doble engañoVéase Doble simulaciónDoble simulación (Double dummy)
Técnica de enmascaramiento que permiteutilizar la formulación farmacéutica del fár-maco de referencia, incluso si la forma depresentación y/o la vía de administración esdiferente. Para ello es necesario disponer delos placebos correspondientes de ambospreparados. Por ejemplo, si se compara unfármaco en forma de comprimidos con unfármaco de referencia en inyectable, seríanecesario disponer de placebo correspon-diente a los comprimidos y a los inyectables.Cada sujeto recibe ambas formulaciones, enunos casos la forma activa y en otros el pla-cebo.Pacientesn/2: Fármaco activo A ●● Placebo B ■n/2: Placebo A ●● Fármaco activo B ■
DOEAcrónimo de denominación oficial española.Véanse Denominación común internacional yMedicamento genérico.
Dosis diaria definida (DDD) (Defined daily dose,DDD)
Estimación de la dosis media de manteni-miento de un fármaco para su indicaciónprincipal. No debe confundirse con la dosisdiaria recomendada por el fabricante, laprescrita por el médico o la ingerida por elpaciente. En ocasiones la DDD es similar a larecomendada en una enfermedad, pero mu-chas veces está alejada de la misma. Se utili-za como unidad de comparación para estu-dios de utilización de medicamentos.
Dosis máxima tolerada (DMT) (Maximum tole-rated dose, MTD)
Es la dosis mayor de un fármaco que ha po-dido administrarse a los participantes en unensayo clínico sin que aparecieran efectosindeseables relevantes. La dosis máxima to-lerada puede variar dependiendo del tipo depoblación de estudio, y es en general másbaja en voluntarios sanos que en enfermos.Por encima de ella se encuentra la dosis mí-nima no tolerada. Su determinación es unode los objetivos de los estudios en fase I y delos estudios puente. Su conocimiento deter-mina las dosis máximas que se emplearánen los estudios de eficacia terapéutica en en-fermos. Véanse Dosis mínima no tolerada,Estudios en fase I y Estudios puente.
Dosis mínima no tolerada (Minimum intolerateddose, MID)
Es la dosis mayor de un fármaco que, tras suadministración a los participantes en un en-sayo clínico, ha provocado efectos indesea-bles que limitan su utilización. En general setrata de la dosis inmediatamente superior ala dosis máxima tolerada. Como su nombreindica, es la dosis más baja que produce unatoxicidad valorable clínica o biológicamente.Véase Dosis máxima tolerada.
Dosis respuesta (Dose response)Relación entre la dosis/concentración de unfármaco y la respuesta observada. VéanseInferencia causal, Relación dosis respuesta,Respuesta gradual y Respuesta del todo onada.
EBMAcrónimo de Evidence-based medicine. Véa-se Medicina basada en la evidencia.
Educación sanitaria (Health education)Proceso por el que determinados individuoso grupos aprenden a adoptar actitudes posi-
28
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

tivas respecto a la promoción, el manteni-miento y el restablecimiento de la salud paramodificar las conductas no saludables. Inclu-ye el suministro de información y el asesora-miento relacionado con la conducta perso-nal.
EEMAcrónimo de Error estándar de la media.
Efectividad (Effectiveness)Grado en el que una intervención produceun resultado beneficioso en las condicionesreales de la práctica habitual. Por este moti-vo, lo que puede resultar eficaz en los parti-cipantes de un ensayo clínico, puede no serefectivo en la población general, debido acondicionantes como el cumplimiento tera-péutico o las características propias de la po-blación. Véase Eficacia.
Efecto (Effect)Resultado de una acción o una causa. Enepidemiología es sinónimo de medida de unefecto. En farmacología, los efectos son el re-sultado del mecanismo de acción de un fár-maco.
Efecto de solapamiento (Overlapping effect)Resultado de la suma de los efectos residua-les de un tratamiento al administrar otro.Puede darse cuando los períodos de lavadoson insuficientes, tanto en el período de in-clusión de los sujetos en cualquier ensayo,como en los ensayos cruzados al iniciar elsegundo período de tratamiento. Véase Efec-to residual.
Efecto del trabajador sano (Healthy worker ef-fect)
Fenómeno observado en los estudios que in-cluyen trabajadores, que generalmente sonmás sanos que la población general, y portanto tienen unas tasas de morbimortalidadinferiores. Por este motivo, el exceso de ries-go asociado a una ocupación específica tien-de habitualmente a ser infraestimado cuandose compara con el de la población general.
Efecto Hawthorne (Hawthorne effect)Efecto, generalmente positivo o beneficioso,que ocurre cuando una persona sabe queestá siendo estudiada. Esta denominaciónderiva del efecto observado en los trabajado-res de la planta Hawthorne de la WesternElectric Company, en Estados Unidos. En és-ta se observó que los cambios de la ilumina-ción, tanto si se aumentaba como si se dis-minuía la intensidad, mejoraba la productivi-dad de los trabajadores, porque éstos sabíanque en cualquiera de los casos estaban sien-do observados.
Efecto indeseableVéase Reacción adversa.
Efecto placebo (Placebo effect)Componente inevitable de la actividad médi-ca por el que los pacientes mejoran tras lainstauración de un tratamiento, sea o no acti-vo en la enfermedad considerada. La exis-tencia de efecto placebo no debe tomarsecomo prueba de la inexistencia de un sínto-ma o enfermedad. Véase Placebo.
Efecto residual (Carry-over effect)Persistencia del efecto después de interrum-pir el tratamiento. Su aparición potencialobliga a la realización de períodos de lavadoentre los distintos tratamientos en los estu-dios de diseño cruzado y/o en los períodosde inclusión. Véase Efecto de solapamiento.
Eficacia (Efficacy)Grado en el que una intervención produceun resultado beneficioso en unas condicio-nes ideales, como puede ser en el marco deun ensayo clínico. Véase Efectividad.
Eficiencia (Efficiency)Grado en que una intervención produce unresultado beneficioso en relación al esfuerzoempleado en términos de recursos humanos,materiales y costes. En general se refiere a lautilización de los recursos estrictamente ne-cesarios que produzcan la máxima efectivi-dad.
Emparejamiento (Matching)Método consistente en seleccionar para cadauno de los sujetos del estudio a otro con ca-racterísticas y factores pronósticos similares.Dentro de cada par, y según la asignaciónaleatoria, un sujeto recibe el fármaco experi-mental y el otro el control. En el caso particu-lar de los estudios de casos y controles, losindividuos son emparejados por aquellas ca-racterísticas diferentes de la propia de inte-rés del estudio. También denominado apare-amiento.
EnmascaramientoVéase Ciego.
Ensayo clínico (Clinical trial)Evaluación experimental de una sustancia omedicamento, a través de su aplicación a se-res humanos, orientada a poner de manifies-to sus efectos farmacodinámicos, su perfilfarmacocinético, establecer su eficacia, co-nocer el perfil de sus reacciones adversas yestablecer su seguridad (RD 561/1993). Enepidemiología, también puede referirse a unestudio en el que se analiza un nuevo proce-dimiento diagnóstico o terapéutico compa-rando dos grupos habitualmente de una ma-
29
GLOSARIO

nera aleatoria. También se denominan estu-dios de intervención. El prototipo de ensayoclínico es aquel en el que los tratamientos seasignan aleatoriamente, existe enmascara-miento (doble ciego) y un grupo control con-currente. Véase Ensayo clínico controlado.
Ensayo clínico a doble ciegoVéase Ciego doble.
Ensayo clínico a ciego simpleVéase Ciego simple.
Ensayo clínico a triple ciegoVéase Ciego triple.
Ensayo clínico abierto (Open, open label, nonblind or non blinded clinical trial)
Ensayo clínico sin enmascaramiento, en elque todas las personas que participan (pa-cientes e investigadores) conocen o son in-formados del tipo de intervención, en contra-posición a un ensayo clínico a simple, dobleo triple ciego.
Ensayo clínico controlado (Controlled clinicaltrial, Randomized controlled trial, RCT, Rando-mized controlled clinical trial)
Ensayo clínico en el que el procedimientoque se evalúa se compara con un controlconcurrente, pudiéndose tratar del procedi-miento estándar o patrón, un placebo en ca-so de ser un tratamiento farmacológico, oninguna intervención. La asignación de cadagrupo de tratamiento o intervención es alea-toria, por lo que algunos autores prefieren eltérmino ensayo clínico controlado y aleatori-zado (Randomized control/controlled clinicaltrial, RCT). Si es posible, los tratamientos de-berán enmascararse. En la mayoría de loscasos el ensayo clínico controlado, aleatori-zado y enmascarado es la única forma cientí-ficamente válida para evaluar la eficacia y se-guridad de una intervención terapéutica.
Ensayo clínico cruzadoVéase Diseño cruzado.
Ensayo clínico de búsqueda de dosisVéase Estudio de búsqueda de dosis
Ensayo clínico de n igual a 1 (N of 1 trial)Ensayo clínico cruzado que se realiza en unsolo individuo. Se utiliza este tipo de ensayosen caso de enfermedades en las que el trata-miento tiene una acción rápida, cuando elefecto termina bruscamente al cesar el trata-miento, y cuando los resultados clínicos sonfácilmente mesurables. En la práctica habi-tual, puede realizarse, por ejemplo, para es-tablecer si determinados síntomas se debena la enfermedad o al tratamiento. En este ca-so, el estudio se realizaría a doble ciego ycomparado con placebo. Por ejemplo, se ha
utilizado este tipo de ensayos en el trata-miento del asma, la artritis reumatoide, lostrastornos del movimiento y la angina de pe-cho.
Ensayo clínico de seguridadVéase Estudio de seguridad.
Ensayo clínico en fase 0 (Phase 0 clinical trial)Denominación que utilizan algunos autorespara referirse a la investigación preclínica ne-cesaria para la realización de los primerosestudios en humanos (Fase I). Véase Fasedel desarrollo de un fármaco.
Ensayo clínico en fase I (Phase I clinical trial)Ensayo diseñado para conocer la farmacoci-nética y/o farmacodinámica de un fármaco, yque proporciona información preliminar sobrelos efectos y la seguridad del producto en su-jetos sanos o en algunos casos en pacientes.Es la primera vez que el fármaco se adminis-tra en humanos. Uno de sus objetivos puedeser la determinación de la dosis máxima tole-rada (la mayor dosis administrada que noocasiona toxicidad relevante). Permite deter-minar la dosis y la pauta de administraciónque se emplearán en los ensayos clínicos defase II/III. No tiene casi nunca finalidad tera-péutica. Recientemente se ha propuesto de-nominarlos Estudios de Farmacología huma-na (CPMP/ICH/291/95). Véanse Dosis máxi-ma tolerada, Estudio puente, Estudio sinfinalidad terapéutica, Fase del ensayo clínicoy Fase del desarrollo clínico de un fármaco.
Ensayo clínico en fase II (Phase II clinical trial)Ensayo que se realiza en individuos que pa-decen la enfermedad o entidad clínica de in-terés, con el objetivo de proporcionar infor-mación preliminar sobre la eficacia del pro-ducto, establecer la relación dosis respuestadel mismo y ampliar los datos de seguridadobtenidos en la fase I. Son los primeros estu-dios terapéuticos en pacientes y uno de susobjetivos primordiales es la búsqueda de la/sdosis para los siguientes ensayos. Habitual-mente son controlados, con asignación alea-toria y enmascarados. Es frecuente el uso deplacebo como control para establecer la efi-cacia real del fármaco. Los pacientes partici-pantes son seleccionados de forma muy es-tricta y su número es limitado (varios cente-nares). Recientemente se ha propuestodenominarlos Estudios terapéuticos explora-torios (CPMP/ICH/291/95). Véanse Ensayoclínico explicativo, Estudio de búsqueda dedosis, Estudio de tolerabilidad, Fase del en-sayo clínico y Fase del desarrollo clínico deun fármaco.
30
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Ensayo clínico en fase III (Phase III clinical trial)Ensayo destinado a evaluar la eficacia y se-guridad del tratamiento experimental en unamuestra de pacientes más representativa dela población general a la que se destinará elmedicamento. Estos estudios son preferen-temente controlados, aleatorizados y enmas-carados. En general, el grupo control loconstituye un fármaco de eficacia conocida(patrón) en esa enfermedad y es menos fre-cuente el uso de placebo. Los pacientes sonmenos seleccionados y más numerosos (va-rios miles) que en los ensayos en fase II. Engeneral se llevan a cabo antes de la comer-cialización del medicamento. Recientementese ha propuesto denominarlos Estudios tera-péuticos confirmatorios (CPMP/ICH/291/95).Véanse Ensayo clínico explicativo, Fase delensayo clínico y Fase del desarrollo clínicode un fármaco.
Ensayo clínico en fase IV (Phase IV clinical trial)Ensayo que se realiza con un medicamentodespués de su comercialización, y que eva-lúa la eficacia y seguridad a largo plazo en laindicación para la que ha sido aprobado. Lafase IV es ideal para estudiar la efectividadde un fármaco. También denominado estu-dio de postcomercialización. Un fármaco co-mercializado (fase IV de desarrollo) que quie-ra estudiarse para una nueva indicación de-berá volver a pasar por las fases previas dedesarrollo (fases II y III). Recientemente seha propuesto denominarlos estudios de usoterapéutico (CPMP/ICH/291/95). Véanse En-sayo clínico pragmático, Fase del ensayo clí-nico y Fase del desarrollo clínico de un fár-maco.
Ensayo clínico explicativo (Explanatory clinicaltrial)
Ensayo clínico cuyo objetivo principal es ob-tener explicaciones biológicas sobre la efica-cia de un fármaco o una intervención. Suelerealizarse en una muestra de participanteshomogénea, representativa sólo de determi-nados grupos de población. Generalmente elanálisis principal suele hacerse de aquellospacientes que completan el ensayo. La ma-yoría de los ensayos clínicos durante el desa-rrollo clínico de un fármaco (fases II y III)pueden considerarse explicativos. VéanseAnálisis explicativo y Ensayo clínico pragmá-tico.
Ensayo clínico multicéntrico (Multicenter clini-cal trial)
Ensayo realizado en dos o más centros conun mismo protocolo y un coordinador que se
encargará del procesamiento de todos losdatos y del análisis de los resultados (RD561/1993 art. 4.2).
Ensayo clínico no controlado (Non-controlledclinical trial)
Ensayo en el que no se compara con un gru-po control. Los ensayos clínicos no controla-dos no parecen útiles para evaluar la eficaciade los fármacos, pero pueden tener interéspara conocer su seguridad. Algunos estudiosde fase I, especialmente los de farmacociné-tica, son no controlados. Véase Estudio deantes y después.
Ensayo clínico paraleloVéase Diseño paralelo.
Ensayo clínico pragmático (Pragmatic clinicaltrial)
Ensayo clínico cuyo objetivo principal es co-nocer la eficacia de un fármaco o de una in-tervención después de aplicarlo en la prácti-ca clínica habitual (efectividad). La muestrade participantes es heterogénea y el análisisse hace por intención de tratar. Suelen reali-zarse en la fase IV del desarrollo de un fár-maco, cuando ya se encuentra comercializa-do. Véanse Análisis por intención de tratar yEnsayo clínico explicativo.
Ensayo clínico promocional (Promotional clini-cal trial)
Ensayo que tiene como única finalidad favo-recer la prescripción de un medicamento.
Ensayo clínico secuencialVéase Diseño secuencial y Análisis secuen-cial.
Ensayo clínico unicéntrico (Unicenter or singlecenter clinical trial)
Ensayo realizado por un solo investigador oequipo de investigación en un centro hospitala-rio o extrahospitalario (RD 561/1993 art. 4.1).
Ensayo comunitario (Community trial)Ensayo que se realiza en comunidades, eva-luando el resultado en el total de la pobla-ción. Dependiendo del número de comuni-dades participantes en el estudio, se puedellevar a cabo la aleatorización o no. Tambiénconocido como Estudio cuasiexperimental(quasi-experimental study) o Ensayo de inter-vención comunitaria. Un ejemplo clásico esel estudio sobre caries dental de Newburgh-Kingston, en Inglaterra, en el que en una co-munidad (Newburgh) se fluoraron las aguasmientras que en la otra (Kingston) se conti-nuó recibiendo agua sin suplementación. Enla comunidad intervenida se observó una re-ducción significativa de la caries y de pérdi-das de piezas dentales.
31
GLOSARIO

Epidemiología (Epidemiology)Tradicionalmente se ha definido como ladisciplina que estudia la distribución de lasenfermedades o los estados relacionadoscon la salud y sus determinantes en pobla-ciones específicas, y su aplicación en elcontrol de los problemas de salud. La epide-miología, aunque cubre un rango muy am-plio de actividades (desde estudios de bro-tes epidémicos hasta ensayos clínicos connuevos fármacos), tiene dos aspectos en co-mún en todas ellas: la unidad de investiga-ción son generalmente grupos de individuos,y la comparación de un grupo de individuoscon otro.
Epidemiología clínica (Clinical epidemiology)Parte de la epidemiología que estudia la apli-cación de la metodología y el razonamientoepidemiológico a los problemas diagnósticosy terapéuticos de la práctica clínica.
Epidemiología molecular (Molecular epidemio-logy)
Parte de la epidemiología que explica lametodología científica y los avances en bio-logía molecular para estudiar los determi-nantes etiológicos, ya sean ambientales ohereditarios, asociados a enfermedades co-mo las neoplásicas, hematológicas o infec-ciosas.
EquivalenciaVéase Estudio de equivalencia.
Error � (α- Error)Probabilidad de rechazar la hipótesis nulacuando ésta es realmente cierta. Tambiéndenominado Error tipo I y Probabilidad α.Véase Significación estadística, nivel de.
Error � (β - Error)Probabilidad de aceptar la hipótesis nulacuando ésta es realmente falsa. También de-nominado Error tipo II y Probabilidad ß. Véa-se Potencia estadística.
Error estándar de la media (EEM) (Standarderror of the mean, SEM)
Medida de dispersión de las medias de repe-tidas poblaciones de tamaños diferentes. Secalcula mediante la fórmula:
δEEM = donde δ = desviación estándar y
√n n = número de individuos.
Algunos autores lo denominan simplementeerror estándar.
Error tipo IVéase Error α.
Error tipo IIVéase Error β.
Especificidad (Specificity)Referido a las pruebas diagnósticas, proba-bilidad de que una prueba resulte negativacuando realmente la enfermedad está au-sente (una prueba altamente específica dapocos resultados falsos positivos). Se cal-cula mediante el cociente entre d y b+d(véase tabla). Cuando se refiere a los crite-rios de causalidad define el grado en queuna sola causa se asocia a un solo efecto.Véanse Causalidad, Inferencia causal ySensibilidad.
EnfermedadPresente + Ausente –
Prueba Positiva + a b a + bdiagnóstica Negativa – c d c + d
a + c b + d
EstandarizaciónVéase Ajuste.
EstocásticoVéase Aleatorio.
Estratificación (Stratification)Técnica para controlar el efecto de las varia-bles de confusión en el análisis de los datos.Consiste en evaluar la asociación en catego-rías homogéneas de la variable de confusión.Por ejemplo, si el sexo se considera una va-riable potencial de confusión, una estimaciónde la asociación entre la exposición y la en-fermedad se debería analizar para hombres ymujeres separadamente.
Estudio analítico (Analytical study)En epidemiología, denominación utilizadapara los estudios observacionales de cohor-tes y estudios de casos y controles, en losque se puede contrastar hipótesis mediantela comparación de dos grupos.
Estudio anidado de casos y controles (Nestedcase control study)
Estudio de casos y controles en el que loscasos y los controles se extraen de un estu-dio de cohortes. Véase Estudio de casos ycontroles y Estudio de cohortes.
Estudio con consentimiento aleatorizado (Ran-domized consent study)
Estudio en el que sólo se pide consentimien-to a aquellos pacientes aleatorizados a losque les corresponde el tratamiento experi-mental. Si les corresponde el tratamientocontrol (terapia estándar) no se les solicita elconsentimiento. Es un diseño muy discutido,tanto desde el punto de vista ético como desu análisis. Su propulsor es M. Zelen. Enocasiones se le denomina método de aleato-rización de Zelen (Fig. 9).
32
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Estudio cruzadoVéase Diseño cruzado.
Estudio cuasiexperimentalVéase Ensayo comunitario.
Estudio de antes y después (Before and afterstudy/design)
Estudio de intervención sin grupo control enel que se comparan los resultados de antesde la intervención y después de la misma.
Estudio de bioequivalenciaVéase Bioequivalencia.
Estudio de búsqueda de dosis (Dose findingstudy, Dose ranging studies)
Estudio en el que se administran distintasdosis de un mismo fármaco y un control agrupos diferentes de pacientes para conocercuál o cuáles de ellas son las más adecua-das en términos de eficacia y seguridad paraproseguir con el desarrollo del fármaco. Engeneral se realizan en la fase II del desarrollode un fármaco. También se denominan estu-dios de titulación de dosis (Dose titration stu-dies) o estudios de escalada de dosis. Véan-se Ensayo clínico en fase II y Relación dosisrespuesta.
Estudio de casos y controles (Case-controlstudy)
Estudio en el que se seleccionan individuoscon una determinada enfermedad y se com-paran con unos controles -apareados poruna serie de características como edad y se-xo- respecto a la exposición previa de posi-bles factores de riesgo asociados a la enfer-medad. La medida que se utiliza para cuanti-ficar la asociación entre la exposición previa
y la enfermedad es la razón de posibilidades(odds ratio). Véanse Estudio retrospectivo yRazón de posibilidades.
Estudio de cohorte (Cohort study)Estudio en el que se selecciona un grupo deindividuos expuestos a unos hipotéticos fac-tores de riesgo y se compara con otro no ex-puesto a los mismos después de un tiempode seguimiento y hasta el desarrollo de la en-fermedad. La medida de asociación que seutiliza en estos estudios es el riesgo relativo yel riesgo absoluto. También llamado estudiolongitudinal. Véanse Estudio prospectivo,Riesgo relativo y Riesgo absoluto.
Estudio de cohorte retrospectivo (Historical co-horts, Retrospective cohort study)
Estudio de cohorte en el que se compara dosgrupos respecto a la exposición en el pasadoa un factor específico y a la presencia de laenfermedad en el presente. Para poder llevareste tipo de estudios es preciso disponer deun buen sistema de registro.
Estudio/método de conexión de registros (Re-cord linkage study/method)
Estudio de farmacovigilancia en el que seutilizan bases de datos donde se registra to-do lo concerniente a la salud de las perso-nas (enfermedades, prescripciones, analíti-cas u hospitalizaciones, entre otras). Estasbases deben contener suficiente informa-ción para poder buscar, seleccionar y extra-er la información deseada: la exposición aun fármaco y la aparición de una enferme-dad o síntoma.
Estudio de controles históricos (Historical con-trol study)
Estudio en el que se compara los resultadosde una nueva intervención con los resultadosobtenidos en una serie previa utilizando la in-tervención antigua. Por definición, no son es-tudios aleatorizados, y se realizan habitual-mente en enfermedades poco frecuentes ocuando no sea ético o práctico llevar a caboun estudio con controles concurrentes. Véa-se Control histórico.
Estudio de correlación (Correlation study)Estudio que utiliza los datos poblacionalespara comparar las frecuencias de una en-fermedad entre diferentes grupos duranteun mismo período de tiempo, o en la mis-ma población en diferentes momentos enel tiempo. Por ejemplo, estudiar la correla-ción entre el consumo per cápita de carney las tasas de cáncer de colon en diferen-tes países. También se denomina estudioecológico.
33
GLOSARIO
Paciente elegible
Aleatorización(A [control] o B [experimental])
ANo pedir
consentimiento
BPedir consentimiento
¿Acepta el tratamiento B?
No Sí
A A B
Figura 9. Estudio con consentimiento aleatorizado.

Estudio de equivalencia (Equivalence trial,Equivalence study)
Ensayo clínico cuyo objetivo es ver si la nue-va intervención es igual de eficaz que la yaconocida. Previamente se tiene que definircon qué márgenes la nueva intervención seconsiderará igual a la ya conocida para po-der hacer la estimación muestral. Obviamen-te las dos intervenciones no pueden seridénticas, ya que el tamaño muestral paradetectar diferencias mínimas es infinito. Unade sus formas es el estudio de bioequivalen-cia. Véase Bioequivalencia.
Estudio de escalada de dosisVéase Estudio de búsqueda de dosis.
Estudio de intervenciónVéase Ensayo clínico.
Estudio de intervención comunitariaVéase Ensayo comunitario.
Estudio de postcomercialización (Postmarke-ting surveillance study)
Tipo de estudio que se realiza cuando losmedicamentos ya se encuentran disponiblespara la población. Forman parte de ellos losensayos clínicos de fase IV y los estudios deutilización de medicamentos. Su objetivo esconocer las consecuencias beneficiosas yperjudiciales del empleo de los medicamen-tos en las condiciones habituales de la prác-tica clínica.
Estudio de seguridad (Safety study)Estudio destinado a evaluar la incidencia, eltipo y la gravedad de las reacciones adversasque aparecen asociadas a tratamientos espe-cíficos. Véase Estudio de tolerabilidad.
Estudio de series de casos clínicos (Case se-ries study)
Estudio que describe las características deun número limitado de pacientes con unadeterminada enfermedad.
Estudio de titulación de dosisVéase Estudio de búsqueda de dosis
Estudio de tolerabilidad (Tolerability, Tolerabi-lity study)
Estudio cuyo objetivo principal es la evalua-ción de los efectos indeseables. En el casode los ensayos clínicos de fase I en volunta-rios sanos, en los que no existe posibilidadde acción terapéutica, los objetivos primariosson siempre la tolerabilidad y la farmacociné-tica/farmacodinámica. En algunos de ellos seintenta definir la dosis máxima tolerada (lamayor dosis administrada que no presentatoxicidad relevante). En ocasiones se le de-nomina estudio de tolerancia, aunque estetérmino puede tener un significado comple-
tamente diferente (disminución del efectopara una misma dosis cuando se administraen varias ocasiones consecutivas o, lo que esequivalente, necesidad de incrementar la do-sis para mantener los mismos efectos). Vé-anse Dosis máxima tolerada y Estudio de se-guridad.
Estudio de toleranciaVéase Estudio de tolerabilidad
Estudio de utilización de medicamentos (EUM)(Drug utilization study, DUS)
Estudio que describe la utilización de medi-camentos en la práctica clínica habitual. Per-mite detectar un uso inadecuado de los mis-mos e iniciar una intervención para corregir-lo.
Estudio descriptivo (Descriptive study)Estudio que describe las características ge-nerales de la distribución de una enferme-dad en relación al individuo (por ejemplo,edad, sexo, raza), lugar y tiempo. Dadas laslimitaciones inherentes al propio diseño, losestudios descriptivos son fundamentalmen-te útiles en la formulación de hipótesis quedespués deberán contrastarse medianteestudios analíticos (ensayos clínicos). Éstospueden ser estudios poblacionales (estu-dios de correlación) e individuales (estu-dios de series de casos o estudios transver-sales).
Estudio ecológicoVéase Estudio de correlación.
Estudio experimental (Experimental study)En epidemiología es sinónimo de ensayo clí-nico controlado aleatorizado. En farmacologíapuede referirse además a estudios realizadosen animales.
Estudio fundamental (Pivotal study)Estudio que se considera esencial para con-seguir el registro de un fármaco para una in-dicación. Son generalmente ensayos clínicosen fase III que demuestran la eficacia del fár-maco frente a un placebo o un control. Algu-nas autoridades aceptan el registro de unfármaco si ha demostrado su eficacia en dosensayos clínicos controlados con gran núme-ro de pacientes.
Estudio longitudinalVéase Estudio de cohorte.
Estudio multicéntrico (Multicenter study)En sentido amplio, cualquier estudio que serealice en dos o más centros con un protoco-lo común. Véanse Comité de expertos y En-sayo clínico multicéntrico y Multicéntrico.
Estudio paraleloVéase Diseño paralelo.
34
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Estudio piloto (Pilot study)Estudio de prueba que se lleva a cabo en unnúmero limitado de sujetos como paso previoa otros estudios más amplios con el fin devalorar aspectos como la idoneidad del dise-ño, así como su factibilidad y viabilidad.También permiten conocer la variabilidadque permitirá el cálculo del tamaño de lamuestra de futuras investigaciones.
Estudio prospectivo (Prospective study)En sentido amplio, se considera como sinóni-mo de estudio de cohorte por algunos auto-res ya que, partiendo de una determinadaexposición, se observa la aparición de unaenfermedad en el futuro. Otros autores, paraevitar confusiones, prefieren utilizar exclusi-vamente esta denominación para los estu-dios de cohorte prospectivos.
Estudio puente (Bridging study)Estudio en el que se determina la dosis máxi-ma tolerada en un grupo pequeño de pa-cientes que padecen la enfermedad en laque se cree que el fármaco va a ser eficaz.Permiten tener una idea preliminar de la toxi-cidad y en ocasiones de la eficacia del fár-maco en su población diana. Se inician pocodespués de conocerse la dosis máxima tole-rada en voluntarios sanos. Permiten una me-jor predicción de las dosis que se emplearándespués en los ensayos clínicos de eficacia.En general, la dosis máxima tolerada de lospacientes resulta mayor que la encontradaen voluntarios sanos. Se han empleado en elestudio de fármacos para el tratamiento de lademencia o la esquizofrenia. Sus defensorespostulan que este diseño permite conocer ladosis máxima tolerada de forma más exacta;así pueden iniciarse los estudios de eficaciacon más precisión. Su nombre proviene desu emplazamiento entre las fases I y II deldesarrollo clínico. Véanse Dosis máxima tole-rada y Ensayos clínicos en fase I.
Estudio retrospectivo (Retrospective study)En sentido amplio, se considera como sinóni-mo de estudio de casos y controles por algu-nos autores ya que, partiendo de la enferme-dad, se observa la exposición en el pasadopara identificar una posible causa. Otros au-tores, para evitar confusiones, prefieren utili-zar exclusivamente esta denominación paralos estudios de cohorte retrospectivos.
Estudio sin finalidad terapéutica (Non-thera-peutic study or trial)
Estudios en los que el objetivo principal noes aliviar, curar o prevenir una enfermedad osíntoma. Puede tratarse de un estudio para
definir la farmacocinética, el mecanismo deacción o los efectos de un fármaco. Porejemplo, evaluar los cambios en las concen-traciones de potasio y en la FEV1 de un nue-vo fármaco agonista de los adrenoceptoresβ2 administrado a dosis única en asmáticos.También pueden serlo aquellos en los que elfármaco se utiliza para diagnosticar una en-fermedad. En general, los estudios en volun-tarios sanos (fase I) no poseen finalidad tera-péutica. En este tipo de estudios es necesa-rio que los participantes consientan enparticipar por escrito. Véanse Ensayo clínicoen fase I y Fase del ensayo clínico.
Estudio transversal (Cross-sectional study)Estudio observacional en el que se mide enun momento del tiempo la presencia de unaenfermedad y de otras variables relacionadascon la misma. También se denominan estu-dios de prevalencia, ya que habitualmenteson útiles para determinar la prevalencia deuna enfermedad o factor de riesgo. Ocasio-nalmente, pueden ser útiles para estableceruna asociación entre un determinado factor yla enfermedad, aunque al haberse registradoambos en el mismo momento del tiempo, nopuede saberse con certeza si la enfermedades realmente la causa o la consecuencia. Porejemplo, una asociación observada entre laartrosis y la obesidad en un estudio transver-sal no nos dice si la artrosis es la causa o laconsecuencia de la obesidad.
Estudio unicéntrico (Unicenter study, Singlecenter study)
Es aquel que se realiza en un solo centro olugar. Véase Ensayo clínico unicéntrico.
Etapas del desarrollo de un fármacoVéase Fase del desarrollo de un fármaco.
Etapas del desarrollo clínico de un fármacoVéase Fase del desarrollo clínico de un fármaco.
EUMAcrónimo de Estudio de utilización de medi-camentos.
Evaluación ciega por terceros (Blind or blindedevaluation)
Procedimiento empleado habitualmente enlos ensayos clínicos por el que el observadorque realiza la medida de la variable principaldesconoce la asignación de los tratamientos.Este método se emplea cuando no es posibleun enmascaramiento completo (doble ciego).Véase Ciego.
Evidencia experimental (Experimental evidence)Demostración mediante métodos experimen-tales de la asociación causal entre los he-chos observados. Véase Inferencia causal.
35
GLOSARIO

Exactitud (Validity)Sinónimo de validez. Se refiere a si un instru-mento está midiendo lo que realmente se de-sea medir. La falta de exactitud es debida aun error sistemático o sesgo, y esencialmen-te es atribuible a aspectos metodológicos di-ferentes al tamaño muestral, como puede serla selección de los sujetos y la calidad de lainformación obtenida. Véanse Precisión y Va-lidez.
Factor (Factor)Acontecimiento, característica u otra entidaddefinible, que puede ocasionar un cambio enel estado de salud o modificar el resultado deuna enfermedad. También se denomina de-terminante. Sinónimo de variable indepen-diente.
Factor de confusión (Confounding, Confoun-ding Factor)
Factor que distorsiona la verdadera relaciónde las variables en estudio en virtud de estarrelacionada con la enfermedad y con la ex-posición o el factor de riesgo del estudio. Porejemplo, la asociación entre el alcohol y elcáncer oral tiene en el tabaco un factor deconfusión, ya que muchos de los pacientesque beben alcohol son asimismo fumadores.Denominado también variable de confusión.
Factor de riesgo (Risk factor)Característica que, según la evidencia epide-miológica, se asocia causalmente con la en-fermedad del sujeto. Por ejemplo el tabacoes un factor de riesgo del cáncer de pulmóny la hipertensión arterial lo es del accidentevascular cerebral. Véase Causalidad.
Factor pronóstico (Prognostic factor)Característica asociada al sujeto que puedepredecir los eventuales resultados en el cur-so de una enfermedad. Puede ser demográ-fica (edad, sexo), específica de enfermedad(estadio del tumor) u otras enfermedadesasociadas (comorbilidad). El factor pronósti-co puede predecir la evolución de un resulta-do, ya sea bueno o malo, y no necesaria-mente se asocia causalmente con la enfer-medad.
Falacia (Fallacy)Según el Diccionario de la Lengua Españolaes Engaño, fraude o mentira con la que seintenta dañar a otro. Véase Sesgo.
Falacia ecológica (Ecological fallacy)Sesgo que puede aparecer al observar unaasociación a partir de un estudio ecológicopero que no representa una asociación cau-sal a nivel individual. Véase Estudio de corre-lación.
Falacia post hoc ergo propter hocVéase Post hoc ergo propter hoc
Falsificación (Counterfeiting, falsification)Acción y efecto de falsificar. Falsedad que secomete en alguna de las partes de una in-vestigación.
Falso negativo, resultado (False negative result)Resultado negativo de una prueba diagnósti-ca en un sujeto que posee realmente el atri-buto o la enfermedad hacia el que iba orien-tado aquélla. Por ejemplo, catalogar de sanaa una persona enferma cuando se practicauna prueba de cribado para detectar la en-fermedad. También se le denomina negativofalso o pseudonegativo. Se expresa general-mente en forma de tasa, y es la proporciónde pacientes con la enfermedad en los quela prueba es negativa. Según la tabla, la tasade falsos negativos sería igual a c/a+c.
EnfermedadPresente + Ausente –
Prueba Positiva + a b a + bdiagnóstica Negativa – c d c + d
a + c b + d
Falso positivo, resultado (False positive result)Resultado positivo de una prueba diagnósti-ca en un sujeto que no posee realmente elatributo o la enfermedad hacia el que ibaorientado aquélla. Por ejemplo, catalogar deenferma a una persona sana cuando sepractica una prueba de cribado para detec-tar la enfermedad. También se le denominapositivo falso o pseudopositivo. Se expresageneralmente en forma de tasa, y es la pro-porción de pacientes sin la enfermedad enlos que la prueba es positiva. Una tasa altade falsos positivos refleja una baja especifici-dad de la prueba. Según la tabla dos por dos(véase Falso negativo, resultado) la tasa defalsos positivos sería b/b+d.
Farmacia (Pharmacy)Ciencia dedicada a la preparación y la dis-pensación de medicamentos. En los ensayosclínicos, el departamento de farmacia es res-ponsable de la conservación, el recuento y elsuministro de las muestras utilizadas enellos.
Fármaco (Drug)En sentido amplio, toda sustancia químicacapaz de interaccionar con un organismo vi-vo. En sentido estricto, toda sustancia quími-ca utilizada en el tratamiento, la curación, laprevención, el diagnóstico de una enferme-dad, o empleada para evitar la aparición deun proceso fisiológico no deseado. Los térmi-
36
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

nos fármaco, medicamento y sustancia me-dicinal se emplean en ocasiones indistinta-mente. Véanse Medicamento y Sustanciamedicinal.
Farmacocinética (Pharmacokinetics)Disciplina que estudia los procesos y factoresque determinan la cantidad de fármaco pre-sente en el organismo. Analiza la absorción,distribución, metabolismo y excreción de unfármaco.
Farmacodinamia (Pharmacodynamics)Disciplina que estudia las acciones y losefectos de los fármacos.
Farmacoeconomía (Pharmacoeconomy)Disciplina dedicada a la descripción y el aná-lisis del coste de un tratamiento farmacológi-co para el sistema sanitario y para la socie-dad.
Farmacoepidemiología(Pharmacoepide-miology)
Disciplina dedicada al estudio del impacto delos fármacos en poblaciones humanas me-diante el método epidemiológico. Resulta dela conjunción de la farmacología clínica y dela epidemiología.
Farmacogenética (Pharmacogenetics)Disciplina que estudia las respuestas farma-cológicas y su modificación por influenciashereditarias. Estas influencias pueden deber-se a alteraciones de base farmacocinética ofarmacodinámica.
Farmacología (Pharmacology)Ciencia biológica que estudia las acciones ypropiedades de los fármacos en los organis-mos.
Farmacología Clínica (Clinical Pharmacology)Disciplina que se ocupa del estudio científicode los medicamentos en el hombre (Informede un grupo de estudio de la OMS. Farmaco-logía clínica: actividades, servicios y ense-ñanza. Ginebra: Organización Mundial de laSalud, serie informes técnicos nº 446, 1970).En España es una especialidad médica conun programa de formación propio.
Farmacopea (Pharmacopoeia)Tratado de referencia que tiene por objetoasegurar la uniformidad de la naturaleza, ca-lidad, composición y riqueza de las sustan-cias medicinales y excipientes. Incluye mo-nografías con los caracteres de las sustan-cias medicinales, excipientes, métodos deensayo y de análisis a utilizar para asegurarsu calidad, los procedimientos de prepara-ción, esterilización, conservación y acondi-cionamiento. Las monografías constituyenexigencias mínimas de obligado cumplimien-
to (Ley del medicamento 25/1990, art. 55, 1y 2). Existen, entre otras, una FarmacopeaEspañola y Europea.
Farmacovigilancia (Pharmacovigilance, Post-marketing Drug Surveillance)
Disciplina dedicada a la identificación y a lavaloración de las reacciones adversas asocia-das al uso agudo o crónico de los medica-mentos en la población o en algún grupo es-pecífico de ella. Entre los principales méto-dos utilizados por la farmacovigilancia seencuentran la notificación espontánea de re-acciones adversas, los estudios de cohorte ylos estudios de casos y controles. Véanse.Notificación espontánea de reacciones ad-versas, Estudio de casos y controles y Estu-dio de cohorte.
Fase 0Véanse Ensayo clínico en fase 0 y Fase deldesarrollo de un fármaco.
Fase IVéanse Ensayo clínico en fase I, Fase del de-sarrollo clínico de un fármaco y Fase del en-sayo clínico.
Fase IIVéanse Ensayo clínico en fase II, Fase deldesarrollo clínico de un fármaco y Fase delensayo clínico.
Fase IIIVéanse Ensayo clínico en fase III, Fase deldesarrollo clínico de un fármaco y Fase delensayo clínico.
Fase IVVéanse Ensayo clínico en fase IV, Fase deldesarrollo clínico de un fármaco y Fase delensayo clínico.
Fase/s del desarrollo clínico de un fármaco(Clinical drug development phases)
Los fármacos se desarrollan en etapas deforma secuencial: primero se inicia la fase I(estudios en sujetos sanos), se pasa a la faseII (búsqueda de dosis en enfermos), despuésa la III (eficacia y seguridad en enfermos) yfinalmente a la IV (comercialización). Algu-nos autores definen una fase II inicial (IIa) yuna posterior (IIb). También se divide la faseIII en dos etapas; así se habla de fase IIIacuando aún no se ha presentado el nuevofármaco al registro sanitario y de fase IIIb alas investigaciones que se realizan cuando yase ha solicitado el registro sanitario.Es frecuente que las fases II, III y IV puedanser simultáneas. Así, un fármaco puede estaren fase IV para una enfermedad (indicaciónaprobada en el registro) y en fase II para otraenfermedad distinta. Además, el desarrollo
37
GLOSARIO

puede ser distinto en diferentes países, en-contrándose comercializado en unos y en fa-ses previas al registro en otros. Para definirexactamente el estado de desarrollo de unfármaco en un momento concreto, deberíaespecificarse la etapa máxima (fase) alcan-zada para cualquier indicación y país.No deben confundirse las fases/etapas dedesarrollo y las fases del ensayo clínico. Eldesarrollo se enmarca en un proceso que tie-ne como finalidad el registro y comercializa-ción de un nuevo fármaco, mientras que lasfases del ensayo clínico nos informan sobrelos objetivos y el diseño de un estudio. Porejemplo un ensayo clínico cuyo objetivo es labúsqueda de dosis lo definiremos como unensayo clínico de fase II, pero es posible queese fármaco esté comercializado (fase IV dedesarrollo clínico). Véanse Ensayo clínico enfase I, Ensayo clínico en fase II, Ensayo clíni-co en fase III, Ensayo clínico en fase IV, Fasedel ensayo clínico y Fase del desarrollo de unfármaco.
Fase/s del desarrollo de un fármaco (Drug de-velopment phases)
Define las distintas etapas en la evolución deun fármaco, desde que surge la idea hastasu comercialización. Existe una fase o etapapreclínica que incluye la síntesis química ylos estudios de farmacología y toxicología. Lesigue una fase o etapa clínica con distintasfases (I, II, III, IV). Además, existe la investi-gación galénica para encontrar la formula-ción farmacéutica más adecuada. Las etapasiniciales suelen ser simultáneas; así, porejemplo, un fármaco en fase II/III está aúnen estudio para determinar su toxicología alargo plazo en animales. Véase Fase del de-sarrollo clínico de un fármaco.
Fase/s del ensayo clínico (Clinical trial phases)Los ensayos clínicos pueden clasificarse se-gún su objetivo y diseño en diferentes fases(I, II, III, IV). Cuando se realizan en sujetossanos para conocer su farmacocinética, far-macodinamia y/o seguridad se denominanen fase I. Cuando tienen por objetivo buscarla dosis eficaz en una enfermedad concretason de fase II. Si lo que pretenden es cono-cer la eficacia de un fármaco frente a otro/sfármaco/s con eficacia ya conocida y en con-diciones de uso no muy restrictivas, son enfase III. Por último, si el medicamento estácomercializado y se quiere estudiar su efecti-vidad (poblaciones poco seleccionadas, usosegún prospecto), nos encontramos con unensayo clínico en fase IV.
Como se ha comentado bajo el epígrafe deFase/s del desarrollo clínico de un fármaco,no debe confundirse la fase del ensayo clíni-co y la fase del desarrollo clínico de un fár-maco.EL CPMP, en su recientemente aprobadaguía Note for guidance on general considera-tions for clinical trials (CPMP/ICH/291/95,ICH Topic E 8), propone una nueva termino-logía para diferenciar claramente las fases dedesarrollo de un fármaco y las fases del ensa-yo clínico. La guía propone el uso exclusivode la palabra fase para definir el desarrolloclínico de un fármaco (fases I, II, II y IV), es-pecifica los distintos tipos de ensayos clínicostípicos de cada fase del desarrollo y proponeuna denominación alternativa a la común-mente utilizada de ensayos clínicos en fase I,II, III y IV. En concreto, los tipos de ensayosclínicos se describen según su objetivo princi-pal, clasificándolos en los de Farmacologíahumana (antes denominados ensayos clíni-cos en fase I, típicos de la fase I del desarrolloclínico de un fármaco), Terapéuticos explora-torios (antes denominados ensayos clínicosen fase II, típicos de la fase II del desarrolloclínico de un fármaco), Terapéuticos confir-matorios (antes denominados ensayos clíni-cos en fase III, típicos de la fase III del desa-rrollo clínico de un fármaco) y de Uso tera-péutico (antes denominados ensayos clínicosen fase IV, típicos de la fase IV del desarrolloclínico de un fármaco). Véanse Ensayo clínicoen fase I, Ensayo clínico en fase II, Ensayo clí-nico en fase III, Ensayo clínico en fase IV, Fa-ses del desarrollo clínico de un fármaco y Fa-ses del desarrollo de un fármaco.
Fase de lavadoVéase Período de lavado.
FDAAcrónimo de Food and Drug Administration.Agencia gubernamental de control de ali-mentos y medicamentos de los EE.UU.
Fenómeno iceberg (Iceberg phenomenon)Expresión empleada para denominar las en-fermedades que permanecen sin registrar,detectar o declarar pese a los esfuerzos diag-nósticos o de vigilancia (porción sumergidadel iceberg). Comprende las enfermedadesque no reciben atención médica, las que síla reciben pero no se diagnostican con preci-sión, y las que sí se diagnostican pero no sedeclaran. La porción visible o punta del ice-berg corresponde a las enfermedades detec-tadas o diagnosticadas. También denomina-do fenómeno de témpano de hielo.
38
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

FiabilidadVéase Precisión.
Forma farmacéutica (Pharmaceutical form)Disposición individualizada a que se adaptanlas sustancias medicinales y excipientes paraconstituir un medicamento (Ley del Medica-mento 25/90 art. 8.5). También se le conocepor forma galénica.
Forma galénicaVéase Forma farmacéutica.
Fórmula magistralMedicamento destinado a un paciente indivi-dualizado, preparado por el farmacéutico, obajo su dirección, para cumplimentar expre-samente una prescripción facultativa detalla-da de las sustancias medicinales que inclu-ye, según las normas técnicas y científicasdel arte farmacéutico, dispensado en su far-macia o servicio farmacéutico y con la debi-da información al usuario (Ley del Medica-mento 25/90 art. 8.10).
FormulaciónVéase Forma farmacéutica.
Fracción atribuibleVéase Riesgo absoluto poblacional.
Fracción etiológicaVéase Riesgo absoluto.
Fraude (Fraud)Engaño o falsedad en alguna de las etapas opartes de una investigación.
Garantía de calidad (GC) (Quality assurance,QA)
Conjunto de métodos para recoger, procesary analizar datos, encaminados a mantener omejorar la validez y reproducibilidad de losmismos, así como de los procedimientos uti-lizados para generarlos.Según la Guía ICH Tripartita y Armonizadapara la Buena Práctica Clínica, son todasaquellas acciones planificadas y sistemáticasque se establecen a fin de asegurar que elensayo sea realizado y los datos sean gene-rados, documentados (registrados) y comu-nicados de acuerdo con las normas de Bue-na Práctica Clínica (BPC) y los requisitos re-guladores pertinentes.
GCPAcrónimo de Good Clinical Practice. VéaseNormas de buena práctica clínica.
GenerabilidadVéase Validez externa.
Generalización de resultadosVéase Validez externa.
GLPAcrónimo de Good Laboratory Practice. Véa-se Normas de buena práctica de laboratorio.
GMPAcrónimo de Good Manufacturing Practice.Véase Normas de buena práctica de fabrica-ción.
Gradiente biológico (Biological gradient)Existencia de una relación dosis-respuestaen el estudio de la causalidad. También seconsidera como tal la existencia de una rela-ción entre la duración de la exposición y larespuesta. Véase Inferencia causal y Rela-ción dosis-respuesta.
Grados de libertad (gl) (Degrees of freedom,df)
Número de comparaciones independientesque pueden efectuarse entre los elementosde una muestra. Para algunos autores, esteextraño término, que no tiene un origen polí-tico sino matemático, se refiere al número defrecuencias esperadas que pueden ser espe-cificadas libremente en una prueba estadísti-ca. En el caso concreto de la prueba de t deStudent, es igual a n-1, y en la de χ2 se cal-cula multiplicando el número de filas menos1 por el número de columnas menos 1.
Grupo comparativoVéase Grupo control y Grupo experimental.
Grupo control (Control group, Comparisongroup, Comparative group)
Conjunto de individuos de un ensayo clínicoque se han asignado al tratamiento de con-trol (referencia), en contraposición a los quehan sido asignados al tratamiento experi-mental (test). El tratamiento control puedeser placebo, un fármaco del que se conocesu actividad o eficacia y se considera un es-tándar para el tratamiento de esa enferme-dad, o ninguna intervención. En los estudiosde cohorte se refiere al grupo de los indivi-duos no expuestos al factor de estudio, y enlos estudios de casos y controles se refiere alos individuos sin la enfermedad. En estos úl-timos estudios también se denomina grupocomparativo o de comparación, grupo de re-ferencia o grupo testigo. Véase Grupo experi-mental.
Grupo experimental (Experimental group, Testgroup)
Conjunto de individuos de un ensayo clínicoque se han asignado al tratamiento que sedesea evaluar (test), en contraposición a losque han sido asignados al grupo control (re-ferencia). Véase Grupo control.
Helsinki, declaración de (Helsinki, declarationof)
Documento de la Asociación Médica Mundialque recoge las recomendaciones que deben
39
GLOSARIO

guiar a los médicos en la investigación bio-médica sobre seres humanos. Consta de in-troducción, principios básicos, un capítulodedicado a la investigación médica asociadaa la asistencia profesional (investigación clíni-ca) y una parte final dedicada a la investiga-ción biomédica no terapéutica con seres hu-manos (investigación biomédica no clínica).Fue adoptada en la 18ª Asamblea MédicaMundial celebrada en 1964 en Helsinki. Seha modificado en asambleas realizadas enTokio (1975), Venecia (1983), Hong-Kong(1989) y Sudáfrica (1996). Este documentosentó las bases del comportamiento ético eninvestigación clínica terapéutica y no terapéu-tica. En la mayoría de protocolos de investiga-ción y en la producción científica derivada, seobliga a los autores a declarar que los experi-mentos se han realizado respetando los prin-cipios de esta declaración (véase la declara-ción de Helsinki en el apartado apéndices dela presente monografía, pág 85).
Heterogeneidad (Heterogeneity)Desigualdad entre los parámetros de dosmuestras.
Heteroscedasticidad (Heteroscedasticity)Desigualdad entre varianzas de diferentesmuestras.
Hipótesis (Hypothesis)Suposición, fundada en observaciones o re-flexiones, que puede conducir a prediccio-nes comprobables o refutables.
Hipótesis alternativa (Alternative hypothesis)Hipótesis que considera la posibilidad de laexistencia de diferencias entre dos o másgrupos respecto a determinadas característi-cas. En los ensayos clínicos habituales consi-dera la existencia de diferencias significativasen la eficacia o la seguridad entre el trata-miento de referencia y el experimental. Enlos ensayos clínicos de equivalencia la hipó-tesis alternativa se formula al revés, conside-rando que no hay diferencias entre las inter-venciones. La hipótesis alternativa consideraque los resultados observados en el estudioson diferentes de los que podrían haberseproducido a consecuencia exclusivamentedel azar.
Hipótesis nula (Null hypothesis)Hipótesis que considera la inexistencia de di-ferencias entre dos o más grupos respecto adeterminadas características. En los ensayosclínicos habituales considera que no existendiferencias significativas en la eficacia o laseguridad entre el tratamiento de referenciay el experimental. En los ensayos clínicos de
equivalencia, la hipótesis nula se formula alrevés, considerando que existen diferenciasentre las intervenciones, y considera que losresultados observados en un estudio no sondiferentes de los que podrían haberse produ-cido a consecuencia exclusivamente delazar.
Historia clínica (Medical record)Relación de los datos con significación médi-ca referentes a un enfermo, al tratamiento aque se le somete y a la evolución de la enfer-medad (Diccionario de la Lengua Española).Las historias clínicas de los pacientes inclui-dos en un ensayo clínico deben conservarseel máximo de tiempo posible que permita elhospital, la institución o la consulta privadadonde se haya realizado el ensayo (RD561/1993 art. 21.3). Pueden ser revisadaspara comprobar la fiabilidad, siempre guar-dando la obligatoria confidencialidad, por elmonitor, el promotor, el Comité Ético de In-vestigación Clínica, e inspeccionadas por lasautoridades sanitarias.
HistóricoVéase Control histórico.
Homogeneidad (Homogeneity)Igualdad o similitud entre parámetros de di-ferentes muestras.
Homoscedasticidad (Homoscedasticity)Igualdad entre varianzas de diferentes mues-tras.
I+DAcrónimo de Investigación y desarrollo.
IcebergVéase Fenómeno iceberg.
ICHAcrónimo de International Conference onHarmonization. Conferencia que tiene comoobjetivo armonizar las exigencias de las auto-ridades sanitarias de la Unión Europea, losEstados Unidos de Norteamérica y Japón pa-ra el registro de nuevos medicamentos. Serefieren a aspectos farmacéuticos, farmaco-lógicos, toxicológicos y clínicos.
Idiosincrasia (Idiosyncrasy)Sensibilidad individual peculiar a los efectosde un fármaco. Generalmente se debe a unaalteración de base genética, por ejemplo,una deficiencia enzimática. Es una de las re-acciones adversas de tipo B de los fármacos.
IMEAcrónimo de Índice de mortalidad estandari-zada. Véase Ajuste indirecto.
Incidencia (Incidence)Medida de frecuencia. Se refiere al númerode casos nuevos de una enfermedad en una
40
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

población definida durante un período detiempo determinado. Se expresa general-mente en forma de tasa, resultado de dividirel número de casos por la población en ries-go. En ocasiones se emplea la expresión tasade incidencia para referirse al mismo con-cepto. Dado que el término incidencia llevaimplícita la definición de tasa, resulta inade-cuado el empleo de la expresión ‘tasa de in-cidencia’. Véanse Densidad de incidencia,Incidencia acumulada y Prevalencia.
Incidencia acumulada (Cumulative incidence)Medida de frecuencia que se calcula divi-diendo el número de nuevos casos de enfer-medad por la población definida y multipli-cando por cien.
INDAcrónimo de Investigational New Drug. Es untérmino utilizado por la FDA y es equivalenteal Producto en fase de investigación clínica(PEI).
INDAAcrónimo de Investigational New Drug Appli-cation. Es un término utilizado por la FDA yse refiere a la solicitud para que pueda con-seguirse la denominación de IND. En Españaes equivalente a la solicitud de PEI.
Independencia (Independence)Cualidad por la que la aparición de un hechono puede predecirse de modo alguno a partirde la presentación de otro. Dos variables sonindependientes si la distribución de los valo-res de una no varía para cada uno de los va-lores de la otra. Es lo opuesto a asociación odependencia.
Indicador (Indicator)Variable susceptible de ser medida directa-mente. En medicina se utiliza como variableque refleja el estado de salud.
Índice de kappa (Kappa index)Medida del grado de concordancia no aleato-ria entre observadores o entre mediciones deuna misma variable. Si las mediciones sóloconcuerdan con la frecuencia del azar, el va-lor es cero; si las mediciones concuerdancon más frecuencia de lo que se esperaríapor azar, el valor de kappa es superior a ce-ro. Si la concordancia es completa, kappa esigual a 1. Véase Concordancia.
Indice de mortalidad estandarizadaVéase Ajuste indirecto.
Inferencia causal (Causation, Causality)Asociación que cumple con los siguientescriterios de Hill: magnitud de la asociación,consistencia, especificidad, temporalidad,gradiente biológico, plausibilidad, coheren-
cia, evidencia experimental y analogía (véasecada uno de estos términos). Puede ser queninguno de estos nueve criterios aporte evi-dencia indiscutible a favor o en contra de lahipótesis de causa-efecto, como tampoco escondición sine qua non que se cumplan to-dos ellos.
Inferencia estadística (Statistical inference)Generalización de los resultados obtenidosen una o más muestras a la población asu-miendo un cierto grado de incertidumbre.
Informe de experto (Expert report)Informe requerido por las autoridades sanita-rias encargadas de la regulación de los medi-camentos, que especifica de forma resumidalas evidencias disponibles de algunos de losaspectos de un medicamento. Puede ser fár-maco-toxicológico, químico-farmacéutico oclínico. El autor del informe debe ser un ex-perto en ese campo particular. Puede ser re-dactado por un empleado de la compañíafarmacéutica o por una persona externa.
Informe final (Final report)Descripción completa de un ensayo clínicouna vez terminado. Debe incluir una descrip-ción del material y métodos, los resultadosobtenidos, el análisis estadístico y la evalua-ción de los resultados. En los ensayos clíni-cos autorizados en España, es obligatorio en-viar una copia del informe final a las autori-dades sanitarias y al CEIC que aprobó elensayo. Existe una propuesta guía de laCPMP e ICH sobre su estructura y contenido(Note for guidance on structure and contentof clinical study reports). En su acepción ge-neral, documento que describe de formapormenorizada una investigación y sus resul-tados
INNAcrónimo de International NonproprietaryName. Véase Denominación común interna-cional.
Inspección (Inspection, Audit)Auditoría realizada de forma oficial por lasautoridades sanitarias competentes. VéaseAuditoría.
Interacción (Interaction)Actuación interdependiente de dos o más va-riables para producir o impedir un efecto.Por ejemplo, según los resultados de un es-tudio epidemiológico, el riesgo relativo deque se produzca un accidente vascular cere-bral en mujeres que toman anticonceptivosorales es de 2,1, y en mujeres con hiperten-sión grave es de 5,9. Cuando coinciden am-bos factores de riesgo -anticonceptivos orales
41
GLOSARIO

e hipertensión-, el riesgo relativo es de 12,6,superior a la simple suma de ambos riesgos.Por tanto, existiría una interacción positiva osinergismo entre ambos factores.
Interacción farmacológica (Pharmacological in-teraction, Drug interaction)
Cambios producidos en las propiedades far-macocinéticas o farmacodinámicas de unfármaco provocados por la administración deotro.
IntermedioVéase Análisis intermedio.
Intervalo de confianza (IC) (Confidence inter-val, CI)
Margen de valores dentro de los cuales cabeesperar el valor real de la población con unadeterminada probabilidad. La probabilidadespecificada se denomina nivel de confianza,y los puntos extremos del intervalo de con-fianza, límites de confianza (superior e infe-rior). Se utilizan en general intervalos de con-fianza con una probabilidad del 95%, aun-que a veces se utilizan del 90% o del 99%.
Intervención (Intervention)Cualquier acción (preventiva, diagnóstica, te-rapéutica) aplicada con el fin de modificar elcurso de un proceso, por ejemplo, una enfer-medad, una situación fisiológica o un hábito.
Investigación (Research)Realización de actividades intelectuales y ex-perimentales de modo sistemático con elpropósito de aumentar los conocimientos so-bre una determinada materia.
Investigación básica (Basic research)Investigación que se realiza en animales o enpreparaciones humanas in vitro. También seconoce con este nombre a la que tiene porobjeto conocer los mecanismos de acción olos efectos de los fármacos en grupos de vo-luntarios o pacientes, sin finalidad terapéuti-ca. En sentido amplio, investigación funda-mental para el progreso del conocimientocientífico, cuya aplicación práctica inmediatano es un objetivo concreto.
Investigación clínica (Clinical research)Investigación que se realiza en humanos, yasean voluntarios sanos o pacientes.
Investigación en servicios sanitarios (Healthservices research)
Investigación que evalúa los servicios sanita-rios integrando análisis epidemiológicos, so-ciológicos y económicos. Los tres componen-tes clásicos que incluye la investigación delos servicios sanitarios son: la evaluación dela estructura (recursos), la evaluación delproceso (dónde, cómo y por quién es provis-
ta la atención sanitaria), y evaluación del re-sultado (beneficios mesurables de salud).
Investigación epidemiológica (Epidemiologicalresearch)
Investigación que permite la cuantificaciónde la magnitud de la relación entre la exposi-ción y la enfermedad en seres humanos, yque ofrece la posibilidad de alterar el riesgo através de la intervención.
Investigación preclínica (Preclinical research)Investigación que se realiza en animales,normalmente previa al inicio del estudio enhumanos, aunque también puede ser simul-tánea (en especial la toxicológica). Proporcio-na datos sobre la farmacodinamia, la farma-cocinética y la toxicidad de los fármacos enanimales, para poder extrapolarla a los hu-manos.
Investigación y desarrollo (I + D) (Researchand development, R & D)
En la industria, dos procesos íntimamente re-lacionados por los que nuevos productos onuevas formas de viejos productos son opti-mizados mediante la innovación tecnológicapara su potencial comercialización.
Investigador (Investigator, Researcher)Persona que se dedica a la investigación oque participa en el diseño y/o la ejecución deuna investigación. Puede ser el investigadorprincipal o un colaborador. Véase Investiga-dor principal e Investigador colaborador.
Investigador colaborador (Subinvestigator)Persona que participa en el desarrollo de lainvestigación sin ser el responsable directo.Sin embargo, su participación debe estable-cerse en el protocolo y debe comprometersea seguir los postulados éticos y mantener laconfidencialidad antes de iniciar el estudio.En algunos documentos se le denomina su-binvestigador. Véanse Investigador e Investi-gador principal.
Investigador principal (Principal investigator)Persona que lidera o que es el responsablede una investigación. Referido al ensayo clí-nico (RD 561/1993 art. 16), el investigadorprincipal es quien dirige su realización prác-tica y firma junto al promotor la solicitud deensayo clínico, corresponsabilizándose conél. El investigador principal será un profesio-nal sanitario suficientemente calificado paraevaluar la respuesta a la sustancia o medica-mento objeto de estudio, con experiencia eninvestigación y en el área clínica del ensayopropuesto y con reconocidos criterios de éti-ca e integridad profesional. Son obligacionesdel investigador principal:
42
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

a) estar de acuerdo y firmar junto con el pro-motor el protocolo del ensayob) conocer a fondo las propiedades de losmedicamentosc) obtener el consentimiento informado delos sujetos antes de su inclusión en el ensayod) recoger, registrar y notificar los datos deforma correctae) notificar inmediatamente los aconteci-mientos graves o inesperados al promotorf) garantizar que todas las personas implicadasrespetarán la confidencialidad de cualquier in-formación acerca de los sujetos del ensayog) informar regularmente al Comite Ético deInvestigación Clínica de la marcha del ensayoh) corresponsabilizarse con el promotor de laelaboración del informe final del ensayo,dando su acuerdo al mismo con su firma.Véanse Investigador e Investigador colaborador.
IRBAcrónimo de Institutional Review Board. Véa-se Consejo Institucional de Revisión.
Ji al cuadrado (Chi-square)Habitualmente se simboliza por χ2. VéansePrueba de χ2 y Distribución de χ2.
Kaplan-Meier, métodoVéase Método de Kaplan-Meier.
KappaVéase Índice de kappa.
Lasagna, ley de (Lasagna’s law)Se refiere al hecho de que sólo uno de cadadiez de los pacientes que un investigadorcree que son candidatos a participar en unensayo clínico acaba siendo incluido. Postulaque la incidencia de la enfermedad en estu-dio se reduciría hasta el 10% del valor habi-tual en el momento del reclutamiento, regre-sando al valor anterior al finalizar el períodode reclutamiento. Se debe al optimismo dealgunos clínicos cuando se les plantea la po-sibilidad de reclutar a sus pacientes en unensayo clínico. Louis Lasagna puede consi-derarse como uno de los padres de la Far-macología Clínica.
LavadoVéase Período de lavado.
Límite de confianza (Confidence limit)Cada uno de los dos valores que constituyenlos límites superior e inferior de un intervalode confianza. Véase Intervalo de confianza.
Log-rankVéase Prueba de log-rank.
Magnitud de la asociación (Strength of the as-sociation)
Grado de asociación observada entre una ex-posición y el riesgo de una enfermedad.
Cuanto mayor sea la asociación, es decir,cuanto mayor sea el grado de aumento o dis-minución del riesgo observado, menos pro-bable será que la relación se deba al efectode un factor de confusión o de un factor in-sospechado. Por ejemplo, los sujetos que fu-man más de 20 cigarrillos al día tienen unriesgo de mortalidad por cáncer de pulmón20 veces superior al de los no fumadores, yesta asociación permanece incluso despuésde ajustarla por otros factores de confusióncomo la edad, el sexo o el consumo de alco-hol. Véase Inferencia causal.
MANOVAAcrónimo de Multivariate analysis ofvariance. Véase Análisis de la varianza multi-variado.
Manual de procedimientos (Manual of proce-dures)
Documento que describe los procedimientosutilizados en un centro o un grupo de cen-tros en una investigación específica. Tienecomo objeto armonizar un procedimiento pa-ra que sus resultados puedan agruparse confinalidad estadística.
Manual del investigador (Investigator’s brochu-re)
Documento que resume toda la informaciónrelevante conocida hasta la fecha sobre unfármaco en el momento de iniciar un ensayoclínico. Comprende datos químicos, biofar-macéuticos, farmacológicos y toxicológicosen animales, así como los resultados de en-sayos cl ínicos previos. Según el RD561/1993, el manual del investigador debeser una versión actualizada de la informaciónpreclínica y clínica relevante para el ensayosobre los productos en estudio.
Marcador de riesgo (Risk marker)Característica que, según la evidencia epide-miológica, se asocia con un aumento de laprobabilidad de padecer una enfermedad,aunque no se trata necesariamente de unaasociación causal. Por ejemplo, la asociaciónobservada entre la calvicie y la enfermedadcoronaria. También denominado señal deriesgo. Véase Factor de riesgo.
Media aritmética (Mean, average)Parámetro de tendencia central de un con-junto de valores de una variable, que resultade la suma de estos valores dividida por elnúmero de observaciones. También se le co-noce como promedio.
Media geométrica (Geometric mean)Parámetro de tendencia central de un con-junto de valores de una variable, que resulta
43
GLOSARIO

de la raíz enésima del producto de las obser-vaciones de la variable siendo n el tamañode la muestra.
Mediana (Median)Parámetro de tendencia central que es igualal valor de la variable que divide una distri-bución de frecuencias o de probabilidadesen dos partes iguales. En distribuciones de npar, la mediana sería la media aritmética delos dos valores centrales.
Medicamento (Drug, medicine)Según la Ley del Medicamento, 25/1990 art.8.1, es toda sustancia medicinal y sus aso-ciaciones o combinaciones destinadas a suutilización en las personas o en los animalesque se presenta dotada de propiedades paraprevenir, diagnosticar, tratar, aliviar o curarenfermedades o dolencias o para afectar afunciones corporales o al estado mental.También se consideran medicamentos lassustancias medicinales o sus combinacionesque pueden ser administradas a personas oanimales con cualquiera de estos fines, aun-que se ofrezcan sin explicita referencia aellos. En ocasiones se considera sinónimo defármaco. Véanse Fármaco y Sustancia medi-cinal.
Medicamento genérico (Generic drug)Especialidad farmacéutica que se vende sinmarca, constando sólo la denominación co-mún internacional (DCI) del principio activoque contiene. En España debe constar ladenominación oficial española (DOE) que esequivalente a la DCI en castellano. Puedecomercializarse cuando ha caducado la pa-tente del producto original. Contiene los mis-mos principios activos que la especialidadcon marca original patentada y compite conla misma en base a su precio más económi-co.Según la OMS, es un producto farmacéuticode origen múltiple e intercambiable al haberdemostrado bioequivalencia (interchangea-ble multi-source pharmaceutical products).
Medicamento huérfano (Orphan drug)Fármaco terapéuticamente útil de difícil de-sarrollo o comercialización por emplearse enel tratamiento de enfermedades muy pocofrecuentes, y que por este motivo puede ca-recer de atractivo para las compañías farma-céuticas. Algunas producen fármacos huér-fanos mediante convenios con las autorida-des sanitarias, subvencionando éstas deforma directa o indirecta esta actividad. Porejemplo, la l-carnitina para el déficit de carni-tina.
Medicina basada en la evidencia (Evidence-ba-sed medicine, EBM)
Utilización consciente y juiciosa de la mejorevidencia proveniente de la investigación clí-nica en el manejo individual de los pacien-tes. Los pasos recomendados a seguir son:elaboración de preguntas que puedan sercontestadas, localización de forma eficientede la mejor evidencia con que responderlas,evaluación crítica de la validez y utilidadpráctica de la evidencia disponible, aplica-ción de los resultados de dicha evaluaciónen la práctica clínica y evaluación de la ac-tuación.
Megaensayo (Megatrial)Ensayo clínico aleatorizado, de muestra muygrande, necesario para demostrar una mag-nitud de efecto pequeño. Aunque no existeuna definición precisa de cuál es el númerode pacientes que debe incluir un megaensa-yo, se consideran así los ensayos clínicoscon más de 10.000 pacientes. Ejemplos demegaensayo son los estudios GISSI, GUSTOo ISIS.
Metaanálisis (Meta-analysis)Análisis estadístico de una combinación delos resultados de varios ensayos clínicos me-diante una metodología estandarizada. Per-mite en ocasiones establecer la eficacia deun tratamiento cuando los ensayos clínicosindividuales tienen pocos pacientes, o los re-sultados son contradictorios. Pueden usarsedirectamente los resultados de los estudiospublicados o bien partir de los datos indivi-duales. Esta técnica también puede aplicarsecon los estudios observacionales. Algunosautores proponen la denominación de meta-nálisis.
Metaanálisis acumulativo (Cumulative meta-analysis)
Se refiere a la práctica de metaanálisis ca-da vez que los resultados de un nuevo en-sayo se publican para así obtener cronoló-gicamente un efecto acumulativo. Porejemplo, mediante este tipo de metaanálisisse observó retrospectivamente que la es-treptocinasa intravenosa era ya eficaz en eltratamiento del infarto agudo de miocardiomuchos años antes de su aprobación por laFDA.
Método actuarial (Actuarial method)Método utilizado para calcular la incidenciacuando la duración del tiempo de seguimien-to de los sujetos de un estudio varía sustan-cialmente. También se conoce como tablasde vida o tablas de mortalidad.
44
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Método bayesiano (Bayesian method)Método que emplea el teorema de Bayes.Véanse Análisis de Bayes y Teorema de Ba-yes.
Método de Kaplan-Meier (Kaplan-Meier met-hod)
Método no paramétrico empleado para esti-mar tasas de eventos en el seguimiento me-diante la utilización de probabilidades condi-cionadas. Es especialmente útil en los estu-dios en que los individuos son incluidos enun período de tiempo y seguidos hasta unafecha determinada. También se denominamétodo del producto límite.
Minimización (Minimisation)Método utilizado en el ensayo clínico paracontrolar las posibles desigualdades respectoa los factores pronósticos de los pacientesasignados a un grupo u otro. Por ejemplo, sila edad es un factor pronóstico, y un grupodel estudio tiene más sujetos ancianos que elotro, la estrategia de la asignación deberá serde tal forma que los próximos sujetos ancia-nos tengan mayor probabilidad de ser asig-nados aleatoriamente al grupo con menossujetos de edad avanzada.
Moda (Mode)Parámetro de tendencia central que es igualal valor más frecuente de la distribución deuna variable.
Monitor (Monitor)Profesional capacitado con la necesaria com-petencia clínica elegido por el promotor, quese encarga del seguimiento directo de la rea-lización de un ensayo. Sirve de vínculo entreel promotor y el investigador principal cuan-do estas condiciones no concurren en lamisma persona (RD 561/1993 art. 15). Elmonitor es el encargado de seleccionar a losinvestigadores y de visitarlos periódicamentepara asegurarse de que el estudio se realizasegún lo especificado en el protocolo. Estasvisitas finalizan con la redacción de un infor-me de monitorización. Véase Asistente de in-vestigación clínica.
Monitorización (Monitoring, surveillance)Supervisión y seguimiento de un ensayo clí-nico por parte del monitor. Se realiza me-diante visitas al centro en el que se realiza elensayo clínico; en ellas se realiza un inter-cambio de opiniones con el investigador so-bre los diferentes aspectos del ensayo y, enocasiones, se verifica o completa la informa-ción de los cuadernos de recogida de datos.
Montecarlo, simulación deVéase Simulación de Montecarlo.
Muestra (Sample)Parte de una población que se utiliza paraobtener información sobre características dela misma. Véase Muestra representativa.
Muestra aleatoria (Random sample)Muestra probabilística en la que todos loselementos de la población tienen por azar lamisma probabilidad de ser seleccionados.
Muestra estratificada (Stratified sample)Muestra seleccionada aleatoriamente a partirde diferentes subgrupos de la población (porejemplo, edad, sexo, clase socioeconómica),de tal manera que la proporción de cada unode los subgrupos de la muestra es la mismaque la proporción en la población general.
Muestra para investigación clínica (Study me-dication)
Muestras de medicamentos o de productosen fase de investigación clínica para su utili-zación en ensayos clínicos. Serán proporcio-nadas gratuitamente por el promotor, aunquepuede haber otras vías de suministro. Lasmuestras sobrantes se devolverán al promotoruna vez finalizado el período de tratamientodel ensayo clínico. Estarán garantizadas porel Director Técnico responsable en cuanto acalidad de fabricación. Cuando se le solicite,deberá remitir a las autoridades competentesmuestras de los productos que serán utiliza-dos en el ensayo clínico. La distribución al in-vestigador se realizará a través del servicio defarmacia del hospital donde se realice la in-vestigación. Si el ensayo se realiza en medioextrahospitalario, las obligaciones podrán serasumidas por los servicios farmacéuticos delas estructuras de atención primaria, y extra-ordinariamente por el investigador principal.Irán envasadas y acondicionadas convenien-temente. Su etiquetado o rotulación permitirá,en cualquier momento, su perfecta identifica-ción (RD 561/1993 art. 18).
Muestra representativa (Representativesample)
Muestra que permite estimar los parámetrosde la población con márgenes de error acep-tables, de acuerdo con el objetivo de la in-vestigación.
Multicéntrico (Multicenter)En un sentido amplio, cualquier estudio quese realice en dos o más centros con un pro-tocolo común. Véanse Comité de Expertos,Ensayo clínico multicéntrico y Estudio multi-céntrico.
nEn estadística, símbolo utilizado para definir elnúmero de pacientes incluidos en un estudio.
45
GLOSARIO

NCEAcrónimo de New Chemical Entity. VéaseNueva entidad química.
NDAAcrónimo de New Drug Application. VéaseSolicitud de autorización de especialidad far-macéutica.
NNHAcrónimo de Number needed to be treatedto harm. Véase Número necesario de pacien-tes a tratar para producir un efecto perjudi-cial.
NNTAcrónimo de Number needed to treat/to betreated. Véase Número necesario de pacien-tes a tratar para evitar un caso y Reducciónabsoluta de riesgo.
No-maleficencia (Non-maleficence)El Diccionario de la Lengua Española definemaleficencia como Hábito o costumbre dehacer el mal. Constituye un precepto bioéticoclásico que obliga a no hacer nada malo a al-guien, aunque lo pidiera. Debe considerarsecomo maleficente un ensayo clínico que ca-rezca de validez científica, porque la hipóte-sis no es plausible o porque el diseño es me-todológicamente incorrecto. Véase Principiosbioéticos.
Nocebo (Nocebo)Término empleado para denominar a las re-acciones adversas que se asocian a la admi-nistración del placebo.
Normalización (Standardization)Procedimiento que permite igualar variables,parámetros o medidas a valores similares.Véase Ajuste.
Normas de buena práctica clínica (BPC) (GoodClinical Practice, GCP)
Aquellas según las cuales los ensayos clíni-cos son diseñados, realizados y comunica-dos, de modo que se asegure que los datosson fiables y que se protegen los derechos yla integridad de los sujetos, manteniendo laconfidencialidad de sus datos. (Real Decreto561/1993 art. 17). Su correcta aplicaciónprecisa de la existencia de los Procedimien-tos Normalizados de Trabajo. En la actuali-dad existe una guía de buena práctica clínicaelaborada por la CPMP/ICH. También se lasconoce por el acrónimo BPC.
Normas de buena práctica de fabricación (BPF)(Good Manufacturing Practices, GMP)
Conjunto de reglas destinadas a asegurar laadecuada formulación y producción de losmedicamentos. Con frecuencia, se utilizan losacrónimos BPF o BPM para referirse a ellas.
Normas de buena práctica de laboratorio (BPL)(Good Laboratory Practice, GLP)
Conjunto de reglas destinadas a asegurar lacalidad, la veracidad y la reproducibilidad delos datos de los estudios de farmacología ytoxicología en animales. También se emple-an en análisis químico y en bioquímica clíni-ca. Son conocidas también por el acrónimoBPL. En España, se describen en parte en elRD 822/1993 y en el RD 2043/1994.
Normas de buena práctica de manufacturaVéase Normas de buena práctica de fabrica-ción.
Notificación espontánea de reacciones adversas(Spontaneous reporting of adverse reactions)
Método de registro de sospechas de reaccio-nes adversas a los medicamentos basado enla comunicación voluntaria por médicos, far-macéuticos o empresas farmacéuticas. Enmuchas ocasiones, se emplean formulariosnormalizados conocidos con el nombre detarjeta amarilla (Yellow card). En Españaexiste un programa específico de este tipoque incluye a casi todas las autonomías. Vé-ase Farmacovigilancia.
Nueva entidad química (New Chemical Entity,NCE)
Expresión empleada en los EE.UU. para de-nominar a los principios activos recientemen-te sintetizados en la confianza de que ten-drán actividad terapéutica. Se utiliza espe-cialmente para describir la documentacióncorrespondiente que se presenta por primeravez a las autoridades sanitarias.
Número aleatorio (Random number)Número generado o extraído a través de unproceso aleatorio. Véase Aleatorio.
Número necesario de pacientes a tratar paraevitar un caso (NNT) (Number needed totreat/to be treated, NNT)
Medida epidemiológica de eficacia clínicaque expresa el esfuerzo que hay que reali-zar para prevenir un caso de enfermedad ode muerte. Se calcula mediante el inversode la reducción absoluta del riesgo (1/RAR).Por ejemplo, se precisa tratar 6 pacientesdurante cinco años con ácido acetilsalicílicopara evitar un caso de muerte o de acciden-te cerebrovascular en pacientes que hanpadecido un accidente isquémico transitorio(véase la tabla de la página siguiente).
Número necesario de pacientes a tratar paraproducir un efecto perjudicial (Number neededto treat/to be treated to harm, NNH)
Medida epidemiológica que expresa el nú-mero de pacientes del grupo experimental a
46
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

los que se producirá un daño o efecto perju-dicial en una persona adicional cuando secompara con los pacientes que reciben eltratamiento control. Se calcula mediante elinverso del aumento absoluto del riesgo(1/AAR). Véase Aumento absoluto del riesgo.
Nuremberg, código de (Nuremberg code)Declaración de la comunidad científica inter-nacional realizada en 1947, en la que se es-tipularon por primera vez los postulados bá-sicos de la ética de la investigación clínica.Su espíritu se materializó en la Declaraciónde Helsinki (1964), revisada posteriormenteen las de Tokio (1975), Venecia (1983),Hong Kong (1989) y Sudáfrica (1997). VéaseHelsinki, declaración de.
Objetivo (Object, Objective)Según el Diccionario de la Lengua Española,Perteneciente o relativo al objeto en sí y no anuestro modo de pensar. En investigaciónclínica, definición de los fines que el estudiopretende alcanzar y, por tanto, la razón quejustifica su realización. Véase Objeto.
Objetivo principal (Primary objective)Objetivo más relevante del estudio; es únicoy condiciona la elección de la variable princi-pal, indispensable para la realización del es-tudio, y el cálculo del tamaño de la muestra.
Objetivo secundario (Secondary objective)Objetivo menos relevante que el principal; noes necesariamente único y generalmenteconsidera cualquier otra variable de interésrelevante, pero no indispensable, para la rea-lización del estudio.
Objeto (Objective)Según el Diccionario de la Lengua Española,Todo lo que puede ser materia de conoci-miento o sensibilidad de parte del sujeto, in-cluso este mismo y también Fin o intento aque se dirige o encamina una acción u ope-ración. Generalmente se emplea el términoobjetivo, aunque el Diccionario de la LenguaEspañola prefiere objeto como primera acep-ción. A lo largo de este glosario, se mantieneel vocablo objetivo porque, en opinión de losautores, su uso se encuentra sancionado porla práctica.
Observancia (Compliance)Para algunos autores este término deberíaemplearse en preferencia a cumplimientopara traducir compliance en su acepción re-lacionada con medicamentos. Véase Cumpli-miento.
Odds ratioVéase Razón de posibilidades.
ORAcrónimo de Odds Ratio. Véase Razón deposibilidades.
Orden de rangos (Rank order)Escala que ordena los elementos de un gru-po de mayor a menor según la magnitud delas observaciones asignando números a loselementos y despreciando las distancias ori-ginales entre éstos. Véase Rango.
Organización de investigación por contrato(Contract Research Organization, CRO)
Persona u organización (académica, comer-cial u otra) que es contratada por el promotor
47
GLOSARIO
TABLAEJEMPLOS DE UTILIDAD DE LOS NNT EN TRES ESTUDIOS DIFERENTES
Estudio y Variable Años detratamiento principal seguimiento IC IT RRR (%) RAR NNT NNT 5+Derivación Muertes 5 0,32 0,14 56% 0,18 6 6aorto-coronaria
Ácido Muerte 2,2 0,23 0,16 31% 0,07 14 6acetilsalicílico o AVCen AIT
Antihipertensivos Muerte, AVC 5,5 0,05 0,043 14% 0,007 143 157frente a placebo o IAMen PAD 90-109 mmHg
IC = Incidencia de la enfermedad en el grupo control (no tratamiento o placebo). IT = Incidencia de laenfermedad en el grupo expuesto al tratamiento. RRR = Reducción relativa del riesgo (1 - IT / IC).RAR = Reducción absoluta del riesgo (IC - IT). NNT = Número necesario de pacientes a tratar para evitar uncaso, según la duración del estudio (1 / RAR). NNT 5+ = Número necesario de pacientes a tratar para evitarun caso, ajustado a un período de 5 años. AIT = Accidente isquémico transitorio. AVC = Accidente vascularcerebral. IAM = Infarto agudo de miocardio. PAD = Presión arterial diastólica.

de un estudio para asumir alguna de las ta-reas, funciones u obligaciones en un ensayoclínico. El contrato puede incluir, por ejem-plo, la realización de la monitorización delensayo clínico o el análisis estadístico.
p, valor de probabilidad (p, Probability value)Probabilidad de que el valor estadístico deltest sea igual o más extremo que el observa-do si la hipótesis nula fuera cierta. Arbitraria-mente se han establecido los niveles de con-fianza en 0,05 o 0,01, de tal manera que va-lores de p inferiores a esos niveles (según loque establezca el investigador) se consideranestadísticamente significativos, o lo que es lomismo, es poco probable que las diferenciasobservadas sean debidas al azar.
Paciente ambulatorio (Outpatient)Paciente que no se encuentra ingresado encentros hospitalarios y que es visitado en lasconsultas externas del centro sanitario o enel ámbito de la asistencia primaria.
Paciente elegible (Eligible patient)Paciente que cumple los criterios de inclu-sión establecidos e incumple los de exclu-sión. Es sinónimo de paciente eligible aun-que el Diccionario de la Lengua Españolaconsidera este término como anticuado.
Paradoja de Simpson (Simpson’s paradox)Situación en la que la presencia de un factorde confusión altera notablemente la direc-ción de una asociación entre una exposicióny un resultado, llegando incluso a revertirla.Para algunos, la paradoja no es tal, ya queconstituye la consecuencia inevitable de noconsiderar la presencia de un factor de con-fusión. Por ejemplo, en un estudio sobremortalidad y diabetes se observó que fallecíasólo el 29% de los pacientes con diabetes in-sulinodependiente comparado con el 40%de los que presentaban diabetes no insulino-dependiente. Este análisis no considerabaque la última se desarrolla normalmente des-pués de los 40 años de edad. Cuando los pa-cientes fueron separados en función de laedad en dos grupos (≤ 40 y >40), se observóque fallecía en ambos grupos una proporciónmenor de pacientes con diabetes no insuli-nodependiente que de los que presentabandiabetes insulinodependiente.
Parámetro (Parameter)Según el Diccionario de la Lengua Española,Variable que, en una familia de elementos,sirve para identificar cada uno de ellos me-diante su valor numérico. En estadística yepidemiología, característica medible de unamuestra o de una población. El término pará-
metro se utiliza de forma inadecuada, ya seacomo sinónimo de variable o también parareferirse a cualquier característica, factor ocosa medible. Ejemplo de parámetros detendencia central son la media, la moda y lamediana, mientras que ejemplos de paráme-tros de dispersión son la desviación estándary el error estándar de la media.
Patrón de comparación (Criterion standard)Medida reconocida que se emplea para esta-blecer las diferencias y semejanzas del pro-cedimiento estudiado.
Patrón de oro (Gold standard)Patrón de comparación reconocido que per-mite determinar la bondad de un procedi-miento. Por ejemplo, en los ensayos clínicoscon analgésicos menores, el patrón de oro esel ácido acetilsalicílico, mientras que conanalgésicos mayores, es la morfina. En prue-bas diagnósticas, es la base de comparaciónestablecida y aceptada para determinar lasensibilidad y especificidad de un diagnósti-co. Por ejemplo, la flebografía de extremida-des inferiores es la prueba considerada comopatrón de oro en el diagnóstico de la trombo-sis venosa profunda. También denominadacriterio estándar o criterio de referencia.
PEIAcrónimo de Producto en fase de investiga-ción clínica.
Percentil (Percentile)Valor que divide la distribución normal encien partes iguales. También valor por deba-jo o encima del cual se encuentran un con-junto de observaciones. Tiene la ventaja,frente a otras medidas de dispersión, de evi-tar la influencia de los valores extremos en lainterpretación de los resultados. Se empleaespecialmente para definir los límites de dis-persión de las medianas. El percentil cin-cuenta se corresponde con la mediana.
Pérdidas postaleatorización (Post-randomiza-tion losses)
Pacientes que dejan el estudio tras ser asig-nados a uno de los grupos de éste. Incluyelos abandonos y las retiradas.
Pérdidas postrandomizaciónAnglicismo debido al empleo incorrecto deltérmino randomización. Véase Pérdidas pos-taleatorización.
Pérdidas prealeatorización (Pre-randomizationlosses)
Pacientes elegibles para participar en el es-tudio pero que finalmente no son incluidospor diferentes motivos (por ejemplo, falta deconsentimiento).
48
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Pérdidas prerandomizaciónAnglicismo debido al empleo incorrecto deltérmino randomización. Véase Pérdidas pre-aleatorización.
Período de blanqueoVéase Período de lavado.
Período de lavado (Wash-out period)Tiempo necesario para excluir cualquierefecto residual de un tratamiento previo du-rante el que se administra placebo o ningúntratamiento. Los períodos de lavado puedenrealizarse al inicio del estudio para descartarla influencia de la terapéutica habitual delpaciente, o tras finalizar uno de los trata-mientos en los estudios de diseño cruzado.
Período de preinclusión (Run-in period)Tiempo en el que los pacientes elegibles re-ciben placebo o un tratamiento activo a finde decidir sobre su idoneidad para ser inclui-dos en el estudio. En ocasiones, también seemplea, por ejemplo, para establecer si lospacientes cumplen con el tratamiento y conlas pautas prescritas, si responden al place-bo, o para obtener una medición basal másválida.
Permutación aleatoriaVéase Asignación aleatoria por bloques.
Piloto, estudioVéase Estudio piloto.
Placebo (Placebo)Según el Diccionario de la Lengua Españolaes Sustancia que, careciendo por sí mismade acción terapéutica, produce algún efectocurativo en el enfermo si éste la recibe con-vencido de que esa sustancia posee real-mente tal acción. También se emplea paradenominar al grupo de pacientes que recibeplacebo. En ensayos clínicos se utiliza con elobjeto de discernir los efectos farmacológicosreales de las expectativas asociadas al trata-miento o de las fluctuaciones de la enferme-dad. En estas situaciones en las que se utili-za el placebo como control de la evaluacióncientífica, algunos autores prefieren emplearel término imitación o simulación (dummy).Véanse Efecto placebo y Sensibilidad al pla-cebo.
Plausibilidad (Plausibility of causal relationship)El Diccionario de la Lengua Española defineplausible como atendible, admisible, reco-mendable. En epidemiología, término emple-ado cuando la asociación encontrada esacorde con los conocimientos experimenta-les. Para algunos autores, plausibilidad es si-nónimo de admisibilidad. Véase Inferenciacausal.
PMSAcrónimo de Postmarketing Surveillance. Vé-ase Estudio de postcomercialización.
PNTAcrónimo de Procedimientos Normalizadosde Trabajo.
Población accesible (Accessible population)Subconjunto de la población diana disponi-ble para ser incluida en el estudio y definidapor las características geográficas y tempora-les del ámbito del estudio.
Población de referenciaVéase Población diana.
Población del estudio (Study population)Grupo de individuos sobre los que se realizael estudio, ya que cumplen los criterios deselección del protocolo.
Población destinatariaVéase Población diana.
Población diana (Target population)Grupo de individuos a los que se pretendegeneralizar los resultados del estudio. El ad-jetivo diana no tiene ninguna acepción en es-te sentido en el Diccionario de la Lengua Es-pañola. Algunos autores prefieren utilizar laexpresión población objeto, pero en españolobjeto es sustantivo y no adjetivo, por lo quedebería emplearse la expresión poblaciónobjetivo. También podría emplearse pobla-ción blanco ya que el Diccionario de la Len-gua Española acepta la acepción Fin u objetoa que se dirigen deseos o acciones. Sin em-bargo, el amplio empleo de la expresión po-blación diana aconseja su reconocimiento ydesaconseja el empleo de otras traduccionesque podrían resultar artificiales y, probable-mente, de escaso reconocimiento y acepta-ción. También denominada población objeti-vo, de referencia o destinataria.
Población objetivoVéase Población diana.
Poder estadístico (Power)Anglicismo debido al empleo incorrecto deltérmino poder. Véase Potencia estadística.
Post hoc, ergo propter hocExpresión latina que puede traducirse porDespués de esto, por lo tanto debido a esto.Se refiere a una falacia de interpretación deresultados en la que la sucesión temporal seconsidera como evidencia de asociacióncausal.
Potencia estadística (Statistical power)Probabilidad de detectar una diferencia esta-dística entre los grupos evaluados cuando re-almente existe. Algunos autores la definencomo la probabilidad de rechazar la hipótesis
49
GLOSARIO

nula cuando ésta es falsa. Se expresa como1-β, y generalmente se acepta un valor supe-rior o igual a 0,8 (80%). En ocasiones, tam-bién se emplea el anglicismo Poder estadísti-co para nombrar el mismo concepto. VéaseError β.
Pragmático, ensayo clínicoVéase Ensayo clínico pragmático.
Precisión (Accuracy, Reliability, Reproducibi-lity)
Grado en el que una medida se realiza sinerror aleatorio y también grado de concor-dancia entre los valores medidos y los verda-deros. Sinónimo de repetibilidad, fiabilidad yconfiabilidad. Se refiere a si un instrumentoestá midiendo algo de forma reproducible. Lafalta de precisión es debida a un error aleato-rio y esencialmente atribuible a la variaciónmuestral, que depende del tamaño muestraly de las características estadísticas del esti-mador (varianza) (Fig. 10). Véanse Exactitudy Validez.
Preferencia de cifrasVéase Preferencia de números.
Preferencia de dígitosVéase Preferencia de números.
Preferencia de números (Digit preference,Number preference)
Error inconsciente que consiste en escogerpreferentemente números determinados (co-mo ‘0’ o ‘5’) al realizar una medida. Puede
evitarse comunicando al observador el riesgoen que puede incurrir. También se utiliza eltérmino preferencia de cifras, dígitos o nú-meros dígitos.
Preferencia entre dos períodos de tratamiento(Preference between two treatment periods)
Forma de medida de la eficacia. Cuando dostratamientos se administran sucesivamenteal mismo paciente, se le puede preguntarqué tratamiento de los que ha recibido pre-fiere, y comparar el número de preferenciaspara cada tratamiento.
Prevalencia (Prevalence)Medida de frecuencia que define el númerode individuos que presenta una determinadacaracterística o enfermedad en una pobla-ción (o en muestra representativa) y en unperíodo de tiempo determinado. Se expresaen forma de porcentaje. En ocasiones se em-plea la expresión tasa de prevalencia para re-ferirse al mismo concepto. Dado que el tér-mino prevalencia lleva implícito la definiciónde tasa, resulta inadecuado el empleo de laexpresión ‘tasa de prevalencia’. Véanse Inci-dencia y Estudio Transversal.
Prevención (Prevention)Cualquier intervención que reduzca el riesgode que una enfermedad o trastorno afecte aun individuo, que interrumpa o detenga suprogreso o evite la muerte.
Prevención primaria (Primary prevention)Cualquier intervención dirigida a individuossanos o sin la enfermedad que se pretendeprevenir para evitar la aparición de la misma.Ejemplos de prevención primaria son las va-cunaciones, la modificación de factores deriesgo como el tabaco para evitar el cáncer ola utilización del ácido acetilsalicílico para pre-venir el infarto de miocardio en sujetos sanos.
Prevención primordial (Primordial prevention)Cualquier intervención dirigida a evitar el in-cremento de la prevalencia de los hábitosnocivos o los factores de riesgo antes de queéstos se produzcan, como, por ejemplo, evi-tar el inicio del consumo del tabaco, o reco-mendar una dieta saludable para evitar laobesidad o la hipercolesterolemia. Este tér-mino ha sido acuñado por algunos autorespara diferenciarlo de la prevención primaria,sobre todo en el campo de las enfermedadescrónicas.
Prevención secundaria (Secondary Prevention)Desde un punto de vista diagnóstico, inter-venciones dirigidas a detectar precozmenteuna enfermedad, como, por ejemplo, la utili-zación de la mamografía para la detección del
50
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA
Precisasy válidas
Imprecisasy válidas
Precisasy no válidas
Imprecisasy no válidas
Figura 10. Comparación de validez y precisión.

cáncer de mama o la prueba de Papanicolaupara el cáncer de cérvix. Desde un punto devista terapéutico, se refiere a la modificacióny control de los factores de riesgo de una en-fermedad cuando ésta ya se ha producidopara evitar su progresión o la muerte. Ejemplode ésta última definición sería el control de lahipercolesterolemia y la abstención del taba-co en los pacientes que han padecido un epi-sodio de infarto agudo de miocardio.
Prevención terciaria (Tertiary prevention)Intervención preventiva en pacientes que yahan padecido una enfermedad relacionadacon la rehabilitación y la mejora de la calidadde vida. Un ejemplo de este tipo de preven-ción sería el tratamiento fisioterápico y reha-bilitador en pacientes que han padecido unaccidente vascular cerebral.
Principios bioéticos (Ethical standards)Normas destinadas a proteger a los sujetosparticipantes en proyectos de investigación yrecogidas por primera vez en el Informe Bel-mont (1978). Son los principios de autono-mía, beneficencia y justicia. Algunos autoresañaden el de no-maleficencia, como una va-riedad del de beneficencia con identidadpropia. Véanse Autonomía, Beneficencia,Justicia y No-maleficencia.
Probabilidad (Probability)Frecuencia relativa de aparición de un acon-tecimiento en una secuencia de n eventosseleccionados al azar y tendiendo al infinito.Se calcula dividiendo la frecuencia de even-tos por n. También es la medida del gradode creencia de una hipótesis o afirmaciónque va de cero a uno.
Probabilidad �Véase Error α.
Probabilidad �Véase Error β.
Probit (Probit)Transformación empleada para representarlinealmente curvas dosis-respuesta cuánti-cas. También se denomina unidad de proba-bilidad.
Procedimientos normalizados de trabajo (PNT)(Standard Operating Procedures, SOP)
Según el RD 561/1993 art. 45, Las normasde buena práctica clínica exigen la existenciade unos procedimientos normalizados de tra-bajo (PNT) que indiquen de forma detalladala conducta a seguir en cada uno de los as-pectos relacionados con la organización, rea-lización, recopilación de datos, documenta-ción y verificación de los ensayos clínicos. Esresponsabilidad del promotor establecerlos y
garantizar que su conocimiento y puesta enpráctica sean obligados para todos aquellosque participen en el ensayo clínico, especial-mente para el monitor del ensayo, antes deiniciar éste. Habitualmente consisten en unaserie de instrucciones detalladas por escritopara la realización adecuada y eficiente delensayo clínico. También se las conoce por elacrónimo de PNT. Los aspectos que, comomínimo, deben regularse en los PNT segúnel RD 561/1993 son identificación, califica-ción e idoneidad del investigador y centro,archivo de la documentación, procedimien-tos de monitorización, regulación del sumi-nistro y dispensación de la medicación, noti-ficación de acontecimientos adversos gravese inesperados e información a los sujetos yobtención del consentimiento informado.
Producto en fase de investigación clínica (PEI)(Investigational new drug, IND)
Según el RD 561/1993 art. 9, es aquel queha sido calificado como tal por la DirecciónGeneral de Farmacia y Productos Sanitariosy se dedica únicamente a ser utilizado, porexpertos calificados por su formación científi-ca y experiencia para la investigación, enpersonas para valorar su seguridad yeficacia. También se le conoce por el acróni-mo PEI. Su definición es similar al IND o alCTX en los EE.UU. y el Reino Unido, respec-tivamente.
PromedioVéase Media aritmética.
Promoción de la salud (Health Promotion)Proceso que permite capacitar a la poblaciónen el control y la mejora de la salud, utilizan-do métodos de educación sanitaria y cambiosorganizativos, económicos y ambientales.
Promotor (Sponsor)Según el RD 561/1993 art. 14, Es promotordel ensayo clínico la persona física o jurídicaque tiene interés en su realización, firma lassolicitudes de autorización dirigidas al Comi-té Ético de Investigación Clínica o a la Direc-ción General de Farmacia y Productos Sani-tarios y se responsabiliza de él, incluyendosu organización, comienzo y financiación. Elcitado Decreto especifica las obligaciones delpromotor. Algunos autores prefieren el térmi-no patrocinador.
Prospectivo, estudioVéase Estudio prospectivo.
Protocolo (Protocol)Según el RD 561/1993 art. 8, Documentoque establece la razón de ser de un estudio,sus objetivos, diseño, metodología y análisis
51
GLOSARIO

previsto de sus resultados, así como las con-diciones bajo las que se realizará y desarro-llará el ensayo. En general se denomina pro-tocolo al documento en el que se describentodos los aspectos que explican cómo se rea-liza el estudio. Es, por tanto, una herramientade trabajo imprescindible que tiene dos fun-ciones principales: describir la estructuracientífica del proyecto y especificar detalla-damente las instrucciones para el personalinvolucrado. El mencionado decreto describecómo debe ser la estructura y el contenidode un protocolo de ensayo clínico con medi-camentos.
Protocolo clínico (Clinical protocol, Clinicalguideline, Clinical practice guideline)
Documento en el que se detalla el procedi-miento que debe seguirse para el diagnóstico otratamiento de una enfermedad determinada.
Prueba bilateral (Two-tailed test or Two-sidedtest)
Referido al error de tipo I (riesgo α), suponela asunción de la probabilidad de error en losdos sentidos, es decir que A > B o que B >A. Por ejemplo, cuando se comparan dostratamientos en un ensayo clínico, si a priorise desconoce si un tratamiento es mejor queotro, la prueba estadística será más exigenteal tener en cuenta ambas posibilidades.También se llama prueba de dos colas.
Prueba de dos colasVéase Prueba bilateral.
Prueba de �2 (Chi-square test)Prueba estadística que se aplica para compa-rar dos o más proporciones cuando se tratade grupos independientes. También se la de-nomina prueba de ji al cuadrado de Pearson.Para las tablas de dos por dos se considerauna prueba estadística aproximada. VéanseDistribución de χ2 y Tabla de dos por dos.
Prueba de la suma de los rangos con signoVéase Prueba de Wilcoxon.
Prueba de log-rank (Log-rank test)Prueba estadística utilizada para comparar ladistribución temporal de los eventos entre losdiferentes grupos de un ensayo clínico o deun estudio observacional prospectivo. Elnombre de log-rank deriva de consideracio-nes matemáticas no muy claras que no valela pena interpretar. Algunos autores tambiénlo denominan prueba de Mantel-Haenszelpara datos de supervivencia.
Prueba de Mantel-Haenszel (Maentel-Haenszeltest)
Prueba estadística utilizada para compararmás de una tabla de dos por dos, calculando
una prueba de ji al cuadrado combinada.Por ejemplo, en un estudio de casos y con-troles, donde se comparan los casos de cán-cer de pulmón con sus controles y la exposi-ción al tabaco, para pacientes menores de50 años y mayores de 50 años.
Prueba de McNemar (McNemar test)Prueba estadística para comparar dos pro-porciones cuando se aplica a grupos empa-rejados (no independientes).
Prueba de significación estadística (Test of sig-nificance)
Evaluación de los datos observados medianteuna prueba estadística determinada y el cál-culo del valor de p correspondiente. VéanseSignificación estadística, Significación clínicay p valor de probabilidad.
Prueba de U de Mann-Whitney (Mann-WhitneyU test)
Prueba estadística no paramétrica que com-para diferencias a favor de uno u otro grupoy se aplica a grupos no emparejados (inde-pendientes).
Prueba de una colaVéase Prueba unilateral.
Prueba de Wilcoxon (Wilcoxon test or Wilcoxonsigned-rank sum test)
Prueba estadística no paramétrica que com-para diferencias a favor de uno u otro grupo,y que se aplica a grupos emparejados (no in-dependientes).
Prueba estadística no paramétrica (Non-para-metric test or non-parametric statistical test)
Prueba que se emplea para comparar gru-pos cuando las variables no se distribuyen si-guiendo un patrón de normalidad. Talespruebas no utilizan la media y la desviaciónestándar para analizar los resultados, sinoque emplean el orden de los datos recogidosy su distribución (o no) aleatoria. Ejemplosde pruebas no paramétricas son la U deMann-Whitney y la prueba de Wilcoxon.
Prueba estadística paramétrica (Parametrictest or parametric statistical test)
Prueba que se emplea para comparar gru-pos cuando los resultados se distribuyen si-guiendo un patrón conocido, habitualmentela distribución normal. Ejemplos de pruebasparamétricas son el análisis de la varianza(ANOVA) y la prueba t de Student.
Prueba exacta de Fisher (Fisher’s exact test)Prueba estadística para comparar dos pro-porciones que se aplica cuando alguno delos valores esperados es inferior a 5. Estaprueba sólo es válida para las tablas de dospor dos. También se llama método exacto
52
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

condicionado porque teóricamente los valo-res marginales están fijados previamente.
Prueba t de Student (Student’s t test)Prueba estadística utilizada para compararuna variable continua que sigue una distribu-ción normal entre muestras de dos poblacio-nes. Puede emplearse para datos indepen-dientes y datos emparejados.
Prueba t para datos emparejados (Paired t-test)Prueba de t para analizar la existencia de di-ferencias entre dos mediciones de una mis-ma variable en un mismo sujeto. También sela conoce por prueba de t para datos aparea-dos. Véase Prueba t de Student.
Prueba t para datos independientes (Unpairedt-test)
Prueba de t para analizar la existencia de di-ferencias entre dos mediciones de una mis-ma variable en sujetos distintos. También sela conoce como prueba de t para datos noemparejados. Véase Prueba t de Student.
Prueba unilateral (One-tailed test or one-sidedtest)
Referido al error de tipo I (riesgo α), asun-ción de la probabilidad de error en un solosentido, es decir, que A > B, pero no consi-dera que B > A. Por ejemplo, cuando secomparan dos tratamientos en un ensayo clí-nico, si a priori se conoce que uno de los fár-macos es más eficaz que el otro, la pruebaestadística será menos exigente y por tantose alcanzarán más fácilmente diferenciassignificativas al tener en cuenta sólo una po-sibilidad. También se denomina Prueba deuna cola.
QAAcrónimo de Quality Assurance. Véase Ga-rantía de calidad.
QALYAcrónimo de Quality-Adjusted Life Year. Véa-se Año de vida ajustado por calidad.
QAUAcrónimo de Quality Assurance Unit. VéaseUnidad de garantía de calidad.
RandomizaciónAnglicismo que se emplea para designar elproceso de asignación aleatoria. Véase Asig-nación aleatoria.
Rango (Range)Término empleado con frecuencia comoequivalente de las palabras inglesas range yrank. El primero significa intervalo, margen orecorrido, mientras que el segundo indicamás bien el orden que se ocupa dentro deuna sucesión. Según el Diccionario de laLengua Española, rango es En estadística,
amplitud de una variación de un fenómenoentre un límite menor y uno mayor claramen-te especificados. Por tanto, define el valormás alto y más bajo de una muestra. El tér-mino inglés rank debe traducirse, en cambio,por ordenación.
RatioAnglicismo que se emplea para designar Ra-zón.
Razón (Ratio)Valor obtenido al dividir una magnitud porotra. Por ejemplo, si la proporción de hom-bres en una población es del 60% y la demujeres del 40%, la razón de sexos en esapoblación será de 1,5. También se utiliza co-mo sinónimo de cociente.
Razón de coste-beneficio (Cost-benefit ratio)Expresión consecuente de considerar de for-ma conjunta los beneficios y los costes eco-nómicos asociados a una intervención médi-ca. Véase Análisis de coste-beneficio.
Razón de coste-efectividad (Cost-efectivenessratio)
Razón en la que el numerador tiene encuenta los costes netos de los recursos utili-zados en la atención sanitaria (los costes deltiempo del médico y de la enfermera, laspruebas de laboratorio y los fármacos), loscostes del tratamiento de cualquier efectoadverso y otros costes inducidos por la inter-vención, mientras que el denominador se re-fiere a la efectividad en términos de saludcomo la mejora en la calidad de vida y en laesperanza de vida (número de años de vidasalvados). Expresado en dólares, se conside-ra que razones de coste-efectividad de me-nos de 20.000 dólares por año de vida gana-do son muy favorables, mientras que por en-cima de 100.000 dólares reflejaría gastosexcesivos y utilización inadecuada de recur-sos. Véase Análisis de coste-efectividad.
Razón de disparidadesVéase Razón de posibilidades.
Razón de mortalidad estandarizadaVéase Ajuste indirecto.
Razón de posibilidades (Odds ratio, OR)Medida epidemiológica de asociación obteni-da en los estudios de casos y controles queresulta de dividir el producto del número decasos expuestos (a) y el número de controlessin exposición (d) por el producto del núme-ro de casos sin exposición (b) y el número decontroles expuestos (c). Así, la razón de posi-bilidades (OR) se calcularía por la expresión:OR = a × d/b × c. La OR es numéricamenteparecida al RR cuando se trata de una enfer-
53
GLOSARIO

medad poco frecuente. También se le deno-mina en ocasiones razón de riesgos, razónde ventajas, razón de disparidades y desi-gualdad relativa.
EnfermedadPresente + Ausente –
Exposición Positiva + a b a + bNegativa – c d c + d
a + c b + dEn los ensayos clínicos o metaanálisis, la ORdescribe la posibilidad (odds) que tiene unpaciente del grupo experimental de padecerun evento en relación con la que tiene unpaciente del grupo control. Por ejemplo, 100pacientes (a + b) reciben tratamiento experi-mental y 20 de ellos (a) desarrollan un even-to, mientras que 80 no lo desarrollan (b).Otro grupo de 100 pacientes (c + d) recibetratamiento control, de ellos 40 desarrollanun evento (c) y 60 no lo desarrollan (d). Laodds del grupo experimental es 0,25 (a/b), yla odds del grupo control es 0,67 (c/d), sien-do la OR ([a/b]/[c/d] = 0,25/0,67)igual a0,37. Véanse Estudio de casos y controles yRiesgo relativo.
Razón de riesgosVéase Razón de posibilidades.
Razón de ventajasVéase Razón de posibilidades.
Razonamiento por analogía (Analogy, reasoningor judgement by analogy)
Procedimiento empleado en epidemiologíapara la evaluación de causalidad, por el quese tiene en cuenta la presencia de asociacio-nes o casos parecidos al que es motivo deestudio. Véase Inferencia causal.
RCTAcrónimo de Randomized controlled trial.Véase Ensayo clínico controlado.
Reacción adversa (Adverse effect, adverse re-action, adverse drug reaction, ADR)
Reacción nociva y no intencionada que seproduce a dosis utilizadas normalmente en elhombre para la profilaxis, el diagnóstico o eltratamiento de enfermedades, o para la mo-dificación de una función fisiológica (RD2000/1995). Algunos autores prefieren la de-nominación de efecto indeseable. Durantelas fases de investigación de un fármaco, esdecir, antes de su comercialización, se em-plea el término acontecimiento adverso. Poranalogía con los acontecimientos adversos,pueden clasificarse en graves e inesperadas.En ocasiones se utiliza el término más con-creto de reacción adversa a medicamento(RAM). Véase Acontecimiento adverso.
Reacción adversa tipo A (Type A adverse reac-tion, Augmented adverse reaction)
Reacción que resulta de un efecto farmaco-lógico exagerado pero previsible. En general,depende de la dosis, es más frecuente y me-nos grave que la de tipo B.
Reacción adversa tipo B (Type B adverse reac-tion, Bizarre adverse reaction)
Reacción que aparece a consecuencia deefectos farmacológicos imprevisibles. En ge-neral, no depende de la dosis, es menos fre-cuente y más grave que la de tipo A.
Reclutamiento (Recruitment)Proceso por el que se selecciona entre la po-blación accesible aquellos individuos quepueden cumplir los criterios de inclusión pa-ra ser considerados como población del es-tudio.
Reducción absoluta del riesgo (RAR) (Absoluterisk reduction, ARR)
Medida epidemiológica obtenida en los estu-dios de intervención, que resulta de restar laincidencia de la enfermedad o efecto obser-vado del grupo control (tratamiento estándar,placebo o no intervención) de la incidenciade la enfermedad o efecto observado del gru-po con intervención. Véase Número necesariode pacientes a tratar para evitar un caso.
Reducción relativa del riesgo (RRR) (Relativerisk reduction, RRR)
Medida epidemiológica obtenida en los estu-dios de intervención, que resulta de restar laincidencia de la enfermedad en el grupocontrol de la incidencia de la enfermedad enel grupo con la nueva intervención, y dividirlopor la incidencia de la enfermedad en el gru-po control. Expresa la reducción de riesgorespecto al grupo control. Véase Número ne-cesario de pacientes a tratar para evitar uncaso.
Registro (Registry)Según el Diccionario de la Lengua Española,Libro a manera de índice, donde se apuntannoticias o datos. En epidemiología, se aplicaa un archivo que recoge los casos de unaenfermedad en un grupo de individuos de-terminado, ya sea hospitalario o poblacional.
Registro de especialidades farmacéuticas(Drug regulatory review or approval)
Proceso que emplean los organismos sanita-rios para establecer la idoneidad de la docu-mentación científica de un medicamento afin de proceder a la autorización de su em-pleo en unas condiciones determinadas.
RegresiónVéase Análisis de regresión.
54
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Regresión a la media (Regression to the meanor regression toward the mean)
Tendencia que tienen los valores de una va-riable, cuando están alejados de la media, aacercarse al valor de la media cuando dichavariable es medida repetidas veces.
Regresión lineal (Lineal regression)Análisis de regresión en el que el valor de unparámetro y (variable dependiente) es igual aa+bx, siendo a la ordenada en el origen y bla pendiente, ambas constantes, y x la varia-ble independiente. Véase Análisis de regre-sión.
Regresión logística (Logistic regression)Análisis de regresión en el que la variable de-pendiente adopta únicamente dos valores(por ejemplo, vivo o muerto, enfermedad ono enfermedad, 0 o 1), mientras que las va-riables independientes pueden ser cualitati-vas o cuantitativas. véase Análisis de regre-sión.
Regresión múltiple (Multiple regression)Análisis de regresión en el que existe más deuna variable independiente que puede expli-car el valor de la variable dependiente (y =b0+b1x1+b2x2+...+bkxk+ε, donde b0,b1,b2,bkson constantes, x1,x2,xk son las variables in-dependientes y å es un término de error. Sesupone que los valores de ε para cada indivi-duo se distribuyen independientemente conmedia cero y varianza δ2. Véase Análisis deregresión.
Relación beneficio-riesgo (Benefit to risk ratio)Relación que existe entre la eficacia de untratamiento, su tolerabilidad y la gravedad dela enfermedad que se está tratando. Consti-tuye uno de los elementos básicos del proce-so de decisión terapéutica, ya que se consi-dera que está comparando los datos de efi-cacia y seguridad del tratamiento con lagravedad y el pronóstico de la enfermedad.En el caso de los medicamentos, tiene encuenta la eficacia y los efectos indeseables.
Relación causalVéase Inferencia causal.
Relación dosis-respuesta (Dose-response rela-tionship)
En farmacología, estudio de la relación exis-tente entre la dosis administrada de un me-dicamento o su concentración y los efectosfarmacológicos observados. En ensayos clíni-cos, este proceso se realiza durante la fase IIpara evaluar la dosis y la posología óptima enel tratamiento de una enfermedad determi-nada (estudios de búsqueda de dosis). Enepidemiología, establecimiento de la presen-
cia de mayor riesgo o gravedad de una enfer-medad conforme se incrementa la intensidado el tiempo de exposición a un factor deter-minado. En estudios de causalidad, se le de-nomina gradiente biológico. Véanse Dosisrespuesta, Respuesta gradual, Respuesta deltodo o nada y Estudio de búsqueda de dosis.
Relación temporalVéase Temporalidad.
Relevancia clínica (Clinical relevance)Cualidad de un tratamiento que permite es-tablecer que su empleo, en una determinadaenfermedad, puede suponer una mejoría im-portante y significativa en los pacientes so-metidos a tal terapéutica. Véase Significaciónclínica y Significación estadística.
Repetibilidad (Reliability)En análisis químico, medida de la precisiónde los resultados de un método efectuado enlas mismas condiciones y en la misma mues-tra por un mismo analista, laboratorio o apa-rato. Es sinónimo de precisión intraensayo.Véase Precisión.
Reproducibilidad (Reproducibility)En análisis químico, medida de la precisiónde los resultados de un método efectuado enla misma muestra, pero en condiciones dife-rentes, por ejemplo, distintos analistas, apa-ratos o días. Es sinónimo de precisión inte-rensayo. Véase Precisión.
RescateVéase Tratamiento de rescate.
Residual, efectoVéase Efecto residual.
Respeto por las personas (Respect for auto-nomy)
Principio bioético básico que obliga a conside-rar a los sujetos como entes autónomos (ca-paces de decidir por ellos mismos) y a prote-ger a los sujetos con autonomía disminuida.Implica la obtención del consentimiento paraparticipar en una investigación. Se emplea au-tonomía o respeto a las personas de forma si-nónima. Véase Principios bioéticos.
Responsable de proyecto de investigación (Cli-nical project leader)
Persona que asume la coordinación de lasdistintas etapas de la investigación clínica deun/os fármaco/s. En ocasiones se responsa-biliza de líneas o áreas terapéuticas, quepueden incluir distintos fármacos.
Respuesta gradual (Graded response)Respuesta que admite valores intermediosdentro de un rango determinado. Por ejem-plo, la presión arterial o la temperatura cor-poral.
55
GLOSARIO

Respuesta del todo o nada (All-or-none respon-se, quantal response)
Respuesta que está presente o ausente, peroque no admite valores intermedios. Porejemplo, muerte o supervivencia. Tambiéndenominada respuesta cuántica.
Respuesta placeboVéanse Efecto placebo, Placebo y Sensibili-dad al placebo.
Resultado finalVéase Variable principal de valoración.
Resultado final intermedioVéase Variable sustitutiva.
Resultado final secundarioVéase Variable secundaria de valoración.
Resultado final sustitutivoVéase Variable sustitutiva.
Retirada (Withdrawal)Separación del paciente del ensayo clínicocuando aparecen situaciones que encierrenun peligro injustificado u otras que así loaconsejen (por ejemplo, reacciones adver-sas, insuficiencia del tratamiento, violación oincumplimiento del protocolo o la falta de se-guimiento de la intervención). Con frecuen-cia también se consideran retiradas losabandonos, aunque estrictamente no soniguales: en los abandonos, la iniciativa partedel paciente, y en las retiradas la iniciativa esdel investigador. En cualquier caso, el proto-colo debería especificar la necesidad de re-coger todas las causas de pérdidas de pa-cientes para proceder a un análisis adecua-do. Véase Abandono.
Retrospectivo, estudioVéase Estudio retrospectivo.
Revisión por colegasVéase Revisión científica.
Revisión científica (Peer review/Refeering)Procedimiento para valorar las propuestas deinvestigación científica, los informes de resul-tados y los artículos científicos mediante laconsulta previa a individuos o grupos con es-pecial cualificación en el tema de que se tra-te. También se la conoce por revisión por co-legas o revisión por iguales.
Revisión por igualesVéase Revisión científica.
Revisión sistemática (Systematic review)Método utilizado para identificar datos tantopublicados como no publicados, determinarla elegibilidad de los datos para su inclusión,y analizar estos datos. A diferencia del meta-análisis,.que prima el método estadístico, enlas revisiones sistemáticas se da ademásmucha importancia a la validez de los datos.
Riesgo (Risk)En un sentido amplio, probabilidad de quealgo ocurra. En investigación clínica, es el re-sultado desfavorable de una actividad, inter-vención o exposición, especialmente referidoa la probabilidad de que aparezca un fenó-meno adverso concreto. En el caso de estu-dios sin beneficio terapéutico, se acepta co-mo lícito el que los voluntarios se encuentrensometidos a riesgo mínimo o insignificante,según la FDA o el Royal College of Physi-cians. Se acepta como tal la probabilidad deentre 1 y 100 por mil de sufrir una complica-ción menor y de entre 10 y 1.000 por millónde sufrir una complicación grave.
Riesgo �Véase Error α.
Riesgo �Véase Error β.
Riesgo absoluto (RA) (Absolute risk, AR)Medida epidemiológica de asociación obte-nida en los estudios de cohortes, que re-sulta de restar la incidencia de enferme-dad en la población expuesta de la inci-dencia de la enfermedad en la poblaciónno expuesta. Cuando el riesgo absoluto sedivide por la incidencia de los expuestos,se habla de la proporción atribuible o frac-ción etiológica, y se refiere a la proporciónde la enfermedad entre los expuestos quees atribuible a la exposición. En los estu-dios de casos y controles no se puede cal-cular el riesgo absoluto, pero sí se puedeestimar el riesgo absoluto porcentual, res-tando el riesgo relativo de 1 y dividiéndolopor el riesgo relativo y multiplicándolo por100. También denominado riesgo atribui-ble y fracción etiológica.
Riesgo absoluto poblacional (Population attri-butable risk)
Medida epidemiológica de asociación obteni-da de los estudios de cohortes que resulta derestar la incidencia total en la población de laincidencia en la población de los no expues-tos. Alternativamente se puede calcular mul-tiplicando el riesgo absoluto por la propor-ción de individuos expuestos en la población.Esta medida es interesante para conocercuál es la fracción atribuible a la exposiciónen el total de la población.
Riesgo atribuibleVéase Riesgo absoluto.
Riesgo insignificanteVéase Riesgo.
Riesgo mínimoVéase Riesgo.
56
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Riesgo relativo (RR) (Relative risk, RR)Medida epidemiológica de asociación obteni-da de los estudios de cohortes que resulta dedividir la incidencia de enfermedad de la po-blación expuesta por la incidencia en la po-blación no expuesta, indicando la probabili-dad de desarrollar una enfermedad en elgrupo expuesto relativo a los no expuestos.Se calcula según la fórmula RR = (a/a +b):(c/c + d), en quea = Expuestos que presentan la enfermedadb = Expuestos que no presentan la enfermedadc = No expuestos que presentan la enfermedadd = No expuestos que no presentan la enfermedad
EnfermedadPresente + Ausente –
Exposición Positiva + a b a + bNegativa – c d c + d
a + c b + d
Véanse Estudios de cohortes y Razón de po-sibilidades.
RMEAcrónimo de Razón de Mortalidad Estandari-zado. Véase Ajuste indirecto.
ROCAcrónimo de Receiver Operating Characteris-tic. Véase Curva de características funciona-les.
RRAcrónimo de Riesgo relativo.
“Salir de pesca” (Fishing expedition analysis/studies)
Expresión empleada irónicamente en aque-llas situaciones que pretenden encontrar di-ferencias estadísticamente significativasmediante el análisis de subgrupos y compa-raciones múltiples no previstas en el diseñodel estudio. También se aplica a los estu-dios en los que se pretende recoger infor-mación sin un objetivo previamente estable-cido.
SDAcrónimo de Standard Deviation. Véase Des-viación estándar.
Secuencia temporalVéase Temporalidad.
Secuencial, análisisVéase Análisis secuencial.
Secuencial, ensayo clínicoVéase Ensayo clínico secuencial.
Seguimiento del paciente (Patient follow-up)Proceso de contacto periódico con los pa-cientes que participan en una investigación afin de administrar el tratamiento, observarsus efectos, seguir su evolución o recoger los
datos necesarios. Véanse Visita de segui-miento, Visita inicial y Visita final.
Seguridad (Safety)Grado en que una intervención está exentade efectos indeseables o reacciones adver-sas.
SEMAcrónimo de Standard Error of the Mean. Vé-ase Error estándar de la media.
Sensibilidad (Sensitivity, responsiveness, sen-sibility)
Según el Diccionario de la Lengua Española,grado o capacidad de la eficacia de ciertosaparatos y también capacidad de respuestaa muy pequeñas excitaciones, estímulos ocausas. Es un término muy empleado comotraducción de diferentes palabras anglosajo-nas que no tienen siempre un mismo signifi-cado. Una primera acepción es la que indicala proporción de individuos realmente enfer-mos que han sido clasificados como talesmediante la utilización de una prueba diag-nóstica, con lo que una prueba altamentesensible daría pocos resultados falsos negati-vos. Se calcula mediante el cociente entrelos pacientes correctamente diagnosticados yel total de pacientes que presentan la enfer-medad (a / a + c) (véase tabla dos por dosen Especificidad).Una segunda acepción es la que considerala sensibilidad como la cualidad de una va-riable que permite detectar pequeños cam-bios en la condición estudiada, ya sea la ca-pacidad de un estudio para detectar diferen-cias entre tratamientos (sensitivity), o la deun instrumento para detectar cambios (res-ponsiveness). Adicionalmente, algunos auto-res la encuentran prácticamente sinónima devalidez frontal. Véanse Análisis de sensibili-dad, Especificidad y Validez frontal.
Sensibilidad, análisis deVéase Análisis de sensibilidad.Sensibilidad al placebo (Placebo reactor)
Propiedad de algunos pacientes por la quepresentan respuesta al placebo. Debe tener-se en cuenta que esta cualidad no es perma-nente y depende de las circunstancias enque se administra el placebo. Véase Placebo.
Serendipitia (Serendipity)Término procedente del inglés y empleadopara describir la facultad de realizar descu-brimientos útiles de forma accidental. Supo-ne la presencia de una buena formacióncientífica para poder apreciar el interés deuna determinada observación. Fue creadopor Horace Walpole inspirándose en el cuen-
57
GLOSARIO

to de hadas Los tres príncipes de Serendip(nombre antiguo de Sri Lanka), en el que loshéroes realizan repetidos descubrimientosinintencionados.
Series de casos, estudio deVéase Estudio de series de casos.
Sesgo (Bias)Error sistemático que puede producirse en lainclusión, asignación, recogida de datos,análisis o interpretación de un estudio, loque origina resultados o interpretaciones in-correctos. Véase Validez.
Sesgo de Berkson (Berkson bias)Sesgo de selección que se produce en losestudios de casos y controles cuando éstosse realizan en población hospitalaria. Porejemplo, en un estudio de casos y controlespara ver la asociación de cáncer de vejiga yla exposición a un tóxico ambiental, los ca-sos se seleccionarían del servicio de oncolo-gía, mientras que los controles pertenecerí-an a un servicio distinto, como el de trauma-tología. El sesgo observado podría ir en losdos sentidos dependiendo de la probabili-dad de hospitalización de las personas ex-puestas al tóxico ambiental, de la probabili-dad de hospitalización de los pacientes concáncer de vejiga y de la probabilidad de hos-pitalización de los pacientes con accidentestraumáticos.
Sesgo de clasificación incorrecta (Missclassifi-cation bias)
Sesgo resultante de considerar los sujetosdel estudio como expuestos cuando no loson, o viceversa. También puede aparecerpor clasificar a los sujetos como enfermoscuando no lo son y viceversa. En ocasionesse le denomina sesgo de mala clasificación.
Sesgo de derivaciónVéase Sesgo de remisión.
Sesgo de detección (Detection bias)Sesgo debido a la existencia de diferenciasen los procedimientos empleados para diag-nosticar o verificar la enfermedad en los gru-pos del estudio.
Sesgo de incidencia/prevalencia (Prevalencebias)
Sesgo de selección en los estudios de casosy controles que consiste en considerar loscasos ya existentes y en despreciar los casosnuevos. Una asociación importante con laprevalencia puede relacionarse con la dura-ción de la enfermedad más que con su inci-dencia, debido a que la prevalencia es pro-porcional tanto a la incidencia como a la du-ración de la enfermedad.
Sesgo de información (Information bias)Sesgo producido por medir con distinto gra-do de intensidad la exposición o la evoluciónen los grupos del estudio.
Sesgo de mala clasificaciónVéase Sesgo de clasificación incorrecta.
Sesgo de medición (Measurement bias)Sesgo que aparece cuando se aplican de for-ma distinta, o con distinto criterio, los instru-mentos o métodos empleados para medir lasvariables biológicas según el grupo al quepertenece el sujeto.
Sesgo de memoria (Memory bias)Sesgo que se debe a las diferencias en el re-cuerdo a la exposición previa en los casos ylos controles, de tal manera que habitual-mente los pacientes con la enfermedad re-cuerdan mejor la exposición previa a fárma-cos que los controles sanos.
Sesgo de observación o del observador (Obser-ver bias)
Sesgo que consiste en aplicar diferencias sis-temáticas entre el valor real y el observado.En un ensayo clínico, el más frecuente seasocia a que el investigador conozca el trata-miento que está recibiendo cada sujeto.Comporta la sobreestimación o la infravalora-ción de uno de los tratamientos.
Sesgo de proveedor (Provider bias)Sesgo que radica en el hecho que los estu-dios de calidad de vida tienden a realizarseprecisamente por investigadores que espe-ran obtener unos resultados positivos, encontraposición a los más escépticos.
Sesgo de publicación (Publication bias)Sesgo resultante de creer que los estudiospublicados son realmente los realizados. Sedebe a que muchos ensayos clínicos no sepublican, con frecuencia por la ausencia dediferencias significativas entre los grupos es-tudiados. Es especialmente relevante en losmetaanálisis que se realizan sólo con los re-sultados publicados, ya que los estudios clí-nicos con resultados negativos tienden aquedar inéditos. Recientemente se ha pro-puesto a través de diferentes revistas biomé-dicas la creación de un registro de ensayosclínicos no publicados.
Sesgo de referencia (Reference bias)Sesgo que radica en la tendencia de algunosautores a citar únicamente los artículos pre-vios que coinciden con su hipótesis o resul-tados, ignorando los que los contradicen.También se aplica cuando existe un exagera-do número de autocitas o un excesivo énfa-sis en los hallazgos propios anteriores.
58
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Sesgo de remisión (Referral bias)Sesgo en los resultados de un estudio cuan-do las razones para remitir un paciente paraatención médica se relacionan con el estadode exposición. También conocido por Sesgode derivación (de casos, de pacientes).
Sesgo de selección (Selection bias)Sesgo que se deriva de que las característi-cas de los pacientes incluidos en un estudioson diferentes de las de los excluidos. Talhecho condiciona el que la muestra de pa-cientes del estudio no sea representativa detoda la población, ya que podrían diferir enalgunos factores pronósticos.
Sesgo del voluntario (Volunteer bias)Sesgo que se produce al incluir sujetos queacuden voluntariamente para participar enun estudio. Estos pueden ser diferentes a lapoblación general, ya que pueden ser mássanos, ser más cumplidores de las medidasterapéuticas, y tener una mortalidad más ba-ja. En otro sentido, los sujetos voluntariostambién pueden ser aquellos individuos queestán preocupados por su salud por identifi-carse ellos mismos como de riesgo alto, yasea por los propios antecedentes personaleso familiares, o por las características del esti-lo de vida. En este caso, estos sujetos podrí-an tener un riesgo mayor de mortalidad quelos de la población general. Por este motivo,la participación de sujetos voluntarios en unestudio puede sesgar los resultados en unadirección que es difícil de predecir.
Sesgo del entrevistador (Interviewer bias)Tipo de sesgo de información causado por larecogida selectiva de datos, consciente o in-consciente. Se reduce con el empleo decuestionarios estructurados.
Sesgo protopático (Protopathic bias)Sesgo consistente en interpretar una variablecomo resultado de una exposición cuando,de hecho, es su determinante. Por ejemplo,establecer una relación entre un analgésico yla polirradiculoneuritis, porque muchos pa-cientes que presentan ésta habían ingeridopreviamente el fármaco para aliviar el dolorque puede aparecer en la primera fase delproceso.
Significación clínica (Clinical significance)Presencia de diferencias entre dos grupos detratamiento de la suficiente envergadura paradecidir la elección de una de las dos alterna-tivas por el beneficio que supone para lospacientes. La significación estadística no im-plica siempre la existencia de significaciónclínica; ésta va más allá del concepto aritmé-
tico y está determinada por el juicio clínico.Las medidas epidemiológicas más útiles paraestablecer la significación clínica son la re-ducción absoluta del riesgo y el número ne-cesario de pacientes a tratar para evitar uncaso. Véanse Relevancia clínica y Significa-ción estadística.
Significación estadística (Statistical significance)Presencia de diferencias estadísticamentesignificativas entre los grupos de tratamiento.Su presencia no supone necesariamente laexistencia de significación clínica. VéanseRelevancia clínica y Significación clínica.
Significación estadística, nivel de (Statisticalsignificance, level of)
Riesgo de equivocarse asumido por el inves-tigador al rechazar la hipótesis nula, cuandoen realidad ésta es verdadera (probabilidadde cometer el error de tipo I). Por conven-ción se acepta habitualmente un riesgo infe-rior al 5% (p<0,05).
Simple ciego (Single blind or blinded, Singlemask or masked)
Procedimiento utilizado habitualmente en losensayos clínicos por el que algunas personas(por ejemplo, los médicos del estudio) cono-cen o son informados del tratamiento o de laenfermedad, mientras que a otros (por ejem-plo, los pacientes) desconocen o se les ocul-ta la información sobre el tratamiento que re-ciben o la enfermedad que padecen. VéaseCiego.
Simulación (Sham, Simulation)Métodos que se utilizan, como tratamientocontrol, para evaluar de forma lo más ciegaposible determinados procedimientos tera-péuticos no farmacológicos. Su uso puedeasimilarse de alguna manera al placebo em-pleado en los estudios con medicamentos.Por ejemplo, un método de simulación en in-tervenciones quirúrgicas sería realizar única-mente la incisión en la piel.
Simulación de Montecarlo (Monte Carlo simu-lation)
Método de simulación de algunos procedi-mientos o procesos estocásticos utilizandonúmeros aleatorios o pseudoaleatorios.
SMRAcrónimo de Standardized Mortality Ratio.Véase Ajuste indirecto.
Solicitud de autorización de especialidad far-macéutica (New Drug Application, NDA, Drugapproval application, Regulatory approval ap-plication)
Petición a las autoridades sanitarias para po-der registrar y comercializar una especialidad
59
GLOSARIO

farmacéutica. Para la autorización se debensatisfacer diferentes condiciones: seguridad,eficacia, calidad y pureza, correcta identifica-ción e información precisa (ver capítulo II, Ley25/1990 del Medicamento). En algunos paí-ses se tiene en cuenta además la necesidad.Su definición es similar al NDA de los EE.UU.En este país, las autoridades reguladoras delos medicamentos (FDA) promueven la pre-sentación de la solicitud en formato informáti-co (computer assisted NDA o CANDA).
Solicitud de producto en fase de investigaciónclínica (Investigational new drug application,INDA)
Petición a las autoridades sanitarias para ca-lificar como PEI una entidad química o bioló-gica, que no sea principio activo de algunaespecialidad farmacéutica registrada en Es-paña, y con la que se pretende iniciar la fasede investigación clínica. (RD 561/1993 deensayos clínicos). En EE.UU. se realiza unasolicitud de IND (INDA).
SOPAcrónimo de Standard OperatingProcedures. Véase Procedimientos normali-zados de trabajo.
Subjetivo (Subjetive)Según el Diccionario de la Lengua Españolarelativo a nuestro modo de pensar o de sentiry no al objeto en sí mismo. En investigaciónclínica, se aplica a todas aquellas variables,medidas o características que dependen dela expresión del sujeto y que no pueden ob-tenerse sin la colaboración activa de éste.Por ejemplo, el dolor o la ansiedad.
Subrogada, variableVéase Variable sustitutiva
Substitución de sujetos (Replacement of sub-jects)
Práctica generalmente incorrecta, que con-siste en incluir un nuevo paciente que reem-plaza a otro que ha abandonado el estudio oha sido retirado del mismo. La sustitución desujetos no es recomendable y la posibilidadde casos de abandono o de retiradas debetenerse en cuenta cuando se calcula el ta-maño de la muestra. Únicamente es justifi-cable en estudios de fase I, por ejemplo far-macocinéticos, después de una investigacióncuidadosa de las causas de abandono o reti-rada. Véase Tamaño de la muestra, cálculo.
Sujeto del ensayo (Trialist)Según el RD 561/1993, Persona sana o en-ferma que participa en un ensayo clínico,después de haber otorgado libremente suconsentimiento informado.
SupervivenciaVéase Tasa de supervivencia.
Sustancia medicinal (Drug, Medicine, Medici-nal substance)
Según la Ley del Medicamento (25/1990 art.8.2) es Toda materia, cualquiera que sea suorigen -humano, animal, vegetal, químico ode otro tipo- a la que se atribuye una activi-dad apropiada para constituir un medica-mento. En ocasiones se utiliza como sinóni-mo de fármaco y medicamento. Véanse Fár-maco y Medicamento.
Sustitutiva, variableVéase Variable sustitutiva.
t de Student, pruebaVéase Prueba t de Student.
Tabla de contingencia (Contingency tables)Clasificación tabular de datos de una mues-tra de población, en la que las subcategoríasde una característica se indican horizontal-mente (en filas) y las de otra verticalmente(en columnas). Tal clasificación permite apli-car pruebas de asociación entre las caracte-rísticas de las filas y las de las columnas.
Tabla de dos por dos (Tabla 2 x 2) (Two by twotable)
Tabla de contingencia en la que se incluyendos categorías de la característica en las filasy dos en las columnas (cuatro valores en to-tal) El análisis estadístico se puede realizarmediante métodos exactos, como la pruebaexacta de Fisher, o con los métodos aproxi-mados, como la prueba de ji al cuadrado.Véanse Razón de posibilidades y Riesgo rela-tivo.
Tabla de mortalidadVéase Método actuarial.
Tabla de vidaVéase Método actuarial.
Tamaño de la muestra (Sample size)Número de sujetos o pacientes que se inclui-rán en una investigación o ensayo clínico.Véase Tamaño de la muestra, cálculo.
Tamaño de la muestra, cálculo (Sample sizecalculation)
Determinación del número de pacientes ne-cesarios para que el estudio planeado asumaconclusiones adecuadas. En el caso de losensayos clínicos, su cálculo se realiza tenien-do en cuenta la diferencia clínicamente sig-nificativa entre tratamientos y los errores α yβ. Para variables continuas se precisa unaestimación de la desviación estándar de lamedia. Habitualmente se aceptan valores deα <0,05 y β= 0,2. En los estudios de preva-lencia, se calcula a partir del error α, una es-
60
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

timación de la prevalencia y el nivel de preci-sión de ésta. En los estudios analíticos (co-horte, casos y controles), se calcula a partirde una estimación del porcentaje de exposi-ción del grupo control y del riesgo relativo.En el momento de su cálculo, debe realizar-se una estimación del porcentaje de pérdidade individuos o pacientes, y ajustar el núme-ro final en consonancia.
Tasa (Rate)Medida de la frecuencia con que ocurre unhecho en una población determinada. Se ex-presa en forma de cociente.
Tasa de falsos negativos o pseudonegativosVéase Falso negativo.
Tasa de falsos positivos o pseudopositivosVéase Falso positivo.
Tasa de letalidad (Case fatality rate)Número de fallecimientos por una enferme-dad en un período determinado dividido porel número de casos diagnosticados de la en-fermedad en ese período, y multiplicado porcien.
Tasa de morbilidad (Morbidity rate)Término confuso que se emplea indiscrimi-nadamente para referirse a las tasas de inci-dencia, de prevalencia de una enfermedad ode altas hospitalarias.
Tasa de mortalidad (Mortality rate)Proporción de una población que fallece enun período determinado. Habitualmente seexpresa como unidades por 100.000 o mi-llón de habitantes. Véase Ajuste directo yAjuste indirecto.
Tasa de supervivencia (Survival rate)Proporción de sujetos de un grupo que seencuentran vivos al inicio de un estudio yque sobreviven tras finalizar el período deduración de éste.
Temporalidad (Temporality)Propiedad de la causalidad por la que la ex-posición de interés precede siempre a la en-fermedad por un período de tiempo que seaconsistente con los conocimientos biológicos.También se denomina secuencia temporal.Véase Inferencia causal.
Teorema de Bayes (Bayes’s rule)Principio que establece la probabilidad depadecer la enfermedad a condición de te-ner un síntoma o una prueba diagnósticapositiva. Considerando el caso de la pre-sencia o ausencia de una enfermedad es-pecífica basada en la aparición de un sínto-ma, la probabilidad de padecer la enferme-dad a condición de tener el síntoma es lasiguiente:
P(E) × P(S/E)P(E/S) =
P(E) × P(S/E) + P(NE) × P(S/NE)
P = probabilidadE = enfermedadS = síntomaNE = ausencia de enfermedad
Tolerabilidad, estudio deVéase Estudio de tolerabilidad.
Tolerancia (Tolerance)Disminución del efecto de una dosis de unfármaco tras la administración repetida, loque obliga a un aumento de la dosis para ob-tener el mismo efecto. También es la necesi-dad de aumentar la dosis para obtener unmismo efecto. Puede tener una base farma-cocinética o farmacodinámica. En ocasionesse emplea la expresión de tolerancia para re-ferirse a tolerabilidad. Véase Estudio de tole-rabilidad.
Tolerancia, estudio deVéase Estudio de tolerabilidad.
Toxicidad (Toxicity)Genéricamente, conjunto de las reaccionesadversas que se presentan tras la adminis-tración de medicamentos. En ocasiones, seutiliza este término para designar a los estu-dios de toxicología, mutagénesis, carcinogé-nesis y teratogénesis realizados en el períodopreclínico.
Toxicología (Toxicology)Clásicamente, ciencia que estudia los vene-nos. Modernamente, ciencia que estudia to-das aquellas acciones producidas por sus-tancias que causan efectos adversos en losseres vivos, como medicamentos, sustanciasvegetales, contaminantes ambientales o tóxi-cos industriales. En humanos, en ocasionesse restringe el objetivo de la toxicología al es-tudio de las reacciones adversas de los me-dicamentos a largo plazo (carcinogénesis,mutagénesis o teratogénesis), mientras quelas que aparecen a corto plazo constituyen elobjetivo de la farmacología clínica. Se em-plea también para definir las reacciones ad-versas que se observan durante la intoxica-ción por una sustancia.
Transversal, estudioVéase Estudio transversal.
Tratamiento controlVéase Grupo control.
Tratamiento de referencia (Standard treatment)Forma de terapia que está reconocida comola más adecuada para esa enfermedad.Constituye un patrón de comparación y seusa con frecuencia como tratamiento control.Véase Grupo control.
61
GLOSARIO

Tratamiento de rescate (Rescue treatment)Terapéutica contemplada en el protocolo deun estudio para administrar a los pacientesque no obtienen un resultado adecuado conlos tratamientos evaluados. Es especialmenterecomendable en las situaciones en que seemplea placebo y existe un tratamiento efi-caz para la enfermedad considerada.
Triple ciego (Triple blind/blinded, Triplemask/masked)
Procedimiento empleado habitualmente enlos ensayos clínicos en el que se utilizan unoscódigos, de tal manera que ni los pacientes,ni el personal clínico, ni los responsables dela monitorización o del análisis estadístico delestudio conocen la asignación del tratamien-to. Véanse Ciego, Simple ciego y Doble ciego.
U de Mann-WhitneyVéase Prueba de U de Mann-Whitney.
Unidad de garantía de calidad (UGC) (Qualityassurance unit, QAU)
Departamento responsable de identificar losproblemas de garantía de calidad, recomen-dar y proporcionar soluciones, y finalmenteverificar si éstas han sido cumplimentadas.Sus miembros deben ser independientes delos que están implicados en una investigaciónespecífica. La unidad de garantía de calidadpuede auditar algunas investigaciones y emi-tir en su caso un certificado de conformidadde que el protocolo se ha realizado conformea los procedimientos normalizados de trabajo(PNT) o que los resultados se ajustan a losdatos originales. Véase Garantía de calidad.
Unidad de probabilidadVéase Probit.
Uso compasivo (Compassionate use)Según el RD 561/1993 art. 23, Utilización,en pacientes aislados y al margen de un en-sayo clínico, de productos en fase de investi-gación clínica, o también la utilización de es-pecialidades farmacéuticas para indicacio-nes o condiciones de uso distintas de lasautorizadas, cuando el médico, bajo su ex-clusiva responsabilidad, considera indispen-sable su utilización.
Utilización de medicamentos, estudioVéase Estudio de utilización de medicamen-tos.
Validación (Validation)Proceso que tiene por objeto demostrar queun método, técnica o instrumento es válido.Véase Validez.
Validación de datos (Data validation)Procedimiento utilizado para asegurar quelos datos primarios, la base de datos, la co-
pia impresa o electrónica del cuaderno derecogida de datos, las impresiones de orde-nador, los análisis estadísticos y las tablas deresultados concuerdan con las observacio-nes originales. Se realiza mediante una ins-pección manual o mediante sistemas decomputación.
Validez (Validity)Sinónimo de exactitud. Grado en que uninstrumento mide realmente lo que quieremedir. Generalmente, la medida de la vali-dez precisa de la existencia de un elemen-to reconocido de medida (patrón de oro).En epidemiología, puede definirse comogrado de ausencia de sesgos o de erroressistemáticos. Véanse Exactitud y Precisión(Fig. 10).
Validez conceptualVéase Validez teórica.
Validez concurrente (Concurrent validity)Tipo de validez de criterio en que la compa-ración con el patrón de oro se realiza de for-ma simultánea. Véase Validez de criterio.
Validez convergenteVéase Validez teórica.
Validez de construcción o de constructoVéase Validez teórica.
Validez de contenido (Content validity)Grado en que la medida considera el rangode características propias del fenómeno es-tudiado. Por ejemplo, en las medidas de cali-dad de vida relacionadas con la salud, se re-fiere al grado en que se miden áreas comomortalidad, función física, psicológica y so-cial, y las percepciones. Consiste en un juiciosobre la capacidad del instrumento para exa-minar todos los aspectos del fenómeno estu-diado. En psicometría, su estudio permitedeterminar si el instrumento incluye unamuestra representativa de las conductas quese pretende evaluar.
Validez de criterio (Criterion validity)Correlación de una escala con cualquier otramedida del fenómeno que queremos estu-diar, idealmente un patrón de oro. Existendos tipos: la validez concurrente y la validezpredictiva. También denominada validez se-gún criterios. Véanse Validez concurrente yValidez predictiva.
Validez diagnóstica (Diagnostic validity)Grado de sensibilidad y especificidad de unaprueba diagnóstica. También se denominaeficacia diagnóstica. Véanse Sensibilidad yEspecificidad.
Validez discriminatoria/discriminativaVéase Validez teórica.
62
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Validez empírica (Empirical validity)En psicometría, su estudio permite compararlos resultados de una prueba con un criterioexterno. Véase Validez de criterio.
Validez externa (External validity)Grado en que las conclusiones extraídas deun estudio se pueden extrapolar o generali-zar más allá de la muestra estudiada. Depen-de del tamaño y características de la mues-tra. También se la denomina generalizacióno generabilidad (esta última no está acepta-da por el Diccionario de la Lengua Española).Véase Validez interna.
Validez frontal (Face validity)Grado por el que, a simple vista, el instru-mento parece valorar las cualidades desea-das. Representa un juicio subjetivo basadoen una revisión de la medida en sí mismapor uno o más expertos y raramente se utili-zan abordajes empíricos. Para algunos auto-res, es un juicio sobre la validez del instru-mento basado en una valoración intuitiva delgrado en que un determinado instrumentoreúne determinados criterios, como aplicabi-lidad, claridad, simplicidad, ausencia de ses-gos, extensión adecuada y ausencia de ítemsredundantes.
Validez interna (Internal validity)Grado en que los resultados de un estudioson aplicables a la población misma del es-tudio y que depende del diseño de éste. Lavalidez interna es independiente de la exter-na, de forma que la presencia de la primerano obliga a la segunda. Véase Validez exter-na.
Validez predictiva (Predictive validity)Tipo de validez de criterio en el cual se com-para el nuevo instrumento con la medida deun patrón que no se encuentra disponiblehasta un tiempo después. Un ejemplo clási-co de validez predictiva se aplica a los instru-mentos utilizados en el ingreso de alumnosen un centro educativo, cuya utilidad paraseleccionar estudiantes sólo puede estable-cerse mucho tiempo después a través de laspruebas de rendimiento académico. VéaseValidez de criterio.
Validez teórica (Construct validity)Grado en que la medición se correspondecon los conceptos teóricos (construcciones)referentes al fenómeno que hay que estu-diar. Por ejemplo, si teóricamente un fenó-meno cambia con la edad, una medición convalidez de construcción ha de reflejar dichocambio. La demostración de la validez teóri-ca puede realizarse mediante la validez con-
vergente y la validez discriminativa. La pri-mera se establece mediante la comparacióncon otra escala que estudia aspectos teóricossimilares y determinando la presencia de co-rrelación entre ambas. La validez discrimina-tiva compara el instrumento con otro de ca-racterísticas totalmente distintas a fin de es-tablecer la ausencia de relación. La validezteórica también se conoce como validez con-ceptual, validez de construcción y validez deconstructo.
Valor atípico (Outlier)Valor que difiere mucho del resto de obser-vaciones. Su presencia genera la sospechade que pueda deberse a un error en su de-terminación o a su procedencia de una po-blación distinta. Aunque se dude de su cre-dibilidad, no es recomendable excluirlo delanálisis y, en todo caso, se pueden analizarlos datos con él y sin él. También denomina-do valor extremo.
Valor de probabilidadVéase p, valor de probabilidad.
Valor extremoVéase Valor atípico.
Valor predictivo negativo (Negative predictivevalue)
Referido a pruebas diagnósticas, probabili-dad de que una persona con un resultadonegativo no padezca la enfermedad. Se cal-cula mediante el cociente entre el número deindividuos con una prueba negativa y que nopresentan la enfermedad (d) y la suma de to-dos los que tienen la prueba negativa (c + d)(según la tabla dos por dos en Especifici-dad). Véanse Especificidad y Sensibilidad.
Valor predictivo positivo (Positive predictive va-lue)
En las pruebas de diagnóstico, probabilidadde que una persona con un resultado positi-vo padezca realmente la enfermedad. Se cal-cula mediante el cociente entre el número deindividuos con una prueba positiva correcta-mente diagnosticados como poseedores dela enfermedad (a) y la suma de todos los quetienen la prueba positiva (a + b) (según la ta-bla dos por dos en Especificidad). VéanseEspecificidad y Sensibilidad.
Variabilidad (Variability)Propiedad por la que las mediciones de unavariable difieren según el tiempo y las cir-cunstancias.
Variabilidad interindividual (Between-subjectsvariability)
Variación entre individuos consecuente consus diferencias biológicas.
63
GLOSARIO

Variabilidad intraindividual (Within-subject va-riability)
Variación de la medida de un parámetro enun mismo individuo según las circunstanciasen que ésta se realice. Por ejemplo, las osci-laciones de la presión arterial, la temperaturacorporal o la secreción de cortisol.
Variable (Variable)Según el Diccionario de la Lengua Española,Magnitud que puede tener un valor cualquie-ra de los comprendidos en un conjunto.
Variable aleatoria (Random variable)Variable que puede asumir un número de di-ferentes valores, siendo este número deter-minado por una distribución probabilística,como puede ser la normal o la de Bernoulli.Véase Aleatorio.
Variable basal (Baseline variable)Variable que se mide, observa o valora en unsujeto o paciente al inicio del estudio antesde la asignación del tratamiento o del iniciode éste. Las variables basales son especial-mente importantes para establecer las dife-rencias absolutas o relativas respecto a susvalores después del tratamiento y para com-parar entre los diferentes grupos del estudio.Permiten así mismo establecer la homoge-neidad de éstos. Véase Características basa-les.
Variable binaria (Binary variable)Variable que es capaz de asumir solamenteuno de dos valores. Por ejemplo, hombre omujer, o también 0 o 1.
Variable blanda (Soft variable)Variable que depende de manifestacionessubjetivas u opiniones, sin posibilidad demedición instrumental exacta, lo que dificul-ta su reproducibilidad.
Variable categórica (Categoric variable)Variable cualitativa que sólo es capaz de to-mar algunos valores dentro de un intervalode variación. Por ejemplo, el sexo. Tambiénllamada variable discreta.
Variable continua (Continous variable)Variable cuantitativa que es capaz de tomarcualquier valor real dentro de un cierto inter-valo de variación. Por ejemplo, la presión ar-terial o la glucemia.
Variable cualitativa (Qualitative variable)Variable que no es medible numérica-mente y que clasifica a los elementos deuna población. Por ejemplo, la clasifica-ción funcional de la sintomatología de lainsuficiencia cardíaca de la New York He-art Association en cuatro categorías (de laI a la IV).
Variable cuantitativa (Quantitative variable)Variable que es medible numéricamente. Porejemplo, la altura o la colesterolemia.
Variable de confusiónVéase Factor de confusión.
Variable de eficacia (Efficacy variable)Variable por la que se mide el resultado deun tratamiento o intervención en términos demejoría, curación o prevención de una enfer-medad.
Variable de estratificación (Stratification variable)Variable utilizada para clasificar las unidadesde observación en estratos. Generalmente seemplea para categorizar variables continuas.Por ejemplo, la división de la muestra en va-rios grupos según la hemoglobina glicosiladao los valores de antígeno prostático específi-co (APE, PSA).
Variable de resultado final primarioVéanse Variable principal de valoración y Va-riable secundaria de valoración.
Variable de resultado final secundarioVéanse Variable secundaria de valoración yVariable principal de valoración.
Variable de resultado final sustitutivaVéase Variable sustitutiva.
Variable dependiente (Dependent variable)Variable cuyo valor viene determinado por elde otra variable y cuyo valor no lo fija el ex-perimentador. Por ejemplo, en un ensayo clí-nico, para evaluar la eficacia de dos medica-mentos antihipertensivos, la variable depen-diente es la medida de la presión arterial. Enun estudio observacional en el que se analizala asociación entre obesidad y factores dieté-ticos, una variable dependiente sería el peso.
Variable discretaVéase Variable categórica.
Variable dura (Hard variable)Variable que depende de mediciones objeti-vas con posibilidad de medición experimen-tal exacta y de fácil reproducibilidad. Porejemplo, la muerte es la variable más dura,aunque no pueda reproducirse.
Variable independiente (Independent variable)Variable cuyo valor determina el de otra uotras variables y que es determinado por elexperimentador. Por ejemplo, en un ensayoclínico para evaluar la eficacia de dos medi-camentos antihipertensivos, una variable in-dependiente sería el medicamento emplea-do. En un estudio observacional en el que seanaliza la asociación entre cáncer de vejiga yfactores exógenos, una variable independien-te sería el grado de exposición a tóxicos in-dustriales. También llamada factor.
64
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Variable intermediaVéase Variable sustitutiva.
Variable ordinal (Ordinal variable)Variable en la que existe una relación de or-den entre sus valores. Por ejemplo, el niveleducativo. Véase Variable categórica.
Variable principal (End point, Primary outcomevariable, Outcome variable)
Variable (parámetro) que se considera másimportante para los objetivos del estudio. Esdeseable que sea relevante desde el puntode vista clínico y que su medición sea lo másobjetiva posible. El cálculo del tamaño de lamuestra se debe realizar teniendo en cuentaesta variable. Por ejemplo, en un ensayo clí-nico con antibióticos en el shock séptico, lavariable principal de evaluación sería la re-ducción de la mortalidad.
Variable secundaria (Secondary outcome va-riable, Secondary variable)
Variable, diferente de la principal, que seevalúa en un estudio. Puede tener mayor omenor relevancia clínica. En ocasiones, losresultados de estas variables pueden generarnuevas hipótesis. Por ejemplo, en un ensayoclínico con antibióticos en el shock séptico,la variable secundaria de evaluación sería lareducción de la estancia en unidades de cui-dados intensivos.
Variable sustitutiva (Surrogate end-point, Su-rrogate marker, Surrogate outcome variable,Surrogate variable)
Variable que se puede medir en lugar de lavariable principal, frecuentemente por la difi-cultad para realizar la medida o la identifica-ción de ésta. Cuando un estudio a corto pla-zo puede correlacionarse de forma fiable conun determinado beneficio terapéutico a largoplazo, la demostración del beneficio a cortoplazo (sustitutivo) se convierte en una evi-dencia suficiente de eficacia terapéutica. Porejemplo, un estudio que evaluara la eficaciade un fármaco en la diabetes podría tener,como variable sustitutiva, el descenso de laglucemia, en lugar de la variable principal deevaluación que sería la reducción de lascomplicaciones debidas a la enfermedad.También se la denomina variable intermediay variable subrogada.
VarianciaVéase Varianza.
Varianza (Variance)Medida de dispersión de una variable res-pecto a su media aritmética. Se calcula me-diante la suma de los cuadrados de las des-viaciones respecto a la media, dividida por
los grados de libertad. La raíz cuadrada de lavarianza se denomina desviación estándar.La fórmula para su cálculo es la siguiente:
n∑i=1 (xi - x)2
S = n-1
donde: xi = cada valorx- = median = número de individuos (grados libertad)S = varianza
Varianza, análisis de laVéase Análisis de la varianza.
Verificación de datos (Data verification)Establecimiento de la corrección y la exacti-tud de los datos de un estudio. Véase Valida-ción de datos.
Verosimilitudes, razón deVéase Cociente de probabilidades.
Violación del protocolo (Protocol violation)Falta de seguimiento o incumplimiento de loespecificado en el protocolo de un estudio.
Visita de aleatorización (Randomization visit)Visita en la que los pacientes seleccionadosque cumplen con todos los criterios de inclu-sión, con ninguno de los de exclusión, y quehan dado su consentimiento a participar enel estudio son asignados al azar a uno de losposibles tratamientos. En el caso de estudioscon medicamentos, se les entrega en esa vi-sita la medicación del estudio.
Visita de prealeatorización (Pre-randomizationvisit)
Cualquier visita realizada a un paciente po-tencialmente elegible con el propósito de es-tablecer su posible idoneidad para ser inclui-do en el estudio y que se realiza antes de lavisita de aleatorización.
Visita de seguimiento (Follow-up visit/control)Cualquier visita realizada a un paciente in-cluido en un ensayo clínico. Se realiza entrela visita inicial y la final.
Visita final (Final visit)Última visita a la que acude un paciente in-cluido en un ensayo clínico. Es la visita en laque se realiza la última evaluación y se cierrala historia clínica referente al estudio.
Visita inicial (Initial visit, Selection visit)Primera visita realizada a un paciente queparticipa en un ensayo clínico. En ella secomprueba la posible elegibilidad del pacien-te y se le solicita su consentimiento para par-ticipar en la investigación.
Voluntario (Volunteer)Sujeto que ha otorgado libremente su con-sentimiento para participar en un estudio
65
GLOSARIO

epidemiológico o un ensayo clínico. El con-sentimiento debe ser informado y quedar re-flejado en un impreso que firmará el volunta-rio o en ocasiones un testigo que documentala voluntariedad. Con frecuencia, se otorgaesta denominación a los voluntarios sanos,es decir, a los participantes en estudios noterapéuticos, como los farmacocinéticos y detolerabilidad de fase I. Sin embargo, el RD561/1993 art 4 considera voluntarios tanto alos pacientes como a los sanos, con lo quelos asimilaría a sujetos del ensayo.
Voluntario sano (Healthy volunteer, on-ill vo-lunteer)
Sujeto que, según la información disponible,no padece ninguna enfermedad significativacon relevancia para el estudio propuesto, cu-yas proporciones corporales y peso estándentro de los límites normales y que tiene unestado mental que le permite comprender y
otorgar su consentimiento válido para el es-tudio. Se utiliza también la denominación devoluntario no enfermo. Véase Sesgo del vo-luntario.
Yates, corrección de (Yate’s correction)Método estadístico utilizado en las tablas dedos por dos para corregir la prueba de ji alcuadrado por el hecho de que una distribu-ción continua de ji al cuadrado es utilizadapara una variable que es discreta. Esta co-rrección resta al valor absoluto de las dife-rencias entre la frecuencias observadas y es-peradas 0,5 unidades y, por tanto, hace dis-minuir el valor del estadístico, aumentando elvalor de p y disminuyendo la significación dela prueba. Habitualmente se utiliza cuandolos tamaños de las muestras son pequeños.
Zelen, método deVéase Estudio con consentimiento aleatoriza-do.
66
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Asociación Española de Toxicología (Repetto M,Sanz P, editores). Glosario de términos toxicológicos.Sevilla: Asociación Española de Toxicología, 1995.
Bakke OM, Carné X, García Alonso F. Ensayos clíni-cos con medicamentos. Barcelona: Doyma, 1994.
Bégaud B, Martín Arias LH. Diccionario de farmaco-epidemiología. Barcelona: Masson, 1997.
Bentzen N. An international glossary for general fa-mily practice. Fam Pract 1995; 12: 342-369.
Burley DM, Clarke JM, Lasagna L. PharmaceuticalMedicine. 2nd ed. London: Edward Arnold, 1993.
Carré MC, Jiménez J. Abreviaturas, siglas y acróni-mos en el mundo de los medicamentos. Farm Clín1995; 12: 62-75.
Committee for Proprietary Medicinal Products. Notefor guidance on general considerations for clinicaltrials (CPMP/ICH/291/95, ICH Topic E 8). London:EMEA, 1997.
Committee for Proprietary Medicinal Products. Notefor guidance on Structure and content of clinicalstudy reports (CPMP/ICH/137/95, ICH Topic E 3).London: EMEA, 1997.
Committee for Proprietary Medicinal Products(CPMP). Guidelines on the efficacy of medicinal pro-ducts for humans issued by the CPMP/efficacy wor-king party 1995-1996. Madrid: Ediciones Ergón,1996.
Dukes MNG (ed). Drug utilization studies. Copenha-gen: WHO Regional Publications, European series,no. 45, 1993.
Fletcher RH, Fletcher SW, Wagner EH. Epidemiolo-gía clínica. Barcelona: Ediciones Consulta, 1989.
Friedman LM, Furberg CD, DeMets DL. Fundamen-tals of clinical trials. 3rd ed. St Louis: Mosby, 1996.
García Alonso F, Bakke OM. Metodología del ensayoclínico. Monografías Dr Antonio Esteve 11. Barcelo-na: Fundación Dr Antonio Esteve, 1991.
Guía de ICH de BPC. Guía ICH tripartita y armoniza-da para la Buena Práctica Clínica (BPC). El Medica-mento I + D (nº 2, febrero), 1997.
Guyatt G, Patrick D, Feeny D. Glossary. ControlledClin Trials 1991; 12: 274S-280S.
Haynes RB, Mulrow CD, Huth EJ, Altman DG, Gard-ner MJ. More informative abstracts revisited. Ann In-tern Med 1990; 113: 69-76.
Hennekens CH, Buring JE. Epidemiology in Medici-ne. Boston: Little, Brown, 1987.
Informe de un grupo de estudio de la OMS. Farma-cología clínica: actividades, servicios y enseñanza.Ginebra: Organización Mundial de la Salud, serie in-formes técnicos nº 446, 1970.
Kleinbaum DG, Kupperr LL, Morgenstern H. Epi-demiology research. Principles and quantitativemethods. New York: Van Nostrand Reinhold,1982.
Laporte JR. Principios básicos de investigación clíni-ca. Madrid: Ediciones Ergón, 1993.
Laporte JR, Tognoni G (ed). Principios de epide-miología del medicamento. 2ª ed. Barcelona: Edi-ciones Científicas y Técnicas (Masson-Salvat),1993.
Last JM. Diccionario de epidemiología. Barcelona:Salvat, 1989.
Laurence DR, Shaw LC. Un glosario para farmacólo-gos. Monografías Dr Antonio Esteve 4. Barcelona:Fundación Dr Antonio Esteve, 1987.
Ley del Medicamento. Ley 25/1990, de 20 de di-ciembre, del medicamento. BOE sábado 22 de di-ciembre 1990.
Lloyd J, Raven A (ed). Handbook of clinical rese-arch. Edinburgh: Churchill Livingstone, 1994.
Mann RD, Rawlins MD, Auty RM. A textbook ofpharmaceutical medicine. Current practice. Carn-forth: The Partenon Publishing Group, 1993.
Bibliografía
67

Matos L (ed). Farmacoepidemiología. Santiago deCompostela: Xunta de Galicia, 1995.
Meinert CL. Clinical trials. Design, conduct andanalysis. New York: Oxford University Press, 1986.
Navarro FA. Traducción y lenguaje en Medicina.Monografías Dr Antonio Esteve 20. Barcelona: Fun-dación Dr Antonio Esteve, 1997.
Norman GR, Streiner DL. PDQ Statistics. Toronto:B.C. Decker, 1986.
Orden de 25 junio de 1985, del Ministerio de Sani-dad y Consumo, por la que se regulan los órganosencargados de farmacovigilancia. BOE de 11 de ju-nio de 1985 (corrección de errores en BOE de 26 dejulio de 1985).
Pocock SJ. Clinical trials. A practical approach. Chi-chester: John Wiley & Sons, 1984.
Raven A. Clinical trials. An introduction. 2nd ed. Ox-ford: Radcliffe Medical Press, 1993.
Real Academia Española. Diccionario de la LenguaEspañola. 21ª ed. Madrid: Espasa Calpe, 1992.
Real Decreto 561/1993, de 16 de abril, por el quese establecen los requisitos para la realización deensayos clínicos con medicamentos. BOE jueves 13de mayo de 1993.
Real Decreto 822/1993, de 28 de mayo, por el quese establecen los principios de buenas prácticas delaboratorio y su aplicación en la realización de estu-dios no clínicos sobre sustancias y productos quími-cos. BOE sábado 29 de mayo de 1993.
Real Decreto 2043/1994, de 14 de octubre sobreinspección y verificación de buenas prácticas de la-boratorio. BOE jueves 24 de noviembre de 1994.
Real Decreto 2000/1995, de 7 de diciembre, por elque se modifica el Real Decreto 767/1993, de 21 demayo, que regula la evaluación, autorización, regis-tro y condiciones de dispensación de especialidades
farmacéuticas y otros medicamentos de uso huma-no fabricados industrialmente. BOE viernes 12 deenero de 1996.
Rothman KL, Greenland S. Modern Epidemiology.2nd ed. Philadelphia: Lippincott-Raven. 1998.
Sackett DL, Richardson WS, Rosenberg W, HaynesRB. Medicina basada en la evidencia. Madrid: Chur-chill Livingstone, 1997.
Sackett DL, Haynes RB, Guyatt GH, Tugwell P. Clini-cal Epidemiology. 2nd ed. Boston: Little Brown,1991.
Serrano MA, García-Alonso F, González MJ, TristánC, Galende I, de Abajo FJ, et al. Ensayos clínicos enEspaña (1982-1988). Dirección General de Farma-cia y Productos Sanitarios. Monografías técnicas nº17. Madrid: Ministerio de Sanidad y Consumo,1990.
Sociedad Española de Bioquímica Clínica y Patolo-gía Molecular (Comisión de terminología). Termino-logía bioquímico-clínica: vocabulario de metrología.Química Clínica 1994; 13: 257-260.
Spilker B. Guide to clinical trials. New York: RavenPress, 1991.
Spilker B. Medical dictionary in six languages. NewYork: Raven Press, 1995.
Spilker B, Cuatrecasas P. Inside the drug industry.Barcelona: Prous Science, 1990.
Spriet A, Dupin-Spriet T, Simon P. Methodology ofclinical drug trials. 2nd ed. Basel: Karger, 1994.
Streiner DL, Norman GR, Munroe Blum H. PDQ Epi-demiology. Toronto: B.C. Decker, 1989.
Strom BL (ed). Pharmacoepidemiology. 2nd ed. Chi-chester: Wiley, 1994.
Termcat. Diccionari d’estadística. Barcelona: Funda-ció Barcelona-Termcat, 1994.
68
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

AAB . . . . .Aumento absoluto del beneficioAAR . . . . .Aumento absoluto del riesgoADR . . . . .Adverse drug reaction, Reacción
adversaAE . . . . . .Adverse event, Acontecimiento
adversoANCOVA .Análisis de la covarianzaANOVA . .Análisis de la varianzaABI . . . . .Absolute benefit increase, Aumento
absoluto del beneficioAR . . . . . .Absolute risk, Riesgo absolutoARI . . . . .Absolute risk increase, Aumento
absoluto del riesgoARB . . . . .Aumento relativo del beneficioARR . . . . .Aumento relativo del riesgoARR . . . . .Absolute risk reduction, Reducción
absoluta del riesgoAVAC . . . .Año/s de vida ajustado/s por
calidadBPC . . . . .Buena Práctica ClínicaBPF . . . . .Buena Práctica de FabricaciónBPL . . . . .Buena Práctica de LaboratorioBPM . . . .Buena Práctica de ManufacturaCANDA . .Computer Assisted New Drug
Application, Solicitud deautorización de especialidadfarmacéutica
CEC . . . . .Comité de Ensayos ClínicosCEIC . . . .Comité Ético de Investigación
ClínicaCI . . . . . .Confidence interval, Intervalo de
confianzaCPMP . . .Committee for Proprietary
Medicinal Products, Comité deEspecialidades Farmacéuticas
CRA . . . . .Clinical Research Assistant,Asistente de investigación clínica
CRD . . . . .Cuaderno de recogida de datosCRF . . . . .Case report form. Cuaderno de
recogida de datosCRO . . . . .Contract Research Organization,
Organización de investigación porcontrato
CV . . . . . .Coefficient of variation, Coeficientede variación
CV . . . . . .Curriculum vitaeCTX . . . . .Clinical Trial Exemption Certificate,
Producto en fase de investigaciónclínica
DCI . . . . .Denominación común internacionalDDD . . . .Defined daily dose, Dosis diaria
definidaDE . . . . . .Desviación estándarDF . . . . . .Degrees of freedom, Grados de
libertadDI . . . . . .Densidad de incidenciaDMT . . . .Dosis máxima toleradaDOE . . . . .Denominación oficial españolaDUS . . . . .Drug utilization study, Estudio de
utilización de medicamentosEBM . . . .Evidence-based medicine,
Medicina basada en la evidencia.EEM . . . . .Error estándar de la mediaEMEA . . .European Agency for the Evaluation
of Medicinal Products, AgenciaEuropea de Evaluación deMedicamentos
EUM . . . .Estudio de utilización demedicamentos
FDA . . . . .Food and Drug AdministrationGC . . . . . .Garantía de calidadGCP . . . . .Good clinical practice, Normas de
buena práctica clínicagl . . . . . . .Grados de libertadGLP . . . . .Good Laboratory Practice, Normas
de buena práctica de laboratorioGMP . . . .Good Manufacturing Practices
Normas de buena práctica defabricación
IC . . . . . .Intervalo de confianzaI+D . . . . .Investigación y desarrolloICH . . . . .International Conference on
HarmonizationIME . . . . .Índice de mortalidad
estandarizadaIND . . . . .Investigational New Drug, Producto
en fase de investigación clínicaINDA . . . .Investigational New Drug
Application, Solicitud de productoen fase de investigación clínica
Lista de siglas y acrónimos
69

INN . . . . .International Nonpropritery Name,Denominación común internacional
IRB . . . . .Institutional Review Board, Comitéde Ética, Comité Ético deInvestigación Clínica
MANOVA .Multivariate analysis of variance,Análisis de la varianza multivariado
MID . . . . .Minimum intolerated dose, Dosismínima no tolerada
MTD . . . .Maximum tolerated dose, Dosismáxima tolerada
NCE . . . . .New Chemical Entity, Nuevaentidad química
NDA . . . .New Drug Application, Solicitud deautorización de especialidadfarmacéutica
NNH . . . .Number needed to be treated toharm, Número necesario depacientes a tratar para producir unefecto perjudicial
NNT . . . .Number needed to treat/to betreated, Número necesario depacientes a tratar para evitar uncaso
OR . . . . . .Odds ratio, Razón de posibilidadesPEI . . . . .Producto en fase de investigación
clínicaPMS . . . .Postmarketing surveillance, Estudio
de postcomercializaciónPNT . . . . .Procedimientos normalizados de
trabajoQA . . . . . .Quality assurance, Garantía de
calidad
QALY . . . .Quality-adjusted life year, Año de vida ajustado por calidad
QAU . . . .Quality Assurance Unit, Unidad deGarantía de Calidad
RA . . . . . .Riesgo absolutoRAM . . . .Reacción adversa a medicamentoRAR . . . . .Reducción absoluta del riesgoR&D . . . .Research and development,
Investigación y desarrolloRBI . . . . .Relative benefit increase, Aumento
relativo del beneficioRCT . . . . .Randomized controlled trial,
Ensayo clínico controladoRME . . . .Razón de Mortalidad EstandarizadaROC . . . . .Receiver Operating Characteristic
Curve, Curva de característicasfuncionales
RR . . . . . .Relative risk, Riesgo relativoRRI . . . . .Relative risk increase, Aumento
relativo del riesgoRRR . . . .Relative risk reduction, Reducción
relativa del riesgoSD . . . . . .Standard deviation, Desviación
estándarSEM . . . . .Standard error of the mean,
Error estándar de la mediaSMR . . . .Standardized Mortality Ratio,
Razón de mortalidad estandarizadaSOP . . . . .Standard operating procedures,
Procedimientos normalizados detrabajo
UGC . . . .Unidad de garantía de calidad
70
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Absolute benefit increase . . . . . . . . . . . . . . . . . . . . .Aumento absoluto del beneficioAbsolute bioavailability . . . . . . . . . . . . . . . . . . . . . . .Biodisponibilidad absolutaAbsolute risk . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Riesgo absolutoAbsolute risk increase . . . . . . . . . . . . . . . . . . . . . . . .Aumento absoluto del riesgoAbsolute risk reduction . . . . . . . . . . . . . . . . . . . . . . .Reducción absoluta del riesgoAccessible population . . . . . . . . . . . . . . . . . . . . . . . .Población accesibleAccuracy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .PrecisiónActuarial method . . . . . . . . . . . . . . . . . . . . . . . . . . .Método actuarialAdherence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .CumplimientoAdverse drug reaction . . . . . . . . . . . . . . . . . . . . . . . .Reacción adversaAdverse effect . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Reacción adversaAdverse event . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Acontecimiento adversoAdverse reaction . . . . . . . . . . . . . . . . . . . . . . . . . . . .Reacción adversaAgreement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .ConcordanciaAlternative hypothesis . . . . . . . . . . . . . . . . . . . . . . . .Hipótesis alternativaAll-or-none response . . . . . . . . . . . . . . . . . . . . . . . . .Respuesta del todo o nadaAnalytical study . . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio analíticoAnalogy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Analogía, Razonamiento por analogíaAnalysis of secular trends . . . . . . . . . . . . . . . . . . . . .Análisis de series temporales (seculares)Analysis of temporal trends . . . . . . . . . . . . . . . . . . . .Análisis de series temporales (seculares)Analysis of covariance . . . . . . . . . . . . . . . . . . . . . . . .Análisis de la covarianza (ANCOVA)Analysis of variance . . . . . . . . . . . . . . . . . . . . . . . . .Análisis de la varianza (ANOVA)Area under the ROC curve . . . . . . . . . . . . . . . . . . . .Área bajo la curva de características
funcionalesAssignment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .AsignaciónAssociation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .AsociaciónAudit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .InspecciónAudit certificate . . . . . . . . . . . . . . . . . . . . . . . . . . . .Certificado de auditoríaAudit statement . . . . . . . . . . . . . . . . . . . . . . . . . . . .Certificado de auditoríaAugmented adverse reaction . . . . . . . . . . . . . . . . . . .Reacción adversa tipo AAutocorrelation . . . . . . . . . . . . . . . . . . . . . . . . . . . . .AutocorrelaciónAutonomy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .AutonomíaAverage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Media aritméticaBaseline characteristics . . . . . . . . . . . . . . . . . . . . . .Características basalesBaseline variable . . . . . . . . . . . . . . . . . . . . . . . . . . .Variable basalBasic research . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Investigación básicaBayes’s rule . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Teorema de BayesBayesian analysis . . . . . . . . . . . . . . . . . . . . . . . . . . .Análisis de BayesBayesian method . . . . . . . . . . . . . . . . . . . . . . . . . . .Método bayesianoBefore and after design . . . . . . . . . . . . . . . . . . . . . . .Diseño de antes y despuésBefore and after study . . . . . . . . . . . . . . . . . . . . . . .Estudio de antes y despuésBelmont report . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Belmont, informeBeneficence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .BeneficenciaBenefit to risk ratio . . . . . . . . . . . . . . . . . . . . . . . . . .Relación beneficio-riesgoBerkson bias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo de Berkson
Términos inglés-castellano
71

Bernouilli distribution . . . . . . . . . . . . . . . . . . . . . . . .Distribución de BernouilliBetween-subjects variability . . . . . . . . . . . . . . . . . . .Variabilidad interindividualBias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .SesgoBinary variable . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Variable binariaBinomial distribution . . . . . . . . . . . . . . . . . . . . . . . . .Distribución binomialBioavailability . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .BiodisponibilidadBioequivalence . . . . . . . . . . . . . . . . . . . . . . . . . . . . .BioequivalenciaBiological gradient . . . . . . . . . . . . . . . . . . . . . . . . . .Gradiente biológicoBiomedical ethics . . . . . . . . . . . . . . . . . . . . . . . . . . .BioéticaBizarre adverse reaction . . . . . . . . . . . . . . . . . . . . . .Reacción adversa tipo BBlind/ed . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .CiegoBlind/ed evaluation . . . . . . . . . . . . . . . . . . . . . . . . . .Evaluación ciega por tercerosBlock/ed randomization . . . . . . . . . . . . . . . . . . . . . .Asignación aleatoria por bloquesBlocking randomization . . . . . . . . . . . . . . . . . . . . . .Asignación aleatoria por bloquesBridging study . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio puenteCalibration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .CalibraciónCarry-over effect . . . . . . . . . . . . . . . . . . . . . . . . . . . .Efecto residualCase control study . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio de casos y controlesCase fatality rate . . . . . . . . . . . . . . . . . . . . . . . . . . . .Tasa de letalidadCase-finding . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Búsqueda de casosCase report form . . . . . . . . . . . . . . . . . . . . . . . . . . . .Cuaderno de recogida de datosCase series study . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio de series de casos clínicosCategoric variable . . . . . . . . . . . . . . . . . . . . . . . . . . .Variable categóricaCausality . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Causalidad, inferencia causalCausation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Causalidad, inferencia causalChance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .AzarChi-square . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Ji al cuadradoChi-square distribution . . . . . . . . . . . . . . . . . . . . . . .Distribución de ji al cuadradoChi-square test . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Prueba de ji al cuadradoClinical drug development phases . . . . . . . . . . . . . . .Fase/s del desarrollo clínico de un fármacoClinical Epidemiology . . . . . . . . . . . . . . . . . . . . . . . .Epidemiología clínicaClinical guideline . . . . . . . . . . . . . . . . . . . . . . . . . . .Protocolo clínicoClinical Pharmacology . . . . . . . . . . . . . . . . . . . . . . . .Farmacología clínicaClinical practice guideline . . . . . . . . . . . . . . . . . . . . .Protocolo clínicoClinical project leader . . . . . . . . . . . . . . . . . . . . . . . .Responsable de proyecto de investigaciónClinical protocol . . . . . . . . . . . . . . . . . . . . . . . . . . . .Protocolo clínicoClinical relevance . . . . . . . . . . . . . . . . . . . . . . . . . . .Relevancia clínicaClinical research . . . . . . . . . . . . . . . . . . . . . . . . . . . .Investigación clínicaClinical Research Assistant . . . . . . . . . . . . . . . . . . . .Asistente de investigación clínicaClinical significance . . . . . . . . . . . . . . . . . . . . . . . . .Significación clínicaClinical trial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínicoClinical trial phase . . . . . . . . . . . . . . . . . . . . . . . . . .Fase/s del ensayo clínicoCochrane collaboration . . . . . . . . . . . . . . . . . . . . . . .Colaboración CochraneCoding dictionary . . . . . . . . . . . . . . . . . . . . . . . . . . .Diccionario de codificaciónCoefficient of correlation . . . . . . . . . . . . . . . . . . . . . .Coeficiente de correlaciónCoefficient of variation . . . . . . . . . . . . . . . . . . . . . . . .Coeficiente de variaciónCoherence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .CoherenciaCohort study . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio de cohorteCommittee for Proprietary Medicinal Products . . . . . .Comité de Especialidades FarmacéuticasCommunity trial . . . . . . . . . . . . . . . . . . . . . . . . . . . .Ensayo comunitarioComparative group . . . . . . . . . . . . . . . . . . . . . . . . . .Grupo controlComparison group . . . . . . . . . . . . . . . . . . . . . . . . . .Grupo controlCompassionate use . . . . . . . . . . . . . . . . . . . . . . . . . .Uso compasivoCompliance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Cumplimiento, observanciaConcurrent control . . . . . . . . . . . . . . . . . . . . . . . . . .Control concurrenteConcurrent validity . . . . . . . . . . . . . . . . . . . . . . . . . .Validez concurrente
72
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Confidence interval . . . . . . . . . . . . . . . . . . . . . . . . . .Intervalo de confianzaConfidence limit . . . . . . . . . . . . . . . . . . . . . . . . . . . .Límite de confianzaConfounding . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Factor de confusiónConsistency . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .ConsistenciaContract Research Organization . . . . . . . . . . . . . . . .Organización de investigación por contratoConstruct validity . . . . . . . . . . . . . . . . . . . . . . . . . . .Validez teóricaContent validity . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Validez de contenidoContingency tables . . . . . . . . . . . . . . . . . . . . . . . . . .Tabla de contingenciaContinous variable . . . . . . . . . . . . . . . . . . . . . . . . . .Variable continuaControl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Control, Grupo controlControl group . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Grupo controlControlled clinical trial . . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico controladoCorrelation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .CorrelaciónCorrelation study . . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio de correlaciónCost-benefit analysis . . . . . . . . . . . . . . . . . . . . . . . . .Análisis de coste-beneficioCost-benefit ratio . . . . . . . . . . . . . . . . . . . . . . . . . . .Razón de coste-beneficioCost-effectiveness analysis . . . . . . . . . . . . . . . . . . . .Análisis de coste-efectividadCost-effectiveness ratio . . . . . . . . . . . . . . . . . . . . . . .Razón de coste-efectividadCost-minimisation analysis . . . . . . . . . . . . . . . . . . . .Análisis de minimización de costesCost-utility analysis . . . . . . . . . . . . . . . . . . . . . . . . . .Análisis de costo-utilidadCounselling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .ConsejoCounterfeiting . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .FalsificaciónCriterion standard . . . . . . . . . . . . . . . . . . . . . . . . . . .Patrón de comparaciónCriterion validity . . . . . . . . . . . . . . . . . . . . . . . . . . . .Validez de criterioCronbach’s alpha . . . . . . . . . . . . . . . . . . . . . . . . . . .Alfa de CronbachCross-over design . . . . . . . . . . . . . . . . . . . . . . . . . . .Diseño cruzadoCross-sectional study . . . . . . . . . . . . . . . . . . . . . . . .Estudio transversalCumulative incidence . . . . . . . . . . . . . . . . . . . . . . . .Incidencia acumuladaCumulative metaanalysis . . . . . . . . . . . . . . . . . . . . . .Metaanálisis acumulativoCurriculum vitae . . . . . . . . . . . . . . . . . . . . . . . . . . . .Curriculum vitaeData entry . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Datos, entrada de Data validation . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Validación de datosData verification . . . . . . . . . . . . . . . . . . . . . . . . . . . .Verificación de datosDecile . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .DecilDeferred consent . . . . . . . . . . . . . . . . . . . . . . . . . . .Consentimiento informado diferidoDefined daily dose . . . . . . . . . . . . . . . . . . . . . . . . . .Dosis diaria definidaDegrees of freedom . . . . . . . . . . . . . . . . . . . . . . . . .Grados de libertadDependent variable . . . . . . . . . . . . . . . . . . . . . . . . . .Variable dependienteDescriptive study . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio descriptivoDetection bias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo de detecciónDiagnostic validity . . . . . . . . . . . . . . . . . . . . . . . . . . .Validez diagnósticaDigit preference . . . . . . . . . . . . . . . . . . . . . . . . . . . .Preferencia de números dígitosDirect standardization . . . . . . . . . . . . . . . . . . . . . . . .Ajuste directoDistribution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .DistribuciónDose finding study . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio de búsqueda de dosisDose ranging study . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio de búsqueda de dosisDose response . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Dosis respuestaDose-response relationship . . . . . . . . . . . . . . . . . . . .Relación dosis-respuestaDouble blind/ed . . . . . . . . . . . . . . . . . . . . . . . . . . . .Doble ciegoDouble dummy . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Doble simulaciónDouble mask/ed . . . . . . . . . . . . . . . . . . . . . . . . . . . .Doble ciegoDrop-out . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .AbandonoDrug . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Fármaco, medicamento, sustancia medicinalDrug Approval Application . . . . . . . . . . . . . . . . . . . .Solicitud de autorización de especialidad
farmacéuticaDrug development phases . . . . . . . . . . . . . . . . . . . .Fase/s del desarrollo de un fármaco
73
TÉRMINOS INGLÉS-CASTELLANO

Drug interaction . . . . . . . . . . . . . . . . . . . . . . . . . . . .Interacción farmacológicaDrug regulatory approval . . . . . . . . . . . . . . . . . . . . . .Registro de especialidades farmacéuticasDrug regulatory review . . . . . . . . . . . . . . . . . . . . . . .Registro de especialidades farmacéuticasDrug utilization study . . . . . . . . . . . . . . . . . . . . . . . .Estudio de utilización de medicamentosEcological fallacy . . . . . . . . . . . . . . . . . . . . . . . . . . .Falacia ecológicaEffect . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .EfectoEffectiveness . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .EfectividadEfficacy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .EficaciaEfficacy variable . . . . . . . . . . . . . . . . . . . . . . . . . . . .Variable de eficaciaEfficiency . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .EficienciaEligible patient . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Paciente elegibleEmpirical validity . . . . . . . . . . . . . . . . . . . . . . . . . . . .Validez empíricaEnd point . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Variable principal de evaluaciónEpidemiological research . . . . . . . . . . . . . . . . . . . . .Investigación epidemiológicaEpidemiology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .EpidemiologíaEquivalence study . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio de equivalenciaEquivalence trial . . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio de equivalenciaα-Error . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Error αβ-Error . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Error βEthical committee . . . . . . . . . . . . . . . . . . . . . . . . . . .Comité de Ética, Comité de Ética AsistencialEthical standards . . . . . . . . . . . . . . . . . . . . . . . . . . .Principios bioéticosEthics Committee . . . . . . . . . . . . . . . . . . . . . . . . . . .Comité de Ética, Comité de Ética AsistencialEthics manual . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Deontología médica, código de European Agency for the Evaluation . . . . . . . . . . . . .Agencia Europea de Evaluación
of Medicinal Products . . . . . . . . . . . . . . . . . . . . . . de MedicamentosExperimental evidence . . . . . . . . . . . . . . . . . . . . . . .Evidencia experimentalExperimental group . . . . . . . . . . . . . . . . . . . . . . . . . .Grupo experimentalExperimental study . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio experimentalExpert report . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Informe de expertoExplanatory analysis . . . . . . . . . . . . . . . . . . . . . . . . .Análisis explicativoExplanatory clinical trial . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico explicativoExternal validity . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Validez externaEvidence-based medicine . . . . . . . . . . . . . . . . . . . . .Medicina basada en la evidenciaFace validity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Validez frontalFactor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .FactorFactorial design . . . . . . . . . . . . . . . . . . . . . . . . . . . .Diseño factorialFallacy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .FalaciaFalse negative rate . . . . . . . . . . . . . . . . . . . . . . . . . .Tasa de falsos negativos o pseudonegativosFalse negative result . . . . . . . . . . . . . . . . . . . . . . . . .Falso negativo, resultadoFalse positive rate . . . . . . . . . . . . . . . . . . . . . . . . . . .Tasa de falsos positivos o pseudopositivosFalse positive result . . . . . . . . . . . . . . . . . . . . . . . . .Falso positivo, resultadoFalsification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .FalsificaciónFinal report . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Informe finalFinal visit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Visita finalFisher’s exact test . . . . . . . . . . . . . . . . . . . . . . . . . . .Prueba exacta de FisherFishing expedition studies . . . . . . . . . . . . . . . . . . . . .“Salir de pesca”Fishing expedition analysis . . . . . . . . . . . . . . . . . . . .“Salir de pesca”Follow-up control . . . . . . . . . . . . . . . . . . . . . . . . . . .Visita de seguimientoFollow-up visit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Visita de seguimientoFraud . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .FraudeFrequency distribution . . . . . . . . . . . . . . . . . . . . . . .Distribución de frecuenciasGeneric drug . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Medicamento genéricoGeometric mean . . . . . . . . . . . . . . . . . . . . . . . . . . . .Media geométricaGold standard . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Patrón de oroGood clinical practices . . . . . . . . . . . . . . . . . . . . . . .Normas de buena práctica clínicaGood laboratory practices . . . . . . . . . . . . . . . . . . . . .Normas de buena práctica de laboratorio
74
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Good manufacturing practices . . . . . . . . . . . . . . . . .Normas de buena práctica de fabricaciónGraded responses . . . . . . . . . . . . . . . . . . . . . . . . . . .Respuesta gradualHard variable . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Variable duraHawthorne effect . . . . . . . . . . . . . . . . . . . . . . . . . . .Efecto HawthorneHealth Education . . . . . . . . . . . . . . . . . . . . . . . . . . .Educación sanitariaHealth Promotion . . . . . . . . . . . . . . . . . . . . . . . . . . .Promoción de la saludHealth-related quality of life . . . . . . . . . . . . . . . . . . .Calidad de vida relacionada con la saludHealth services research . . . . . . . . . . . . . . . . . . . . . .Investigación en servicios sanitariosHealthy volunteer . . . . . . . . . . . . . . . . . . . . . . . . . . .Voluntario sanoHealthy worker effect . . . . . . . . . . . . . . . . . . . . . . . .Efecto del trabajador sanoHelsinki, declaration of . . . . . . . . . . . . . . . . . . . . . . .Helsinki, declaración deHeterogeneity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .HeterogeneidadHeteroscedasticity . . . . . . . . . . . . . . . . . . . . . . . . . .HeteroscedasticidadHistorical cohorts . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio de cohorte retrospectivoHistorical control . . . . . . . . . . . . . . . . . . . . . . . . . . . .Control históricoHistorical control study . . . . . . . . . . . . . . . . . . . . . . .Estudio de controles históricosHomogeneity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .HomogeneidadHomoscedasticity . . . . . . . . . . . . . . . . . . . . . . . . . . .HomoscedasticidadHypothesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .HipótesisIceberg phenomenon . . . . . . . . . . . . . . . . . . . . . . . .Fenómeno icebergIdiosincracy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .IdiosincrasiaIncidence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .IncidenciaIncidence density . . . . . . . . . . . . . . . . . . . . . . . . . . .Densidad de incidenciaIndependence . . . . . . . . . . . . . . . . . . . . . . . . . . . . .IndependenciaIndependent Ethics Commitee . . . . . . . . . . . . . . . . .Comité de ÉticaIndependent variable . . . . . . . . . . . . . . . . . . . . . . . .Variable independienteIndicator . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .IndicadorIndirect standardization . . . . . . . . . . . . . . . . . . . . . .Ajuste indirectoInformation bias . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo de informaciónInformed consent . . . . . . . . . . . . . . . . . . . . . . . . . . .Consentimiento informadoInitial visit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Visita inicialInspection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .InspecciónIntention to treat analysis . . . . . . . . . . . . . . . . . . . . .Análisis por intención de tratarInteraction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .InteracciónInterim analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . .Análisis intermedioInternal consistency . . . . . . . . . . . . . . . . . . . . . . . . .Consistencia internaInternal validity . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Validez internaInternational Nonproprietary Name . . . . . . . . . . . . . .Denominación común internacionalIntervention . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .IntervenciónIntervention study . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio de intervención, Ensayo clínicoInterviewer bias . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo del entrevistadorIntraclass correlation . . . . . . . . . . . . . . . . . . . . . . . . .Correlación intraclaseInvestigational New Drug . . . . . . . . . . . . . . . . . . . . .Producto en fase de investigación clínicaInvestigational New Drug Application . . . . . . . . . . . .Solicitud de producto en fase
de investigación clínicaInvestigator . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .InvestigadorInvestigator brochure . . . . . . . . . . . . . . . . . . . . . . . .Manual de investigadorJudgement by analogy . . . . . . . . . . . . . . . . . . . . . . .Razonamiento por analogíaKaplan-Meier method . . . . . . . . . . . . . . . . . . . . . . . .Método de Kaplan-MeierKappa index . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Índice de KappaKurtosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .CurtosisLasagna’s law . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Lasagna, ley de Latin square . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Cuadrado latinoLineal regression . . . . . . . . . . . . . . . . . . . . . . . . . . . .Regresión linealLikelihood ratio . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Cociente de probabilidadesLogistic regression . . . . . . . . . . . . . . . . . . . . . . . . . .Regresión logística
75
TÉRMINOS INGLÉS-CASTELLANO

Log-rank test . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Prueba de log-rankMann-Whitney U test . . . . . . . . . . . . . . . . . . . . . . . .Prueba U de Mann-WhitneyManual of procedures . . . . . . . . . . . . . . . . . . . . . . . .Manual de procedimientosMask/ed . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .CiegoMatching . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .EmparejamientoMaximal tolerated dose . . . . . . . . . . . . . . . . . . . . . . .Dosis máxima toleradaMcNemar test . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Prueba de McNemarMean . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Media aritméticaMeasurement bias . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo de mediciónMedian . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .MedianaMedical record . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Historia clínicaMedicinal substance . . . . . . . . . . . . . . . . . . . . . . . . .Sustancia medicinalMedicine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Medicamento, sustancia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . medicinal
Memory bias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo de memoriaMegatrial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .MegaensayoMeta-analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .MetaanálisisMinimisation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .MinimizaciónMinimal intolerated dose . . . . . . . . . . . . . . . . . . . . . .Dosis mínima no toleradaMissclassification bias . . . . . . . . . . . . . . . . . . . . . . . .Sesgo de clasificación incorrectaMode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .ModaMolecular epidemiology . . . . . . . . . . . . . . . . . . . . . .Epidemiología molecularMonitor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .MonitorMonitoring . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .MonitorizaciónMontecarlo simulation . . . . . . . . . . . . . . . . . . . . . . . .Simulación de MontecarloMorbidity rate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Tasa de morbilidadMortality rate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Tasa de mortalidadMulticenter . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .MulticéntricoMulticenter clinical trial . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico multicéntricoMulticenter study . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio multicéntricoMultiple comparison test . . . . . . . . . . . . . . . . . . . . . .Comparaciones estadísticas múltiplesMultiple regression . . . . . . . . . . . . . . . . . . . . . . . . . .Regresión múltipleMultivariate analysis of variance . . . . . . . . . . . . . . . .Análisis de la varianza multivariadoN of 1 trial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico de n igual a 1Negative predictive value . . . . . . . . . . . . . . . . . . . . .Valor predictivo negativoNested case control study . . . . . . . . . . . . . . . . . . . . .Estudio anidado de casos y controlesNew Chemical Entity . . . . . . . . . . . . . . . . . . . . . . . . .Nueva entidad químicaNew Drug Application . . . . . . . . . . . . . . . . . . . . . . . .Solicitud de autorización de especialidad
farmacéuticaNocebo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .NoceboNon-blind/ed clinical trial . . . . . . . . . . . . . . . . . . . . .Ensayo clínico abiertoNon-controlled clinical trial . . . . . . . . . . . . . . . . . . . .Ensayo clínico no controladoNon-ill volunteer . . . . . . . . . . . . . . . . . . . . . . . . . . . .Voluntario sanoNon maleficence . . . . . . . . . . . . . . . . . . . . . . . . . . .No maleficenciaNon-parametric statistical test . . . . . . . . . . . . . . . . . .Puebra estadística no paramétricaNon-parametric test . . . . . . . . . . . . . . . . . . . . . . . . .Prueba estadística no paramétricaNon-therapeutic study . . . . . . . . . . . . . . . . . . . . . . .Estudio sin finalidad terapéuticaNon-therapeutic trial . . . . . . . . . . . . . . . . . . . . . . . . .Estudio sin finalidad terapéuticaNormal distribution . . . . . . . . . . . . . . . . . . . . . . . . . .Distribución normalNull hypothesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Hipótesis nulaNumber needed to be treated . . . . . . . . . . . . . . . . .Número necesario de pacientes
a tratar para evitar un casoNumber needed to treat . . . . . . . . . . . . . . . . . . . . . .Número necesario de pacientes
a tratar para evitar un casoNumber needed to be treated to harm . . . . . . . . . . .Número de pacientes a tratar para producir
un efecto perjudicial
76
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Number needed to treat to harm . . . . . . . . . . . . . . . .Número de pacientes a tratar para producir un efecto perjudicial
Number preference . . . . . . . . . . . . . . . . . . . . . . . . .Preferencia de númerosNuremberg code . . . . . . . . . . . . . . . . . . . . . . . . . . .Nuremberg, código deObject . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .ObjetoObjective . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Objetivo, ObjetoObserver bias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo de observación
o del observadorOdds ratio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Razón de posibilidadesOne sided test . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Prueba unilateralOne-tailed test . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Prueba unilateralOpen clinical trial . . . . . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico abiertoOpen label clinical trial . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico abiertoOrdinal variable . . . . . . . . . . . . . . . . . . . . . . . . . . . .Variable ordinalOrphan drug . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Medicamento huérfanoOutcome variable . . . . . . . . . . . . . . . . . . . . . . . . . . .Variable principal de evaluaciónOutlier . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Valor atípicoOutpatient . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Paciente ambulatorioOverlapping effect . . . . . . . . . . . . . . . . . . . . . . . . . . .Efecto de solapamientop, Probability value . . . . . . . . . . . . . . . . . . . . . . . . . .p, valor de probabilidadPaired t-test . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Prueba t para datos emparejadosParallel clinical trial . . . . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico paraleloParallel design . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Diseño paraleloParameter . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .ParámetroParametric statistical test . . . . . . . . . . . . . . . . . . . . .Prueba estadística paramétricaParametric test . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Prueba estadística paramétricaPatient follow-up . . . . . . . . . . . . . . . . . . . . . . . . . . . .Seguimiento del pacientePearson’s correlation . . . . . . . . . . . . . . . . . . . . . . . .Correlación de PearsonPeer review . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Revisión científicaPercentile . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .PercentilPilot study . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio pilotoPharmaceutical form . . . . . . . . . . . . . . . . . . . . . . . .Forma farmacéuticaPharmacodynamics . . . . . . . . . . . . . . . . . . . . . . . . .FarmacodinamiaPharmacoeconomy . . . . . . . . . . . . . . . . . . . . . . . . . .FarmacoeconomíaPharmacoepidemiology . . . . . . . . . . . . . . . . . . . . . . .FarmacoepidemiologíaPharmacogenetics . . . . . . . . . . . . . . . . . . . . . . . . . .FarmacogenéticaPharmacokinetics . . . . . . . . . . . . . . . . . . . . . . . . . . .FarmacocinéticaPharmacological interaction . . . . . . . . . . . . . . . . . . .Interacción farmacológicaPharmacology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .FarmacologíaPharmacopoeia . . . . . . . . . . . . . . . . . . . . . . . . . . . .FarmacopeaPharmacovigilance . . . . . . . . . . . . . . . . . . . . . . . . . .FarmacovigilanciaPharmacy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .FarmaciaPhase 0 trial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico en fase 0Phase I trial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico en fase IPhase II trial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico en fase IIPhase III trial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico en fase IIIPhase IV trial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico en fase IVPilot study . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio pilotoPivotal study . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio fundamentalPlacebo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .PlaceboPlacebo effect . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Efecto placeboPlacebo reactor . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sensibilidad al placeboPlausibility of causal relationship . . . . . . . . . . . . . . . .PlausibilidadPlay the winner design/trial . . . . . . . . . . . . . . . . . . . .Diseño play the winner
(regla de jugar al ganador)Poisson distribution . . . . . . . . . . . . . . . . . . . . . . . . .Distribución de Poisson
77
TÉRMINOS INGLÉS-CASTELLANO

Population atributable risk . . . . . . . . . . . . . . . . . . . .Riesgo absoluto poblacionalPositive predictive value . . . . . . . . . . . . . . . . . . . . . .Valor predictivo positivoPostmarketing drug surveillance study . . . . . . . . . . .FarmacovigilanciaPostmarketing surveillance study . . . . . . . . . . . . . . .Estudio de postcomercializaciónPost-randomization losses . . . . . . . . . . . . . . . . . . . . .Pérdidas postaleatorizaciónPower . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Poder estadístico, Potencia estadísticaPragmatic clinical trial . . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico pragmáticoPreclinical research . . . . . . . . . . . . . . . . . . . . . . . . .Investigación preclínicaPredictive validity . . . . . . . . . . . . . . . . . . . . . . . . . . .Validez predictivaPreference between two treatment periods . . . . . . . .Preferencia entre dos períodos de tratamientoPre-randomization losses . . . . . . . . . . . . . . . . . . . . .Pérdidas prealeatorizaciónPre-randomization visit . . . . . . . . . . . . . . . . . . . . . . .Visita prealeatorizaciónPrevalence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .PrevalenciaPrevalence bias . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo de incidencia/prevalenciaPrevention . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .PrevenciónPrimary objective . . . . . . . . . . . . . . . . . . . . . . . . . . .Objetivo principalPrimary outcome variable . . . . . . . . . . . . . . . . . . . . .Variable principal de evaluaciónPrimary Prevention . . . . . . . . . . . . . . . . . . . . . . . . . .Prevención primariaPrimordial Prevention . . . . . . . . . . . . . . . . . . . . . . . .Prevención primordialPrincipal investigator . . . . . . . . . . . . . . . . . . . . . . . . .Investigador principalProbability . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .ProbabilidadProbability value . . . . . . . . . . . . . . . . . . . . . . . . . . . .p, valor de probabilidadProbit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .ProbitPrognostic factor . . . . . . . . . . . . . . . . . . . . . . . . . . . .Factor pronósticoPromotional clinical trial . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico promocionalProspective study . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio prospectivoProtocol . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .ProtocoloProtocol violation . . . . . . . . . . . . . . . . . . . . . . . . . . .Violación del protocoloProtopathic bias . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo protopáticoProvider bias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo del proveedorPublication bias . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo de publicaciónQualitative variable . . . . . . . . . . . . . . . . . . . . . . . . . .Variable cualitativaQuality assessment . . . . . . . . . . . . . . . . . . . . . . . . . .Garantía de calidadQuality assessment . . . . . . . . . . . . . . . . . . . . . . . . . .Control de calidadQuality assurance . . . . . . . . . . . . . . . . . . . . . . . . . . .Garantía de calidadQuality assurance unit . . . . . . . . . . . . . . . . . . . . . . .Unidad de garantía de calidadQuality control . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Control de calidadQuality of life . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Calidad de vidaQuantal response . . . . . . . . . . . . . . . . . . . . . . . . . . .Respuesta del todo o nadaQuantitative variable . . . . . . . . . . . . . . . . . . . . . . . . .Variable cuantitativaQuartile . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .CuartilQuestionnaire . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .CuestionarioRandom . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .AleatorioRandom . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .AzarRandom number . . . . . . . . . . . . . . . . . . . . . . . . . . .Número aleatorioRandom sample . . . . . . . . . . . . . . . . . . . . . . . . . . . .Muestra aleatoriaRandom variable . . . . . . . . . . . . . . . . . . . . . . . . . . .Variable aleatoriaRandomization . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Aleatorización, Asignación aleatoriaRandomization code . . . . . . . . . . . . . . . . . . . . . . . . .Código de aleatorizaciónRandomization visit . . . . . . . . . . . . . . . . . . . . . . . . . .Visita de aleatorizaciónRandomized assignment . . . . . . . . . . . . . . . . . . . . . .Asignación aleatoriaRandomized controlled trial . . . . . . . . . . . . . . . . . . .Ensayo clínico controladoRandomized controlled clinical trial . . . . . . . . . . . . . .Ensayo clínico controladoRandomized consent study . . . . . . . . . . . . . . . . . . . .Estudio de consentimiento aleatorizadoRange . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .RangoRank . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Ordenación (véase Rango)
78
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Rank order . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Orden de rangosRate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .TasaRatio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .RazónReasoning by analogy . . . . . . . . . . . . . . . . . . . . . . . .Razonamiento por analogíaReceiver Operating Characteristic . . . . . . . . . . . . . . .Curva de características funcionalesReceiver Operating Characteristic Curve . . . . . . . . . .Curva de características funcionalesRecord-linkage study/method . . . . . . . . . . . . . . . . . .Estudio/método de conexión de registrosRecruitment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .ReclutamientoRefeering . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Revisión científicaReference bias . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo de referenciaReferral bias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo de remisiónRegistry . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .RegistroRegression analysis . . . . . . . . . . . . . . . . . . . . . . . . .Análisis de regresiónRegression to the mean . . . . . . . . . . . . . . . . . . . . . .Regresión a la mediaRegression toward the mean . . . . . . . . . . . . . . . . . . .Regresión a la mediaRegulatory approval application . . . . . . . . . . . . . . . .Solicitud de autorización de especialidad
farmacéuticaRelative benefit increase . . . . . . . . . . . . . . . . . . . . . .Aumento relativo del beneficioRelative bioavailability . . . . . . . . . . . . . . . . . . . . . . . .Biodisponibilidad relativaRelative risk . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Riesgo relativoRelative risk increase . . . . . . . . . . . . . . . . . . . . . . . .Aumento relativo del riesgoRelative risk reduction . . . . . . . . . . . . . . . . . . . . . . .Reducción relativa del riesgoReliability . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Fiabilidad, precisión, repetibilidadRemote data entry . . . . . . . . . . . . . . . . . . . . . . . . . .Datos, entrada a distanciaReplacement of subjects . . . . . . . . . . . . . . . . . . . . . .Substitución de sujetosRepresentative sample . . . . . . . . . . . . . . . . . . . . . . .Muestra representativaReproducibility . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Fiabilidad, precisión, reproducibilidadRescue treatment . . . . . . . . . . . . . . . . . . . . . . . . . . .Tratamiento de rescateResearch . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .InvestigaciónResearch Committee . . . . . . . . . . . . . . . . . . . . . . . .Comité de InvestigaciónResearcher . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .InvestigadorResearch and development . . . . . . . . . . . . . . . . . . .Investigación y desarrolloRespect for autonomy . . . . . . . . . . . . . . . . . . . . . . . .Respeto por las personasResponsiveness . . . . . . . . . . . . . . . . . . . . . . . . . . . .SensibilidadRetrospective cohort study . . . . . . . . . . . . . . . . . . . .Estudio de cohorte retrospectivoRetrospective study . . . . . . . . . . . . . . . . . . . . . . . . .Estudio retrospectivoRisk . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .RiesgoRisk factor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Factor de riesgoRisk marker . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Marcador de riesgoRun-in period . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Período de preinclusiónSafety . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .SeguridadSafety study . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio de seguridadSample . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .MuestraSample size . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Tamaño de la muestraSample size calculation . . . . . . . . . . . . . . . . . . . . . . .Cálculo del tamaño de la muestraScreening . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .CribadoSecondary objective . . . . . . . . . . . . . . . . . . . . . . . . .Objetivo secundarioSecondary prevention . . . . . . . . . . . . . . . . . . . . . . . .Prevención secundariaSecondary outcome variable . . . . . . . . . . . . . . . . . . .Variable secundaria de evaluaciónSecondary variable . . . . . . . . . . . . . . . . . . . . . . . . . .Variable secundaria de evaluaciónSelection bias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo de selecciónSelection visit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Visita inicialSensibility . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .SensibilidadSensitivity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .SensibilidadSensitivity analysis . . . . . . . . . . . . . . . . . . . . . . . . . .Análisis de sensibilidadSequential analysis . . . . . . . . . . . . . . . . . . . . . . . . . .Análisis secuencial
79
TÉRMINOS INGLÉS-CASTELLANO

Sequential design . . . . . . . . . . . . . . . . . . . . . . . . . . .Diseño secuencialSerendipity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .SerendipitiaSerious adverse event . . . . . . . . . . . . . . . . . . . . . . . .Acontecimiento adverso graveSham . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .SimulaciónSimple mask/ed . . . . . . . . . . . . . . . . . . . . . . . . . . . .Simple ciegoSimple randomization . . . . . . . . . . . . . . . . . . . . . . . .Asignación aleatoria simpleSimpson’s paradox . . . . . . . . . . . . . . . . . . . . . . . . . .Paradoja de SimpsonSimulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .SimulaciónSingle blind/ed . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Simple ciegoSingle center clinical trial . . . . . . . . . . . . . . . . . . . . .Ensayo clínico unicéntricoSingle center study . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio unicéntricoSingle clinical trial . . . . . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico unicéntricoSkewness . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .AsimetríaSoft variable . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Variable blandaSpearman’s correlation . . . . . . . . . . . . . . . . . . . . . . .Correlación de SpearmanSpecificity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .EspecificidadSpontaneous reporting of adverse reactions . . . . . . .Notificación espontánea de reacciones
adversasSponsor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .PromotorStandard deviation . . . . . . . . . . . . . . . . . . . . . . . . . .Desviación estándarStandard error of the mean . . . . . . . . . . . . . . . . . . . .Error estándar de la mediaStandardization . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Ajuste Standardization . . . . . . . . . . . . . . . . . . . . . . . . . . . . .NormalizaciónStandard Operating Procedures . . . . . . . . . . . . . . . .Procedimientos normalizados de trabajoStandard treatment . . . . . . . . . . . . . . . . . . . . . . . . . .Tratamiento de referenciaStandarized mortality ratio . . . . . . . . . . . . . . . . . . . . .Índice de mortalidad estandarizadaStatistical inference . . . . . . . . . . . . . . . . . . . . . . . . . .Inferencia estadísticaStatistical power . . . . . . . . . . . . . . . . . . . . . . . . . . . .Potencia estadísticaStatistical significance . . . . . . . . . . . . . . . . . . . . . . . .Significación estadísticaStatistical significance, level of . . . . . . . . . . . . . . . . .Significación estadística, nivel deSteering committee . . . . . . . . . . . . . . . . . . . . . . . . . .Comité de expertosStratification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .EstratificaciónStratification variable . . . . . . . . . . . . . . . . . . . . . . . . .Variable de estratificaciónStratified randomization . . . . . . . . . . . . . . . . . . . . . .Asignación aleatoria estratificadaStratified sample . . . . . . . . . . . . . . . . . . . . . . . . . . . .Muestra estratificadaStrength of the association . . . . . . . . . . . . . . . . . . . .Magnitud de asociaciónStudent’s t test . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Prueba t de Student Study medication . . . . . . . . . . . . . . . . . . . . . . . . . . .Muestra para investigación clínicaStudy population . . . . . . . . . . . . . . . . . . . . . . . . . . . .Población del estudioSubgroup analysis . . . . . . . . . . . . . . . . . . . . . . . . . .Análisis de subgruposSubinvestigator . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Investigador colaboradorSubjective . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .SubjetivoSurrogate end-point . . . . . . . . . . . . . . . . . . . . . . . . .Variable sustitutivaSurrogate marker . . . . . . . . . . . . . . . . . . . . . . . . . . .Variable sustitutivaSurrogate outcome variable . . . . . . . . . . . . . . . . . . . .Variable sustitutivaSurrogate variable . . . . . . . . . . . . . . . . . . . . . . . . . . .Variable sustitutivaSurveillance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .MonitorizaciónSurvival analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . .Análisis de supervivenciaSurvival rate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Tasa de supervivenciaSystematic review . . . . . . . . . . . . . . . . . . . . . . . . . . .Revisión sistemáticaTarget population . . . . . . . . . . . . . . . . . . . . . . . . . . .Población dianaTemporality . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .TemporalidadTertiary prevention . . . . . . . . . . . . . . . . . . . . . . . . . .Prevención terciariaTest group . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Grupo experimentalTest of significance . . . . . . . . . . . . . . . . . . . . . . . . . .Prueba de significación estadísticaTolerability . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio de tolerabilidad
80
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Tolerability study . . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio de tolerabilidadTolerance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .ToleranciaToxicity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .ToxicidadToxicology . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .ToxicologíaTrialist . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sujeto del ensayoTriple blind/ed . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Triple ciegoTriple mask/ed . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Triple ciegoTwo by two tables . . . . . . . . . . . . . . . . . . . . . . . . . . .Tablas de dos por dos (2 x 2)Two-sided test . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Prueba bilateralTwo-tailed test . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Prueba bilateralType A adverse reaction . . . . . . . . . . . . . . . . . . . . . .Reacción adversa tipo AType B adverse reaction . . . . . . . . . . . . . . . . . . . . . .Reacción adversa tipo BU Mann-Whitney test . . . . . . . . . . . . . . . . . . . . . . . .Prueba U de Mann-WhitneyUnexpected adverse event . . . . . . . . . . . . . . . . . . . .Acontecimiento adverso inesperadoUnicenter clinical trial . . . . . . . . . . . . . . . . . . . . . . . .Ensayo clínico unicéntricoUnicenter study . . . . . . . . . . . . . . . . . . . . . . . . . . . .Estudio unicéntricoUnpaired t-test . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Prueba t para datos independientesValidation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .ValidaciónValidity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Exactitud, ValidezVariability . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .VariabilidadVariable . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .VariableVariance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .VarianzaVolunteer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .VoluntarioVolunteer bias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Sesgo de voluntarioWash-out period . . . . . . . . . . . . . . . . . . . . . . . . . . . .Período de lavadoWilcoxon test . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Prueba de WilcoxonWilcoxon signed-rank sum test . . . . . . . . . . . . . . . . .Prueba de WilcoxonWithdrawal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .RetiradaWithin-subject variability . . . . . . . . . . . . . . . . . . . . . .Variabilidad intraindividualYate’s correction . . . . . . . . . . . . . . . . . . . . . . . . . . . .Yates, corrección de
81
TÉRMINOS INGLÉS-CASTELLANO

Apéndices
83

85
Adoptada por la 18ª Asamblea Médica Mun-dial, Helsinki, Finlandia (junio de 1964) y en-mendada por la 29ª Asamblea Médica Mun-dial, Tokio, Japón, octubre de 1975, la 35ªAsamblea Médica Mundial, Venecia, Italia (oc-tubre de 1983), la 41ª Asamblea Médica Mun-dial, Hong-Kong (septiembre de 1989) y la 48ªAsamblea General, Somerset West, Repúblicade Sudáfrica (octubre de 1996).
Introducción
La misión del médico es la de salvaguardarla salud de la gente. Sus conocimientos y con-ciencia estarán dedicados al cumplimiento deesta misión.
La Declaración de Ginebra de la AsociaciónMédica Mundial obliga al médico con las pala-bras “La salud de mi paciente será mi primeraconsideración” y el Código Internacional deÉtica Médica declara que: “El médico actuarásolamente en interés del paciente cuando alproporcionar cuidados médicos pudieran tenerel efecto de debilitar las condiciones físicas ymentales del paciente”.
El objetivo de la investigación biomédicaque implique seres humanos, debe ser mejo-rar el diagnóstico, procedimientos terapéuticosy profilácticos y el entendimiento de la etiolo-gía y patogénesis de la enfermedad.
En la práctica médica diaria, la mayoría delos diagnósticos, procedimientos terapéuticosy profilácticos implican riesgos. Esto es aplica-ble, especialmente, a la investigación biomédi-ca.
El progreso médico está basado en la inves-tigación, que en última instancia, deberá apo-yarse en la experimentación en la que partici-pen seres humanos.
En el campo de la investigación biomédicadebe reconocerse una distinción fundamentalentre investigación médica, en la que el objeti-vo es esencialmente diagnóstico o terapéuticopara un paciente, y la investigación médicacuyo objetivo esencial es puramente científicoy sin un valor directo diagnóstico o terapéuticopara la persona objeto de la investigación.
Debe tenerse un cuidado especial en la in-vestigación que pueda afectar al medio am-biente y deberá respetarse el bienestar de losanimales de investigación.
Debido a que es esencial que los resultadosde los experimentos de laboratorio sean apli-cados a humanos para el ulterior conocimientocientífico y para ayudar a la humanidad quesufre, la Asociación Médica Mundial ha prepa-rado las siguientes recomendaciones comoguía para cada uno de los médicos participan-tes en investigación biomédica en la que parti-cipen seres humanos. Deberán someterse arevisión en el futuro. Deberá enfatizarse quelos patrones descritos son sólo una guía paralos médicos de todo el mundo. Los médicosno están exentos de las responsabilidades cri-minales, civiles y éticas según las leyes de suspropios países.
I. Principios básicos
1. La investigación biomédica en la que parti-cipen seres humanos deberá estar deacuerdo con los principios científicos ge-neralmente aceptados y deberá basarseen una adecuada experimentación de la-boratorio y animal, realizada adecuada-mente, así como en un perfecto conoci-miento de la literatura científica.
2. El diseño y cumplimiento de cada uno delos procedimientos experimentales en losque participen seres humanos, deberá es-tar claramente definido en un protocolo
Declaración de Helsinki*
Recomendaciones para guía de médicos en investigación biomédica en la que participan sujetos humanos.
* Autorización de reproducción solicitada a la WorldMedical Association.

experimental que deberá ser transmitidopara consideración, comentarios y guía, aun comité especialmente designado e in-dependiente del investigador y del patroci-nador, asegurando que este comité inde-pendiente está en conformidad con las le-yes y regulaciones del país en el que serealiza la investigación experimental.
3. La investigación biomédica en la que parti-cipen seres humanos deberá realizarse so-lamente por personas cualificadas científi-camente y bajo la supervisión de un médi-co cl ínicamente competente. Laresponsabilidad del sujeto deberá recaersiempre en una persona médica cualifica-da y nunca en el sujeto de la investigación,aun a pesar de que ésta haya otorgado suconsentimiento.
4. La investigación médica, en la que partici-pen humanos, no podrá realizarse legíti-mamente a menos que la importancia delobjetivo guarde proporción con el riesgoinherente al sujeto.
5. Cada proyecto de investigación biomédicaen el que participen humanos deberá estarprecedido por una cuidadosa valoración delos posibles riesgos en comparación conlos beneficios que se buscan en el pacien-te o para otros. Deberá prevalecer, en todomomento, el interés del sujeto sobre cual-quier otro interés científico y social.
6. Siempre se ha de respetar la integridad yseguridad del sujeto participante en la in-vestigación. Se tomarán todas las precau-ciones para respetar la confidencialidaddel sujeto y minimizar el impacto que pu-diera tener el estudio sobre su integridadfísica y mental, así como sobre su perso-nalidad.
7. Los médicos deberían abstenerse de parti-cipar en proyectos de investigación a me-nos que estén seguros de que los riesgosson predecibles. Los médicos deberánabandonar cualquier investigación en laque se observe que los riesgos son supe-riores a los beneficios potenciales.
8. En lo referente a la publicación de los re-sultados de su investigación, el médico es-tá obligado a mantener la exactitud de losresultados. Los informes de experimenta-ción que no estén de acuerdo con los prin-cipios establecidos en esta Declaración, nodeberán ser aceptados para su publica-ción.
9. En cualquier investigación con seres hu-manos, cada sujeto debe ser informado
adecuadamente de los objetivos, métodos,posibles beneficios y riesgos potencialesdel estudio, así como también de las inco-modidades que conllevará. Deberán serinformados de que tienen total libertad pa-ra abstenerse de participar en el estudio yde que son libres para revocar su consen-timiento libre e informado, preferiblementepor escrito.
10. Al obtener el consentimiento informadopara un proyecto de investigación, el mé-dico deberá ser especialmente cauto de siel sujeto tiene una relación de dependen-cia con él o de si accede a otorgar su con-sentimiento bajo presión. En este caso, elconsentimiento informado deberá ser soli-citado por un médico que no esté ligado ala investigación y que no tenga relaciónoficial con el proyecto.
11. En caso de incompetencia legal, el con-sentimiento informado deberá solicitarse alresponsable legal, de acuerdo con la legis-lación nacional. Cuando la incapacidad fí-sica o mental hace imposible el otorga-miento del consentimiento informado, ocuando el sujeto sea un menor, el permisodel familiar responsable sustituye al delsujeto, de acuerdo con la legislación na-cional. Siempre que el menor pueda otor-gar su consentimiento, se deberá solicitarel consentimiento del menor, además delconsentimiento del responsable legal delmismo.
12. El protocolo de investigación siempre de-berá contener una declaración de las con-sideraciones éticas apropiadas y deberáindicar que se cumplen los principiosenunciados en esta Declaración.
II. Investigación médica combinada con atención profesional (Investigación Clínica)
1. En el tratamiento de la persona enferma,el médico deberá tener libertad para utili-zar cualquier nuevo diagnóstico o medidaterapéutica, si, en su criterio, ofrece unaesperanza de salvar la vida, restablecer lasalud o aliviar el sufrimiento.
2. Los posibles beneficios, peligros e incomo-didades de un nuevo método, deberán va-lorarse frente a las ventajas del mejor diag-nóstico y del mejor método terapéutico.
3. En cualquier estudio médico –incluyéndo-se a los sujetos del grupo control – deberá
86
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

III. Investigación biomédica no terapéuticaen la que participen humanos(Investigación Biomédica no Clínica)
1. En la pura aplicación científica de la inves-tigación médica realizada en seres huma-nos, es obligación y deber del médico serel protector de la vida y de la salud deaquella persona en la que esté realizandodicha investigación.
2. Los sujetos deberán ser voluntarios –biensean personas sanas o pacientes- paraquienes el diseño del experimento no estárelacionado con su enfermedad.
3. El investigador o el equipo de investigacióndeberá terminar dicho estudio si en su cri-terio puede, de continuarse, ser lesivo pa-ra el individuo.
4. En la investigación en humanos, el interésde la ciencia y la sociedad nunca prevale-cerá sobre cualquier consideración rela-cionada con el bienestar del sujeto.
87
DECLARACIÓN DE HELSINKI
asegurárseles el mejor diagnóstico y méto-do terapéutico disponible. Esto no excluyela utilización de un placebo inerte en losestudios en los que no se disponga de mé-todos diagnósticos o terapéuticos de efica-cia demostrada.
4. La negativa del paciente a participar en unestudio nunca deberá interferir en la rela-ción médico-paciente.
5. Si el médico considera esencial no solicitarel consentimiento informado, deberá mani-festar las razones concretas de esta deci-sión en el protocolo experimental, para sutransmisión al comité independiente (I.2).
6. El médico puede combinar la investigaciónmédica con la práctica profesional, ya queel objetivo es la adquisición de nuevos co-nocimientos médicos, solamente hasta elextremo de que la investigación médicaestá justificada en base a su potencial va-lor diagnóstico o terapéutico para el pa-ciente.

89
La Ley 14/1986, de 25 de abril, General de Sanidad, dispone en su artículo 95, apartado 2, quepara la circulación y uso de los medicamentos y productos sanitarios que se les asimilen, se requeri-rá autorización previa. Para los demás productos y artículos sanitarios se podrá exigir autorizaciónprevia individualizada o el cumplimiento de condiciones de homologación, señalando en el apartado4 del mismo artículo que el procedimiento de autorización asegurará que se satisfacen las garantíasde eficacia, tolerancia, pureza y estabilidad que marquen la legislación sobre medicamentos y de-más disposiciones que sean de aplicación. En especial se exigirá la realización de ensayos clínicoscontrolados.
Por su parte, la Ley 25/1990, de 20 de diciembre, del Medicamento, dedica el Título III a regularla realización de ensayos clínicos de medicamentos.
Partiendo de los conceptos básicos sobre el ensayo clínico contenidos en la citada Ley del Medi-camento, se hace necesario determinar mediante el presente reglamento las funciones y responsa-bilidades concretas de los agentes implicados en la realización de ensayos clínicos, así como los re-quisitos necesarios para su autorización, en el sector de los medicamentos, que actualiza y sustituyelo expresado en el Real Decreto 944/1978, de 14 de abril, y la Orden ministerial de 3 de agosto de1982.
La calificación como producto en fase de investigación clínica de las nuevas entidades químicas obiológicas por parte de la Administración antes de permitir la realización de ensayos clínicos en hu-manos, pretende velar por la seguridad e integridad física y proteger los derechos de los individuosque participen en el programa de investigación, y que ésta se realice de acuerdo con la metodologíaadecuada para garantizar la validez interna y externa de los ensayos clínicos. De este modo los re-sultados obtenidos podrán ser utilizados para avalar la solicitud de autorización para la comercializa-ción de dichos productos.
De acuerdo con la directiva 91/507/CEE, de 19 de julio, que modifica el anexo de la directiva75/318/CEE, de 20 de mayo, todos los ensayos clínicos en todas las fases, incluyendo aquellos debiodisponibilidad y bioequivalencia, se realizarán según las normas de buena práctica clínica. Estasnormas pretenden garantizar que los ensayos clínicos sean diseñados, realizados y comunicados demodo que se asegure que los datos sean fiables y que se protejan los derechos de los sujetos. El se-guimiento de dichas normas mejorará la calidad de la investigación clínica y permitirá el mutuo re-conocimiento entre las Administraciones Sanitarias de los diferentes Estados con respecto a los re-sultados de los ensayos clínicos realizados. Por ello, en este reglamento se hace referencia a la ne-cesidad de seguir las normas de buena práctica clínica y se recogen los aspectos mínimos queserán de obligado cumplimiento para los ensayos clínicos con medicamentos.
El presente Real Decreto se dicta al amparo de lo dispuesto en el artículo 1491.1.1.ª y 16.ª de laConstitución, en concordancia con el artículo 2.1 y 2 de la Ley 25/1990, de 20 de diciembre, delMedicamento.
En su virtud, a propuesta del Ministro de Sanidad y Consumo, oídos los sectores afectados, deacuerdo con el Consejo de Estado y previa deliberación del Consejo de Ministros en su reunión de16 de abril de 1993,
BOLETÍN OFICIAL DEL ESTADOJueves 13 de mayo de 1993. BOE número 114. Fascículo Primero. Páginas 14346-14364.
MINISTERIO DE SANIDAD Y CONSUMO
12483 REAL DECRETO 561/1993, de 16 de abril, por el que se establecen los requisitos para la realización
de ensayos clínicos con medicamentos

DISPONGO:
TITULO I
Consideraciones generales y principios básicos
Artículo 1. Ambito de aplicación.Este Real Decreto se refiere a todos los ensayos clínicos con medicamentos o productos en fase
de investigación clínica que se realicen en España, incluyendo radiofármacos, hemoderivados, aler-genos, plantas medicinales y todas aquellas sustancias consideradas como medicamentos en el artí-culo 8 de la Ley 25/1990 del Medicamento.
Artículo 2. Definición de ensayo clínico con medicamentos.1. De acuerdo con el artículo 59 de la Ley 25/1990 del Medicamento, se considera ensayo clínico
toda evaluación experimental de una sustancia o medicamento, a través de su aplicación a sereshumanos, orientada hacia alguno de los siguientes fines:
a) Poner de manifiesto sus efectos farmacodinámicos o recoger datos referentes a su absorción,distribución, metabolismo y excreción en el organismo humano.
b) Establecer su eficacia para una indicación terapéutica, profiláctica o diagnóstica determinada.c) Conocer el perfil de sus reacciones adversas y establecer su seguridad.2. Se considerará siempre evaluación experimental aquel estudio en el que los sujetos sean asig-
nados a uno u otro grupo de intervención terapéutica de forma aleatoria, o bien se condicione, di-recta o indirectamente, el proceso de prescripción médica habitual.
3. Se considerará siempre evaluación experimental aquel estudio en que se utilice una sustanciano autorizada como especialidad farmacéutica o bien se utilice una especialidad farmacéutica encondiciones de uso distinto de las autorizadas.
4. No se considera ensayo clínico la administración de la sustancia o medicamento a un solo pa-ciente en el ámbito de la práctica médica habitual con el único propósito de conseguir un beneficiopara el mismo, de acuerdo con lo previsto en el artículo 23 de este Real Decreto referente al usocompasivo de medicamentos. La práctica médica y la libertad profesional de prescripción del médi-co no ampararán, en ningún caso, ensayos clínicos no autorizados, ni la utilización de remedios se-cretos o no declarados a la autoridad sanitaria.
Artículo 3. Tipos de ensayos clínicos según sus objetivos.De acuerdo con los objetivos perseguidos y la información disponible se distinguen los siguientes
tipos de ensayos clínicos en el desarrollo de un fármaco:1. Ensayos clínicos en fase I: constituyen el primer paso en la investigación de una sustancia o me-
dicamento nuevo en el hombre. Son estudios de farmacocinética y farmacodinámica que proporciona-rán información preliminar sobre el efecto y la seguridad del producto en sujetos sanos o en algunoscasos en pacientes, y orientarán la pauta de administración más apropiada para ensayos posteriores.
2. Ensayos clínicos en fase II: representan el segundo estadio en la evaluación de una nueva sus-tancia o medicamento en el ser humano. Se realizan en pacientes que padecen la enfermedad oentidad clínica de interés. Tienen como objetivo: proporcionar información preliminar sobre la efica-cia del producto, establecer la relación dosis-respuesta del mismo, conocer las variables empleadaspara medir eficacia y ampliar los datos de seguridad obtenidos en la fase I. Por lo general, estos en-sayos clínicos serán controlados y con asignación aleatoria a los tratamientos.
3. Ensayos clínicos en fase III: son ensayos clínicos destinados a evaluar la eficacia y seguridaddel tratamiento experimental intentando reproducir las condiciones de uso habituales y consideran-do las alternativas terapéuticas disponibles en la indicación estudiada. Se realizan en una muestrade pacientes más amplia que en la fase anterior y representativa de la población general a la queiría destinado el medicamento. Estos estudios serán preferentemente controlados y aleatorizados.
4. Ensayos clínicos en fase IV: son ensayos clínicos que se realizan con un medicamento despuésde su comercialización. Estos ensayos podrán ser similares a los descritos en las fases I, II y III si es-tudian algún aspecto aún no valorado o condiciones de uso distintas de las autorizadas como podríaser una nueva indicación. Estos estudios serán preferentemente controlados y aleatorizados.
90
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Artículo 4. Tipos de ensayos clínicos según el número de centros participantes.1. Ensayo clínico unicéntrico: es aquel realizado por un solo investigador o equipo de investiga-
ción en un centro hospitalario o extrahospitalario.2. Ensayo clínico multicéntrico: es aquel realizado en dos o más centros con un mismo protocolo
y un coordinador que se encargará del procesamiento de todos los datos y del análisis de los resul-tados.
Artículo 5. Tipos de ensayos clínicos en función de su metodología.1. Ensayo clínico controlado: es el que comporta una comparación con un grupo control o testigo.
El ensayo clínico controlado aleatorizado incluye al menos dos grupos de voluntarios, pacientes o sa-nos, cuya asignación a un tratamiento experimental o control se realiza al azar de forma que ni elsujeto ni el médico responsable de su selección o tratamiento puedan influir en su asignación. Tantola selección de sujetos como los períodos de tratamiento y seguimiento han de tener lugar simultá-neamente en todos los grupos. En la gran mayoría de los casos es la única forma científicamente vá-lida para evaluar la eficacia y seguridad de una intervención terapéutica. Estos ensayos pueden ser:
a) Ensayo clínico con grupos cruzados: ensayo clínico en el que los tratamientos experimental ycontrol son administrados a cada individuo en períodos sucesivos que han sido determinados alea-toriamente, lo que permite a cada sujeto ser su propio control.
b) Ensayo clínico con grupos paralelos: ensayo clínico en el cual uno o varios grupos de sujetosson asignados a recibir el tratamiento experimental al mismo tiempo que otro grupo recibe el trata-miento control.
c) Ensayo clínico secuencial: es aquel en el que poniendo a prueba una hipótesis específica, elnúmero de sujetos no está prefijado de antemano, sino que depende de los resultados que se vanobteniendo a lo largo del mismo.
2. Ensayo clínico no controlado: es el que no comporta una comparación con un grupo control otestigo.
Artículo 6. Tipos de ensayos clínicos según su grado de enmascaramiento.Según las medidas que se tomen para evitar la subjetividad de los resultados se distinguen los si-
guientes tipos de ensayos clínicos:1. Abierto o no ciego: son aquellos ensayos en los que tanto el sujeto como el investigador cono-
cen el grupo de tratamiento al que aquél ha sido asignado.2. Simple ciego: son aquellos ensayos en los que el sujeto desconoce el grupo de tratamiento al
que pertenece.3. Doble ciego: son aquellos ensayos en los que tanto el sujeto como el investigador desconocen
la asignación a los grupos de tratamiento.4. Evaluación ciega por terceros: en estos ensayos clínicos se recurre, para evaluar la respuesta, a
una tercera persona que desconoce el tratamiento que está recibiendo cada sujeto.
Artículo 7. Ensayo clínico piloto.Es aquel que se realiza como paso previo a otros estudios más amplios con el fin de conocer da-
tos que permitan un diseño más adecuado, establecer su viabilidad, así como determinar el tamañode la muestra para posteriores estudios. Debe especificarse, siempre que proceda, esta característi-ca.
Artículo 8. Protocolización de un ensayo clínico.a) Las características de un ensayo clínico estarán íntegramente definidas en un protocolo y la re-
alización del ensayo se ajustará al contenido del protocolo autorizado tal y como se especifica en elapartado 2 del artículo 66 de la Ley del Medicamento.
b) Se define como protocolo el documento que establece la razón de ser del estudio, sus objeti-vos, diseño, metodología y análisis previsto de sus resultados así como las condiciones bajo las quese realizará y desarrollará el ensayo.
c) Todo protocolo de ensayo clínico estará redactado, al menos, en la lengua española oficial delEstado. Incluirá los siguientes apartados básicos:
1. Resumen.2. Índice.
91
REAL DECRETO 561/1993

3. Información general.4. Justificación y objetivos.5. Tipo de ensayo clínico y diseño del mismo.6. Selección de los sujetos.7. Descripción del tratamiento.8. Desarrollo del ensayo y evaluación de la respuesta.9. Acontecimientos adversos.
10. Aspectos éticos.11. Consideraciones prácticas.12. Análisis estadístico.Anexo I. Cuaderno de recogida de datos.Anexo II. Manual del investigador.Anexo III. Procedimientos normalizados de trabajo.Anexo IV. Memoria analítica de las muestras a utilizar.El contenido de cada uno de estos apartados queda descrito a título orientativo en el anexo 1 de
este Real Decreto.
Artículo 9. Producto en fase de investigación clínica (PEI).1. Se denomina producto en fase de investigación clínica aquel que ha sido calificado como tal
por la Dirección General de Farmacia y Productos Sanitarios y se destina únicamente a ser utilizado,por expertos calificados por su formación científica y experiencia para la investigación, en personaspara valorar su seguridad y eficacia.
2. La calificación de productos en fase de investigación clínica se otorgará mediante Resolución dela Dirección General de Farmacia y Productos Sanitarios en la que se enumerarán las indicacionesconcretas que pueden ser objeto de investigación clínica y con las limitaciones, plazos, condiciones,requisitos y garantías que, en su caso, se establezcan. Contra esta Resolución podrá interponerse re-curso ordinario en el plazo de un mes, de conformidad con lo establecido en el artículo 114 de la Leyde Régimen Jurídico de las Administraciones Públicas y del Procedimiento Administrativo Común.
3. Necesitan obtener la calificación de producto en fase de investigación clínica antes de poderser utilizados en investigación clínica en nuestro país las entidades químicas o biológicas que no se-an principio activo de especialidades farmacéuticas registradas en España.
4. Para obtener la calificación de producto en fase de investigación clínica se garantizará la cali-dad del producto y que éste es apto para la investigación clínica en las indicaciones propuestas me-diante los estudios preclínicos necesarios para establecer su perfil farmacológico y toxicológico. Si lafase de investigación lo justifica serán necesarios además datos de estudios clínicos previos.
5. La calificación de producto en fase de investigación clínica tendrá una validez de dos años, sal-vo que en la resolución se indique otro plazo menor, y sin perjuicio de su ulterior renovación o pró-rroga.
6. La extensión de la información requerida dependerá del plan de investigación propuesto, de lanovedad del producto, de los riesgos previsibles y de los conocimientos previos sobre el producto ola indicación en estudio.
7. La documentación que avala la autorización del producto en fase de investigación clínica seconcibe como una información científica que se actualizará de forma periódica en base a los hallaz-gos preclínicos y clínicos obtenidos en cada fase del programa de investigación propuesto. En cual-quier momento, la documentación que obra en poder de la Dirección General de Farmacia y Pro-ductos Sanitarios será suficiente para justificar cada ensayo clínico propuesto.
8. El contenido de la documentación presentada para la solicitud de producto en fase de investi-gación clínica será tratado de forma confidencial, sin perjuicio de la información que resulte necesa-ria para las actuaciones propias de las inspecciones de las Administraciones Sanitarias sobre ensa-yos clínicos.
Artículo 10. Respeto a postulados éticos.1. Todos los ensayos clínicos habrán de contar, antes de poder ser realizados, con el informe pre-
vio del correspondiente Comité Ético de Investigación Clínica (CEIC).2. Los ensayos clínicos se realizarán en condiciones de respeto a los derechos fundamentales de
la persona y a los postulados éticos que afectan a la investigación biomédica con seres humanos,
92
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

siguiéndose a estos efectos los contenidos en la declaración de Helsinki y sucesivas actualizacio-nes. Se obtendrá y documentará el consentimiento informado, libremente expresado, de cada unode los sujetos del ensayo antes de su inclusión, en los términos del artículo 12 del presente RealDecreto.
3. Sólo se podrán realizar ensayos clínicos cuando se cumplan todos los principios siguientes:a) Los datos preclínicos sobre el producto en estudio sean razonablemente suficientes para ga-
rantizar que los riesgos para el sujeto en quien se realiza el ensayo son admisibles.b) El estudio se base en los conocimientos actuales disponibles, la información buscada suponga,
presumiblemente, un avance en el conocimiento científico sobre el ser humano o para mejorar suestado de salud y su diseño minimice los riesgos para los sujetos participantes en el mismo.
c) La importancia de la información buscada justifique el riesgo al que se exponen los sujetos par-ticipantes en el ensayo clínico.
Artículo 11. Sujetos del ensayo.1. Es sujeto del ensayo la persona sana o enferma que participa en un ensayo clínico, después de
haber otorgado libremente su consentimiento informado. En los ensayos clínicos sin beneficio direc-to para la salud de los voluntarios participantes, el riesgo que estos sujetos asuman estará justificadoen razón del beneficio esperado para la colectividad.
2. En menores de edad e incapaces y en personas con la autonomía o competencia disminuidapara dar su consentimiento, sólo podrán realizarse ensayos de interés para su salud particular cuan-do no puedan ser efectuados en sujetos no afectados por estas condiciones especiales, debido aque la patología en estudio sea la propia de aquéllos. En estos casos, el consentimiento se obtendráde la forma que se indica en el apartado 5 del artículo 12.
3. No obstante, en los sujetos referidos en el apartado anterior podrán realizarse ensayos sin fi-nes terapéuticos si el Comité Ético de Investigación Clínica determina que se cumple todo lo si-guiente:
a) La adopción de las medidas necesarias que garanticen que el riesgo sea mínimo.b) Las experiencias a que van a ser sometidos son equivalentes a las que corresponden a su si-
tuación médica, psicológica, social o educacional.c) Del ensayo se obtendrán conocimientos relevantes sobre la enfermedad o situación objeto de
investigación, de vital importancia para entenderla,paliarla o curarla.d) Estos conocimientos no pueden ser obtenidos de otro modo.e) Existen garantías sobre la correcta obtención del consentimiento informado, de acuerdo con lo
contemplado en el artículo 12 del presentado Real Decreto.4. En mujeres gestantes o en período de lactancia sólo se podrán realizar ensayos clínicos sin fi-
nalidad terapéutica cuando el Comité Ético de Investigación Clínica concluya que no suponen nin-gún riesgo previsible para su salud ni para la del feto o niño y que se obtendrán conocimientos útilesy relevantes sobre el embarazo o la lactancia.
5. Los sujetos participantes en ensayos sin interés terapéutico particular recibirán del promotor lacompensación pactada por las molestias sufridas. La cuantía de la compensación económica estaráen relación con las características del ensayo, pero en ningún caso será tan elevada como para in-ducir a un sujeto a participar por motivos distintos del interés por el avance científico. En los casosextraordinarios de investigaciones sin fines terapéuticos en menores e incapaces o personas con lacompetencia o autonomía disminuidas, se tomarán las medidas necesarias para evitar la posible ex-plotación de estos sujetos.
6. La contraprestación que se hubiere pactado por el sometimiento voluntario a la experiencia sepercibirá en todo caso, si bien se reducirá equitativamente según la participación del sujeto en la ex-perimentación, en el supuesto de que desista.
7. Todas las partes implicadas en un ensayo clínico guardarán la más estricta confidencialidad deforma que no se viole la intimidad personal ni familiar de los sujetos participantes en el mismo. Asi-mismo deberán tomarse las medidas apropiadas para evitar el acceso de personas no autorizadas alos datos del ensayo.
8. El tratamiento de los datos de carácter personal de los sujetos participantes en el ensayo seajustará a lo establecido en la Ley Orgánica 5/1992, de 29 de octubre, de Regulación del tratamien-to automatizado de los datos de carácter personal, en especial en lo que al consentimiento del afec-tado se refiere.
93
REAL DECRETO 561/1993

Artículo 12. Consentimiento informado.1. Es imprescindible que el sujeto otorgue libremente su consentimiento informado antes de po-
der ser incluido en su ensayo clínico.2. Todas las personas implicadas en un ensayo clínico evitarán cualquier influencia sobre el suje-
to participante en el ensayo.3. El consentimiento informado es el procedimiento que garantiza que el sujeto ha expresado vo-
luntariamente su intención de participar en el ensayo clínico, después de haber comprendido la in-formación que se le ha dado acerca de los objetivos del estudio, beneficios, incomodidades y ries-gos previstos, alternativas posibles, derechos y responsabilidades, tal como se recoge en el anexo 6,apartado 1. El documento de consentimiento informado (anexo 6, apartados 2 ó 3) acredita que di-cho consentimiento ha sido otorgado.
4. El sujeto expresará su consentimiento preferiblemente por escrito (anexo 6, apartado 2) o, ensu defecto, de forma oral ante testigos independientes del equipo investigador que lo declararán porescrito bajo su responsabilidad (anexo 6, apartado 3). En aquellos ensayos sin interés terapéuticoparticular para el sujeto, su consentimiento constará necesariamente por escrito.
5. En los casos de sujetos menores de edad e incapaces, el consentimiento lo otorgará siemprepor escrito su representante legal (anexo 6 apartado 4), tras haber recibido y comprendido la infor-mación mencionada. Cuando las condiciones del sujeto lo permitan y, en todo caso, cuando el me-nor tenga doce o más años, deberá prestar además su consentimiento (anexo 6, apartado 2) paraparticipar en el ensayo, después de haberle dado toda la información pertinente adaptada a su nivelde entendimiento. El consentimiento del representante legal y del menor, en su caso, será puesto enconocimiento del Ministerio Fiscal, previamente a la realización del ensayo.
6. En el caso excepcional en que por la urgencia de la aplicación del tratamiento no fuera posibledisponer del consentimiento del sujeto o de su representante legal en el momento de su inclusiónen el ensayo clínico,este hecho será informado al Comité Ético de Investigación Clínica y al promotorpor el investigador, explicando las razones que han dado lugar al mismo. En cualquier caso, esta si-tuación estará prevista en el protocolo del ensayo clínico aprobado por el correspondiente ComitéÉtico de Investigación Clínica y únicamente procederá cuando tenga un específico interés terapéuti-co particular para el paciente. El sujeto o su representante legal será informado en cuanto sea posi-ble y otorgará su consentimiento para continuar en el ensayo si procediera. Esta circunstancia ex-cepcional sólo podrá aplicarse a ensayos clínicos con interés terapéutico particular para el paciente.
7. El sujeto participante en un ensayo clínico o su representante podrán revocar su consentimien-to en cualquier momento, sin expresión de causa y sin que por ello se derive para él responsabilidadni perjuicio alguno.
Artículo 13. Del seguro de los sujetos del ensayo.1. La iniciación de un ensayo clínico con productos en fase de investigación clínica o para nuevas
indicaciones de medicamentos ya autorizados o cuando no exista interés terapéutico para el sujetodel ensayo, sólo podrá realizarse si previamente se ha concertado un seguro que cubra los daños yperjuicios que como consecuencia del mismo pudieran resultar para la persona en que hubiere derealizarse.
2. El promotor del ensayo es el responsable de la contratación de dicho seguro de responsabili-dad civil y éste cubrirá las responsabilidades del promotor, del investigador y sus colaboradores ydel titular del hospital o centro donde el ensayo se realice.
3. Cuando por cualquier circunstancia el seguro no cubra enteramente los daños, el promotor delensayo clínico, el investigador principal y el titular del hospital o centro donde se realice el ensayo,son solidariamente responsables, sin necesidad de que medie culpa, del daño que en su salud su-fra el sujeto sometido al ensayo clínico, así como de los perjuicios económicos que de dicho dañodirectamente se deriven, siempre y cuando éste sea consecuencia del tratamiento con la sustanciao producto objeto del ensayo o de las medidas terapéuticas o diagnósticas que se adopten durantela realización del mismo.
4. Ni la autorización administrativa ni el informe del Comité Ético de Investigación Clínica eximiránde responsabilidad al promotor del ensayo clínico, al investigador principal y sus colaboradores o altitular del hospital o centro donde se realice el ensayo.
5. Se presume, salvo prueba en contrario, que los daños que afecten a la salud de la persona su-jeta a ensayo, durante la realización del mismo y en el año siguiente a la terminación del tratamien-
94
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

to, se han producido como consecuencia del ensayo. Sin embargo, una vez concluido el año, el su-jeto del ensayo está obligado a probar el nexo entre el ensayo y el daño producido.
6. A los efectos del régimen de responsabilidad previsto en el presente artículo, se considerará ob-jeto de resarcimiento todos los gastos derivados del menoscabo en la salud del sujeto sometido al en-sayo, así como los perjuicios económicos que de dicho menoscabo directamente se deriven, siempreque éste sea consecuencia del sometimiento al ensayo clínico. No será objeto de resarcimiento bajoel régimen de responsabilidad previsto en el presente artículo, el daño que en su salud sufra el sujetosometido al ensayo cuando éste sea inherente a la patología objeto de estudio, o se incluya dentro delos efectos secundarios propios de la medicación prescrita para dicha patología, así como de la evolu-ción propia de su enfermedad como consecuencia de la ineficacia del tratamiento.
7. El importe mínimo que en concepto de responsabilidad civil estará asegurado será de 30 millo-nes de pesetas por sujeto sometido a ensayo clínico, en concepto de indemnización a tanto alzado.En caso de que dicha indemnización se fije como renta anual constante o creciente, el límite de co-bertura de dicho seguro será de al menos tres millones de pesetas anuales por sujeto sometido aensayo clínico. Se autoriza al Ministro de Sanidad y Consumo para revisar los límites anteriormenteestablecidos.
Artículo 14. Promotor.1. Es promotor del ensayo clínico la persona física o jurídica que tiene interés en su realización,
firma las solicitudes de autorización dirigidas al Comité Ético de Investigación Clínica o a la DirecciónGeneral de Farmacia y Productos Sanitarios y se responsabiliza de él, incluyendo su organización,comienzo y financiación.
2. Las obligaciones del promotor son la siguientes:a) Establecer unos procedimientos normalizados de trabajo.b) Firmar junto con el investigador el protocolo y cualquier modificación del mismo.c) Seleccionar al investigador más adecuado según su calificación y medios disponibles y asegu-
rarse de que éste llevará a cabo el estudio tal como está especificado en el protocolo.d) Proporcionar toda la información básica y clínica disponible del producto en investigación y ac-
tualizar la misma a lo largo del ensayo.e) Solicitar el informe del ensayo por parte del Comité Ético de Investigación Clínica y la autoriza-
ción de la Dirección General de Farmacia y Productos Sanitarios e informarles o solicitar su autoriza-ción, según proceda y sin perjuicio de la comunicación a las Comunidades Autónomas, en caso demodificaciones, violaciones del protocolo o interrupción del ensayo y las razones para ello.
f) Suministrar el medicamento que se va a investigar, garantizar que se han cumplido las normasde correcta fabricación y que las muestras están adecuadamente envasadas y etiquetadas. Tambiénes responsable de la conservación de muestras y sus protocolos de fabricación y control, del registrode las muestras entregadas y de asegurarse que en el centro donde se realiza el ensayo existirá unprocedimiento correcto de manejo, conservación y uso de dichas muestras.
g) Designar el monitor que vigilará la marcha del ensayo.h) Comunicar a las autoridades sanitarias y a los Comités Éticos de Investigación Clínica involu-
crados en el ensayo:1.º Los acontecimientos adversos graves e inesperados que puedan estar relacionados con los tra-
tamientos en investigación, ocurridos dentro o fuera de España.2.º Cualquier información derivada de estudios realizados en animales, que sugiera un riesgo sig-
nificativo para los seres humanos, incluyendo cualquier hallazgo de teratogenicidad o carcinogenici-dad. El promotor junto con el investigador tomarán las medidas necesarias para la protección de lossujetos del ensayo.
i) Proporcionar al investigador y al Comité Ético de Investigación Clínica cuando proceda, cual-quier información de importancia inmediata a la que tenga acceso durante el ensayo.
j) Proporcionar compensación económica a los sujetos en caso de lesión o muerte relacionadascon el ensayo. Proporcionar al investigador cobertura legal y económica en estos casos exceptocuando la lesión sea consecuencia de negligencia o mala práctica del investigador.
k) Acordar con el investigador las obligaciones en cuanto a procesamiento de datos, elaboraciónde informes y publicación de resultados. En cualquier caso, el promotor es responsable de elaborarlos informes finales o parciales del ensayo y comunicarlos a la Dirección General de Farmacia y Pro-ductos Sanitarios.
95
REAL DECRETO 561/1993

Artículo 15. Monitor.1. Es monitor del ensayo clínico el profesional capacitado con la necesaria competencia clínica
elegido por el promotor, que se encarga del seguimiento directo de la realización del ensayo. Sirvede vínculo entre el promotor y el investigador principal cuando estas condiciones no concurran en lamisma persona.
2. Las obligaciones del monitor son las siguientes:a) Trabajar de acuerdo con los procedimientos normalizados de trabajo del promotor, visitar al in-
vestigador antes, durante y después del ensayo para comprobar el cumplimiento del protocolo, ga-rantizar que los datos son registrados de forma correcta y completa, así como asegurarse de que seha obtenido el consentimiento informado de todos los sujetos antes de su inclusión en el ensayo.
b) Cerciorarse de que los investigadores y el centro donde se realizará la investigación son ade-cuados para este propósito.
c) Asegurarse de que tanto el investigador principal como sus colaboradores han sido informadosadecuadamente y garantizar en todo momento una comunicación rápida entre investigador y pro-motor.
d) Comprobar que el almacenamiento, distribución, devolución y documentación de los medica-mentos en investigación es seguro y adecuado.
e) Remitir al promotor informes de las visitas de monitorización y de todos los contactos relevan-tes con el investigador.
Artículo 16. Investigador.1. El investigador principal es quien dirige la realización práctica del ensayo y firma junto con el
promotor la solicitud, corresponsabilizándose con él.2. Solamente podrá actuar como investigador principal un profesional sanitario suficientemente
calificado para evaluar la respuesta a la sustancia o medicamento objeto de estudio, con experienciaen investigación y en el área clínica del ensayo propuesto y con reconocidos criterios de ética e inte-gridad profesional.
3. En todo caso, los ensayos clínicos en humanos se realizarán bajo la vigilancia de un médicocon la necesaria competencia clínica.
4. Son obligaciones del investigador:a) Estar de acuerdo y firmar junto con el promotor el protocolo del ensayo.b) Conocer a fondo las propiedades de los medicamentos.c) Obtener el consentimiento informado de los sujetos antes de su inclusión en el ensayo.d) Recoger, registrar y notificar los datos de forma correcta.e) Notificar inmediatamente los acontecimientos adversos graves o inesperados al promotor.f) Garantizar que todas las personas implicadas respetarán la confidencialidad de cualquier infor-
mación acerca de los sujetos del ensayo.g) Informar regularmente al Comité Ético de Investigación Clínica de la marcha del ensayo.h) Corresponsabilizarse con el promotor de la elaboración del informe final del ensayo, dando su
acuerdo al mismo con su firma.
Artículo 17. Normas de buena práctica clínica.1. Son aquellas normas según las cuales los ensayos clínicos son diseñados, realizados y comuni-
cados de modo que se asegure que los datos son fiables y que se protegen los derechos y la integri-dad de los sujetos, manteniendo la confidencialidad de sus datos.
2. Las normas de buena práctica clínica señalan las responsabilidades de los diferentes implica-dos en cada una de las fases de planificación y ejecución de un ensayo clínico y requieren la exis-tencia de unos procedimientos preestablecidos por escrito que se apliquen de forma sistemática enla organización, dirección, recogida de datos, documentación y verificación de los ensayos clínicos(procedimientos normalizados de trabajo).
Artículo 18. Muestras para investigación clínica.1. Las muestras de medicamentos o productos en fase de investigación clínica para utilización en
ensayos clínicos serán proporcionadas gratuitamente por el promotor. En situaciones especiales po-drán autorizarse ensayos en los que se contemplen otras vías de suministro. Todas las muestras so-brantes serán devueltas al promotor una vez finalizado el período de tratamiento del ensayo clínico.
96
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

2. El Director Técnico responsable de las muestras de un ensayo clínico garantizará la fabricacióny adecuada calidad de las mismas según las normas de correcta fabricación. En caso de que lasmuestras sean productos de importación avalará la calidad de las mismas, debiendo para ello adop-tar las comprobaciones y controles adecuados. Asimismo remitirá a las autoridades competentesmuestras de los productos que serán utilizados en el ensayo clínico cuando le sean solicitadas.
3. Las muestras para un ensayo clínico irán envasadas y acondicionadas convenientemente. Suetiquetado o rotulación permitirá, en cualquier momento, su perfecta identificación. En la etiquetaconstarán, como mínimo, los siguientes datos:
a) Código del protocolo.b) Número de unidades y forma galénica.c) Vía de administración.d) Nombre y dirección de la entidad farmacéutica elaboradora.e) Nombre del Director Técnico responsable.f) Número de lote.g) Fecha de caducidad, si la hubiera.h) Condiciones especiales de conservación, si las hubiera.i) La inscripción «Muestra para investigación clínica».En los ensayos de carácter doble ciego, el número de lote, el nombre y dirección de la entidad farma-
céutica elaboradora y el nombre del técnico responsable de las muestras no se incluirán en la etiqueta,sino en el documento que contenga la identificación del tratamiento, con el fin de no romper la igualdadentre las muestras. Con este mismo fin, cuando difieran las fechas de caducidad o las condiciones deconservación de los productos en comparación, figurará en las etiquetas la más restrictiva de ellas.
4. La distribución al investigador de las muestras para ensayo se realizará a través del servicio defarmacia del hospital donde se realice la investigación. Dichos servicios acusarán recibo por escritode la entrega de los productos y se responsabilizarán de su correcta conservación y dispensación;asimismo controlarán la medicación sobrante al final del ensayo. Si el ensayo se realiza en el medioextrahospitalario, las obligaciones fijadas en este punto serán asumidas por los servicios farmacéuti-cos de las estructuras de atención primaria o, en caso de no existir, por los servicios de farmacia delos hospitales de referencia y, de forma extraordinaria, por el investigador principal del ensayo.
5. El promotor conservará en el archivo principal del ensayo los protocolos de fabricación y controlde los lotes de productos fabricados para el ensayo clínico.
Asimismo las muestras de cada lote se conservarán hasta doce meses después de la fecha de ca-ducidad.
Artículo 19. Acontecimientos adversos.1. Definiciones:a) Acontecimiento adverso: es cualquier experiencia no deseable que ocurra a un sujeto durante
un ensayo clínico, se considere o no, relacionada con los productos en investigación.b) Acontecimiento adverso grave: es aquel que produce la muerte, amenaza la vida, produce in-
capacidad permanente o da lugar a hospitalización o prolongación de la misma.Además se considerarán siempre graves las anomalías congénitas y los procesos malignos.c) Acontecimiento adverso inesperado: es una experiencia no descrita (en naturaleza, gravedad o
frecuencia) en el manual del investigador.2. El investigador está obligado a notificar de forma inmediata al promotor del estudio los aconte-
cimientos adversos graves o inesperados. El promotor notificará a la Dirección General de Farmaciay Productos Sanitarios y a los Comités Éticos de Investigación Clínica involucrados en el ensayo losacontecimientos graves e inesperados que puedan estar relacionados con los tratamientos en inves-tigación que hayan ocurrido tanto en España como en otros países, en el formulario de notificaciónrecogido en el anexo 8 en los plazos que se establezcan, sin perjuicio de la comunicación a las Co-munidades Autónomas. Los acontecimientos adversos graves esperados, los no graves y aquellosque se consideren no relacionados con los tratamientos en estudio serán incluidos de forma tabula-da en el informe anual o final del ensayo clínico.
Artículo 20. Aspectos económicos.1. Todos los aspectos económicos relacionados con el ensayo clínico quedarán reflejados en un
contrato entre cada centro donde se vaya a realizar el ensayo y el promotor. Las Administraciones
97
REAL DECRETO 561/1993

Sanitarias competentes para cada Servicio de Salud especificarán los requisitos comunes y condi-ciones de financiación y el Consejo Interterritorial del Sistema Nacional de Salud podrá acordar losprincipios generales de coordinación.
2. En el contrato constará el presupuesto inicial del ensayo especificando los costes indirectosque aplicará el centro, así como los costes directos extraordinarios, considerando como tales aque-llos gastos ajenos a los que hubiera habido si el sujeto no hubiera participado en el ensayo,comoanálisis y exploraciones complementarias añadidas, cambios en la duración de la atención a los en-fermos, reembolso por gastos a los pacientes, compra de aparatos y compensación para los sujetosdel ensayo e investigadores. También constarán los términos y plazos de los pagos, así como cual-quier otra responsabilidad subsidiaria que contraigan ambas partes.
3. Los Comités Éticos de Investigación Clínica podrán conocer el presupuesto del ensayo y evalua-rán algunos de los contenidos de los presupuestos tales como las compensaciones para los sujetosparticipantes en el ensayo.
Artículo 21. Archivo de la documentación del ensayo.1. El promotor del ensayo es responsable del archivo de la documentación del ensayo.2. El investigador se ocupará de que los códigos de identificación de los sujetos se conserven du-
rante al menos quince años después de concluido o interrumpido el ensayo.3. Las historias clínicas de los pacientes y demás datos originales se conservarán el máximo perí-
odo de tiempo que permita el hospital, la institución o la consulta privada donde se haya realizado elensayo.
4. El promotor o el propietario de los datos conservará toda la restante documentación relativa alensayo durante el período de validez del medicamento. Estos documentos incluirán:
a) El protocolo, incluyendo su justificación, objetivos, diseño estadístico y metodología del ensayo,con las condiciones en las que se efectúe y gestione, así como los pormenores de los productos deinvestigación que se empleen.
b) Los procedimientos normalizados de trabajo.c) Todos los informes escritos sobre el protocolo y los procedimientos.d) El manual del investigador.e) El cuaderno de recogida de datos de cada sujeto.f) El informe final.g) El certificado de auditoría, cuando proceda.5. El promotor o el propietario subsiguiente conservará el informe final hasta cinco años después
de haberse agotado el plazo de validez del medicamento.6. Se documentará todo cambio que se produzca en la posesión de los datos.7. Todos los datos y documentos se pondrán a disposición de las autoridades competentes si és-
tas así lo solicitan.8. Se asegurará, en todo caso, la confidencialidad de los datos y documentos contenidos en el ar-
chivo.
Artículo 22. Publicaciones.1. La publicación de los ensayos clínicos autorizados se realizará en revistas científicas y con
mención del Comité Ético de Investigación Clínica correspondiente.2. Cuando se hagan públicos estudios y trabajos de investigación sobre medicamentos dirigidos a
la comunidad científica, se harán constar los fondos obtenidos por el autor por o para su realizacióny la fuente de financiación.
3. Se mantendrá en todo momento el anonimato de los sujetos participantes en el ensayo.4. Los resultados o conclusiones de los ensayos clínicos se comunicarán prioritariamente en pu-
blicaciones científicas antes de ser divulgados al público no sanitario. No se darán a conocer de mo-do prematuro o sensacionalista procedimientos de eficacia todavía no determinada o exagerar ésta.
5. La publicidad de productos en fase de investigación clínica está absolutamente prohibida, talcomo se recoge en la Ley 34/1988, de 11 de noviembre (RCL 1988\2279), General de Publicidad.
Artículo 23. Uso compasivo.1. Se entiende como uso compasivo la utilización, en pacientes aislados y al margen de un ensa-
yo clínico, de productos en fase de investigación clínica, o también la utilización de especialidades
98
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

farmacéuticas para indicaciones o condiciones de uso distintas de las autorizadas, cuando el médi-co, bajo su exclusiva responsabilidad, considera indispensable su utilización.
2. Para utilizar un medicamento bajo las condiciones de uso compasivo se requerirá el consenti-miento informado por escrito del paciente o de su representante legal, un informe clínico en el queel médico justifique la necesidad de dicho tratamiento, la conformidad del Director del centro dondese vaya a aplicar el tratamiento y la autorización de la Dirección General de Farmacia y ProductosSanitarios para cada caso concreto.
3. El médico responsable comunicará a la Dirección General de Farmacia y Productos Sanita-rios los resultados del tratamiento, así como los acontecimientos adversos que puedan ser debi-dos al mismo, sin perjuicio de la comunicación de reacciones adversas a las Comunidades Autó-nomas.
TITULO II
De la intervención administrativa sobre ensayos clínicos con medicamentos
Artículo 24. Autorización de ensayos clínicos.1. De acuerdo con lo dispuesto en el artículo 65 de la Ley del Medicamento, los ensayos clínicos
con sustancias o medicamentos contarán, para ser realizados en territorio nacional, con la autoriza-ción de la Dirección General de Farmacia y Productos Sanitarios, además del informe previo del co-rrespondiente Comité Ético de Investigación Clínica (CEIC) acreditado.
2. La solicitud de autorización de un ensayo clínico será formulada por el promotor del estudio (orepresentante autorizado) mediante solicitud dirigida al Director general de Farmacia y ProductosSanitarios de acuerdo con el modelo que figura en el anexo 2A de este Real Decreto. En caso de en-sayos con medicamentos registrados en España como especialidades farmacéuticas para condicio-nes distintas de las que fueron autorizadas, la solicitud se dirigirá al Comité Ético de InvestigaciónClínica, el cual trasladará la documentación al Ministerio de Sanidad y Consumo.
3. La solicitud irá acompañada de la siguiente documentación:a) Protocolo del ensayo (anexo 1).b) Compromiso del investigador (anexo 3).c) Informe de la realización y seguimiento del ensayo por parte del Comité Ético de Investigación
Clínica debidamente acreditado según proceda (anexo 4).d) Conformidad de la Dirección del centro en que se realizará el ensayo (Dirección médica del
hospital o Director-gerente de atención primaria) o, en su caso, de la Dirección de la institución sa-nitaria de que depende dicho centro (anexo 5).
4. La Dirección General de Farmacia y Productos Sanitarios comunicará al promotor y a las Co-munidades Autónomas correspondientes la recepción del ensayo y su posterior autorización expre-sa, cuando proceda.
Artículo 25. Autorización de ensayos clínicos cuando el investigador actúa como promotor.Cuando el investigador sea a la vez el promotor que solicita la realización de un ensayo clínico con
un preparado en trámite de registro o con un producto en fase de investigación clínica solicitado oconcedido, podrá hacer referencia a la documentación presentada por el promotor del PEI o solici-tud de registro con una autorización expresa del mismo.
Artículo 26. Autorización previa de ensayos clínicos.1. Serán sometidos a un régimen de autorización previa por la Dirección General de Farmacia y
Productos Sanitarios:a) El primer ensayo clínico con una sustancia calificada como producto en fase de investiga-
ción clínica. Cuando la solicitud de autorización de dicho ensayo se realice conjuntamente conla solicitud de calificación de la sustancia en estudio como producto en fase de investigaciónclínica, la autorización del ensayo se producirá en unidad de acto con la calificación del pro-ducto.
99
REAL DECRETO 561/1993

b) El primer ensayo clínico de un promotor con un principio activo contenido en una especialidadfarmacéutica registrada en España, que se refiera a una nueva indicación terapéutica.
c) Ensayos clínicos sin interés terapéutico para el sujeto que no estén incluidos en el plan de in-vestigación de un producto en fase de investigación clínica autorizado, excepto los ensayos de bioe-quivalencia con genéricos.
2. El régimen de autorización previa requiere la autorización explícita del ensayo por la DirecciónGeneral de Farmacia y Productos Sanitarios que tendrá lugar en los plazos siguientes: a partir deldía de entrada de la solicitud de autorización del ensayo clínico en el Ministerio de Sanidad y Consu-mo, la Dirección General de Farmacia y Productos Sanitarios dispondrá de un plazo de sesenta díasnaturales para hacer las comprobaciones y observaciones correspondientes, solicitar la informaciónpertinente o autorizar o denegar el ensayo clínico. A partir del día de entrada de la última informa-ción solicitada, la Dirección General de Farmacia y Productos Sanitarios dispondrá de treinta díaspara pronunciarse. Se considerarán desestimadas las solicitudes respecto de las cuales no haya re-caído resolución expresa dentro de los mencionados plazos.
Artículo 27. Procedimiento abreviado de autorización de ensayos clínicos.1. Serán sometidos a este régimen de autorización.a) Los ensayos clínicos con una sustancia calificada como producto en fase de investigación clíni-
ca en las indicaciones previstas en la autorización como producto en fase de investigación clínica,una vez que se hubiere autorizado el primero de acuerdo con lo establecido en el artículo 26 delpresente Real Decreto.
b) Ensayos clínicos con un principio activo contenido en una especialidad farmacéutica registradaen España que se refieran a una nueva indicación, cuando ya se haya autorizado para el mismopromotor algún ensayo clínico en esas condiciones.
c) Los ensayos clínicos con principios activos de especialidades farmacéuticas registradas en Es-paña que contemplen nuevas dosificaciones, nuevas combinaciones o en general, condiciones deuso distintas de las autorizadas.
d) Los ensayos clínicos con especialidades farmacéuticas registradas en España que se refieran alas condiciones de uso que figuran en su autorización sanitaria.
e) Estudios de bioequivalencia con genéricos.2. Las solicitudes de autorización de los ensayos referidos en el apartado anterior se podrán
entender estimadas si no ha recaído resolución expresa en el plazo de sesenta días naturales, apartir de la fecha de entrada de la solicitud de autorización en el Ministerio de Sanidad y Consu-mo.
3. En los casos en que la Dirección General de Farmacia y Productos Sanitarios formule objecio-nes, ésta autorizará o denegará explícitamente el ensayo en el plazo de treinta días, contados a par-tir de la fecha de entrada en el Ministerio de Sanidad y Consumo de la última información solicitada.Se considerarán desestimadas las solicitudes respecto de las cuales no haya recaído resolución ex-presa dentro de los referidos plazos.
Artículo 28. Condiciones de la autorización.La autorización de un ensayo clínico fijará el plazo y las condiciones temporales de su realización.
La prolongación de la validez de la autorización será solicitada y justificada por el promotor a la Di-rección General de Farmacia y Productos Sanitarios o al Comité Ético de Investigación Clínica, en sucaso, y se tramitará en los mismos plazos y con los mismos efectos establecidos para el otorgamien-to de la autorización inicial.
Artículo 29. Denegación de ensayos clínicos.1. La Dirección General de Farmacia y Productos Sanitarios formulará propuesta de resolución
denegatoria de un ensayo clínico, cuando proceda, previo trámite de audiencia al promotor en lostérminos que establece la Ley de Régimen Jurídico de las Administraciones Públicas y del Procedi-miento Administrativo Común.
2. La resolución definitiva únicamente se producirá una vez se hayan examinado las alegaciones,en su caso, presentadas por el interesado.
3. La resolución definitiva se comunicará al Comité Ético de Investigación Clínica, a las Comunida-des Autónomas y al promotor, el cual podrá interponer recurso ordinario en el plazo de un mes,
100
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

conforme a lo establecido en el artículo 114 de la Ley de Régimen Jurídico de las AdministracionesPúblicas y del Procedimiento Administrativo Común.
Artículo 30. Modificaciones a los protocolos de ensayos clínicos autorizados.1. Cualquier modificación de un protocolo de ensayo clínico previamente autorizado será notifica-
da a los Comités Éticos de Investigación Clínica involucrados en el mismo, a la Dirección General deFarmacia y Productos Sanitarios y a las Comunidades Autónomas.
2. Cuando la modificación sea relevante requerirá el informe previo de los Comités Éticos de In-vestigación Clínica involucrados en el mismo y la autorización por parte de la Dirección General deFarmacia y Productos Sanitarios. Se consideran modificaciones relevantes aquellas que suponganun aumento del riesgo para los sujetos participantes. La ampliación del número de centros inicial-mente previstos necesitará autorización de la Dirección General de Farmacia y Productos Sanitarios.
3. La solicitud de autorización de cualquier cambio relevante en las condiciones del ensayo ini-cialmente previstas será debidamente justificada. Para ello se utilizará el modelo especificado en elanexo 2B, acompañado de un resumen del protocolo (anexo 1), en el que se haya incluido la modi-ficación propuesta, fechada y firmada por el promotor y el investigador, así como del informe previodel correspondiente Comité Ético de Investigación Clínica.
4. Las modificaciones se entenderán autorizadas si en el plazo de treinta días naturales siguientesa la entrada de la solicitud en el Ministerio de Sanidad y Consumo, la Dirección General de Farma-cia y Productos Sanitarios no ha formulado objeciones a las mismas.
Artículo 31. Suspensión de un ensayo clínico autorizado.1. La realización de un ensayo clínico se suspenderá por petición justificada del promotor o por
decisión de la Dirección General de Farmacia y Productos Sanitarios en los siguientes supuestosprevistos en el apartado 5 del artículo 65 de la Ley del Medicamento:
a) Si se viola la Ley.b) Si se alteran las condiciones de su autorización.c) No se cumplen los principios éticos recogidos en este Real Decreto.d) Para proteger a los sujetos del ensayo, o,e) En defensa de la salud pública.2. Las Comunidades Autónomas, por propia iniciativa o a propuesta del Comité Ético de Investiga-
ción Clínica correspondiente, podrán realizar una suspensión cautelar del ensayo clínico cuando secumpla alguno de los supuestos previstos en el punto anterior, comunicándolo de inmediato a la Di-rección General de Farmacia y Productos Sanitarios.
Artículo 32. Informe final del ensayo clínico.Una vez terminada la realización del ensayo el promotor enviará a la Dirección General de Farma-
cia y Productos Sanitarios el informe final sobre los resultados del mismo, responsabilizándose consu firma junto con la del investigador de la veracidad de los datos reflejados en la comunicación yde su concordancia con los datos originales obtenidos. Cuando la duración del ensayo sea superiora un año, será necesario además que el promotor remita un informe anual sobre la marcha del mis-mo. En el caso de que la investigación no llegue a su fin, el promotor enviará a la Dirección Generalde Farmacia y Productos Sanitarios un informe que incluya los datos obtenidos hasta su suspensióny los motivos de ésta. Asimismo, notificará la finalización del ensayo al Comité Ético de InvestigaciónClínica y a las Comunidades Autónomas.
Artículo 33. Importación de productos para ensayos clínicos.La autorización para la importación de los productos a utilizar en ensayos clínicos se ajustará a la
normativa legal aplicable en cada caso y podrá ser concedida en unidad de acto con la autorizacióndel ensayo clínico y su período de validez será el mismo. El promotor deberá llevar un registro de losproductos importados.
Artículo 34. Continuación del tratamiento tras la finalización del ensayo.Toda continuación de la administración de los productos en ensayo clínico una vez finalizado és-
te, mientras no esté autorizado el medicamento para esas condiciones de uso, se regirá por las nor-mas establecidas para el uso compasivo en el artículo 23 de este Real Decreto.
101
REAL DECRETO 561/1993

Artículo 35. Autorización de un producto en fase de investigación clínica (PEI).1. El promotor solicitará la calificación de producto en fase de investigación clínica cuando pre-
tenda realizar investigación clínica con alguna entidad química o biológica que no sea principio acti-vo de alguna especialidad farmacéutica registrada en España.
2. La solicitud de producto en fase de investigación clínica se presentará de acuerdo con el conte-nido y formato especificados en el anexo 7.
3. Sólo se podrá iniciar el primer ensayo con un producto de los contemplados en el punto ante-rior cuando la Dirección General de Farmacia y Productos Sanitarios haya autorizado la calificaciónde producto en fase de investigación clínica para las indicaciones solicitadas.
4. Dicha autorización tendrá lugar en los plazos siguientes: a partir del día de entrada de la soli-citud de autorización del producto en fase de investigación clínica en el Ministerio de Sanidad yConsumo, la Dirección General de Farmacia y Productos Sanitarios dispondrá de un plazo de se-senta días naturales para hacer las comprobaciones y observaciones correspondientes, solicitar lainformación pertinente o conceder o denegar la calificación de producto en fase de investigaciónclínica. A partir del día de entrada de la última información solicitada, la Dirección General de Far-macia y Productos Sanitarios dispondrá de treinta días para pronunciarse. Se considerarán desesti-madas las solicitudes respecto de las cuales no haya recaído resolución expresa dentro de los refe-ridos plazos.
5. Los protocolos definitivos de ensayos clínicos contemplados en el plan de investigación que nose hayan presentado simultáneamente con la solicitud del producto en fase de investigación clínicapodrán adjuntarse con posterioridad. En cualquier caso la autorización de un producto en fase deinvestigación clínica obliga al inicio en España de, al menos, un ensayo clínico de los previstos en elplan de investigación dentro del plazo de dos años naturales, a partir de la fecha de autorización deun producto en fase de investigación clínica.
Artículo 36. Renovación de la calificación de producto en fase de investigación clínica.1. La autorización de un producto en fase de investigación clínica será renovada cada dos años
hasta que se autorice la comercialización del producto o por el contrario, quedará sin efecto.2. Ante la solicitud de renovación, la Dirección General de Farmacia y Productos Sanitarios, a la
vista del expediente y plan de investigación actualizados, de los informes finales de los ensayos rea-lizados y de los informes de los ensayos en curso, podrá renovar la autorización de producto en fasede investigación clínica o denegarla, previa audiencia del interesado, en los plazos establecidos enel artículo 35.4 de este Real Decreto.
Artículo 37. Suspensión de la calificación de un producto en fase de investigación clínica.La calificación de un producto en fase de investigación clínica puede suspenderse a petición del
promotor, si no se solicita la renovación a los dos años naturales de su autorización o por decisiónmotivada de la Dirección General de Farmacia y Productos Sanitarios si se cumple alguno de los su-puestos del artículo 26 de la Ley del Medicamento que le sean aplicables o siempre que deje de es-tar justificada dicha calificación.
Artículo 38. Autorización de centros para la realización de ensayos sin finalidad terapéutica.1. Los ensayos clínicos sin interés terapéutico para los sujetos de la investigación sólo podrán ser
realizados en centros de investigación que autorice, para cada ensayo, el Ministerio de Sanidad yConsumo.
2. A tal efecto, el responsable del centro o unidad de investigación deberá solicitar la oportunaautorización de la Dirección General de Farmacia y Productos Sanitarios, la cual recabará un in-forme, en su caso, de la Administración sanitaria titular del centro, previo a la concesión de la au-torización.
3. Contra la Resolución de la Dirección General de Farmacia y Productos Sanitarios podrá interpo-nerse recurso ordinario en el plazo de un mes, de conformidad con lo establecido en el artículo 114de la Ley de Régimen Jurídico de las Administraciones Públicas y del Procedimiento AdministrativoComún.
102
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

TITULO III
De los Comités Éticos de Investigación Clínica
Artículo 39. Acreditación de los Comités Éticos de Investigación Clínica.1. Los Comités Éticos de Investigación Clínica serán acreditados por la autoridad sanitaria compe-
tente en cada Comunidad Autónoma que habrá de comunicarlo al Ministerio de Sanidad y Consu-mo.
2. El Ministerio de Sanidad y Consumo quedará encargado de la coordinación y establecimientode criterios comunes para la acreditación de los Comités.
3. La acreditación del Comité será renovada periódicamente por la autoridad sanitaria competentesegún los procedimientos y plazos que ésta determine, debiendo comunicarlo a la Dirección Generalde Farmacia y Productos Sanitarios.
Artículo 40. Ambito de actuación y sistema de elección de miembros.El ámbito geográfico e institucional de actuación de cada Comité, así como el sistema de elección
del Presidente, Secretario y miembros del Comité, será determinado por la Comunidad Autónomacorrespondiente, que lo comunicará a la Dirección General de Farmacia y Productos Sanitarios.
Artículo 41. Requisitos mínimos para la acreditación de un Comité Ético de Investigación Clínica.1. Estar formado como mínimo por siete miembros, de los cuales, dos al menos, deben ser ajenos
a las profesiones sanitarias, debiendo ser uno de ellos Licenciado en Derecho.2. Entre los miembros del citado Comité figurarán médicos, uno de los cuales será Farmacólogo
Clínico, un Farmacéutico de hospital y algún miembro del personal de enfermería.3. Garantía explícita, por parte del titular del centro, de que el Comité cuenta con los medios ne-
cesarios para poder realizar su cometido.4. Ni el Comité Ético de Investigación Clínica ni ninguno de sus miembros podrán percibir directa
ni indirectamente remuneración alguna por parte del promotor del ensayo.
Artículo 42. Funciones de los Comités Éticos de Investigación Clínica.El Comité Ético de Investigación Clínica ponderará los aspectos metodológicos, éticos y legales del
protocolo propuesto, así como el balance de riesgos y beneficios. Para ello:1. Evaluará la idoneidad del protocolo en relación con los objetivos del estudio, su eficiencia cien-
tífica (la posibilidad de alcanzar conclusiones válidas, con la menor exposición posible de sujetos) yla justificación de los riesgos y molestias previsibles, ponderadas en función de los beneficios espe-rados para los sujetos y la sociedad.
2. Evaluará la idoneidad del equipo investigador para el ensayo propuesto. Tendrá en cuenta suexperiencia y capacidad investigadora para llevar adelante el estudio, en función de sus obligacio-nes asistenciales y de los compromisos previamente adquiridos con otros protocolos de investiga-ción.
3. Evaluará la información escrita sobre las características del ensayo que se dará a los posiblessujetos de la investigación, o en su defecto, a su representante legal, la forma en que dicha informa-ción será proporcionada y el tipo de consentimiento que va a obtenerse.
4. Comprobará la previsión de la compensación y tratamiento que se ofrecerá a los sujetos partici-pantes en caso de lesión o muerte atribuibles al ensayo clínico, y del seguro o indemnización paracubrir las responsabilidades especificadas en el artículo 13.2
5. Conocerá y evaluará el alcance de las compensaciones que se ofrecerán a los investigadores ya los sujetos de la investigación por su participación.
6. Realizará el seguimiento del ensayo clínico desde su inicio hasta la recepción del informe final.
Artículo 43. Normas generales de funcionamiento de los Comités Éticos de Investigación Clínica.1. Sus miembros respetarán el principio de la confidencialidad, en lo que respecta a la documen-
tación recibida para la evaluación del protocolo y la identidad de los pacientes.2. Para que las decisiones sobre un protocolo concreto sean válidas, se requerirá la participación
de como mínimo la mitad más uno de sus miembros, de los que, al menos, uno será ajeno a la pro-fesión sanitaria.
103
REAL DECRETO 561/1993

3. En los casos que exista Comisión de Investigación o Comité de Etica Asistencial, será necesariala presencia de, al menos, un miembro de cada una de ellas, para que la decisión del Comité Éticode Investigación Clínica sobre cada uno de los protocolos sea válida.
4. Cuando el Comité evalúe protocolos de investigación clínica con procedimientos quirúrgicos,técnicas diagnósticas o productos sanitarios, contará, además, con al menos una persona expertaen el procedimiento o tecnología a evaluar.
5. Cuando lo estime oportuno, recabará el asesoramiento de personas expertas no pertenecientesal Comité, que respetarán el principio de confidencialidad.
6. El investigador principal o los colaboradores de un ensayo clínico, no podrán participar ni en laevaluación ni en el dictamen de su propio protocolo, aun cuando sean miembros del Comité.
7. Establecer un sistema que garantice que el protocolo aprobado por el Comité Ético de Investi-gación Clínica, es idéntico al enviado a la Dirección General de Farmacia y Productos Sanitarios, y elmismo que finalmente se llevará a cabo.
8. Establecer un sistema de comunicación con los investigadores, que le permita conocer cuándose ha producido un acontecimiento adverso mortal o grave e inesperado.
9. Elaborar y seguir para su funcionamiento unos procedimientos de trabajo específicos, marcaruna periodicidad de reunión y un tiempo máximo de respuesta. Estos procedimientos deberán ha-cerse públicos.
10. Cada reunión del Comité quedará recogida en el acta correspondiente, en la que sedetallarán los miembros asistentes. El acta reflejará, explícitamente, que para cada estudioevaluado se han ponderado los aspectos contemplados en el artículo 42 del presente RealDecreto.
11. El Comité Ético de Investigación Clínica seguirá los procedimientos que las Comunidades Au-tónomas señalen en materia de comunicaciones a las autoridades así como el resto de disposicio-nes que éstas desarrollen en cuanto a funcionamiento.
TITULO IV
Del cumplimiento de las normas de buena práctica clínica y de las inspecciones de buena práctica clínica
Artículo 44. Cumplimiento de las normas de buena práctica clínica.En todos los ensayos clínicos con medicamentos se seguirán las normas de buena práctica clínica
(BPC).
Artículo 45. Procedimientos normalizados de trabajo.1. Las normas de buena práctica clínica exigen la existencia de unos procedimientos normaliza-
dos de trabajo (PNT) que indiquen de forma detallada la conducta a seguir en cada uno de los as-pectos relacionados con la organización, realización, recopilación de los datos, documentación y ve-rificación de los ensayos clínicos. Es responsabilidad del promotor establecerlos y garantizar que suconocimiento y puesta en práctica sean obligados para todos aquellos que participan en un ensayoclínico, especialmente para el monitor del ensayo, antes de iniciar éste.
2. Los aspectos que, como mínimo, serán regulados por los procedimientos normalizados de tra-bajo son los siguientes:
a) Identificación y calificación del investigador principal y sus colaboradores, determinación de laidoneidad del centro donde se realice el estudio, del laboratorio que determine los datos biológicos yclínicos complementarios, y del Comité Ético de Investigación Clínica.
b) Procedimiento de archivo de la documentación esencial del ensayo clínico: protocolo y en-miendas, cuadernos de recogida de datos e informes complementarios, notificación de aconteci-mientos adversos e informes de monitorización. Se definirán los tiempos de archivo, tanto en el cen-tro del investigador como en el del promotor.
c) Procedimientos de monitorización, incluyendo periodicidad mínima, correcciones posibles enel cuaderno de recogida de datos y verificación de los datos originales.
104
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

d) Regulación de los procedimientos de suministro de medicación en estudio, registro de dispen-sación a los sujetos del ensayo y destino de ésta.
e) Procedimiento de notificación de acontecimientos adversos graves e inesperados.f) Procedimiento para proporcionar la información adecuada al sujeto del ensayo y comprobar
que éste ha otorgado su consentimiento informado para participar en el ensayo.3. Cada uno de los aspectos aquí reseñados, se adaptarán a lo establecido en las «Normas de
buena práctica clínica para ensayos clínicos con medicamentos en la Comunidad Europea» elabora-das por la Comisión de las Comunidades Europeas, así como a posteriores revisiones de estas nor-mas.
Artículo 46. Inspecciones por las autoridades sanitarias.1. Las autoridades sanitarias de las Comunidades Autónomas tendrán facultades inspectoras en
materia de ensayos clínicos, pudiendo investigar incluso las historias clínicas individuales de los su-jetos del ensayo,guardando siempre su carácter confidencial.
2. Con objeto de verificar la observancia de las normas de buena práctica clínica, las Comunida-des Autónomas realizarán inspecciones a los Comités Éticos de Investigación Clínica, al centro deinvestigación o al promotor, sin perjuicio de lo previsto en el apartado 3 de este artículo.
3. Corresponden a la Administración General del Estado las funciones de inspección en los casosseñalados en los apartados a) y c) del apartado 2 del artículo 105 de la Ley 25/1990, de 20 de di-ciembre, del Medicamento.
4. La Administración General del Estado podrá suscribir convenios con las Comunidades Autó-nomas para el establecimiento de criterios y procedimientos para la realización de estas inspec-ciones.
Artículo 47. Procedimiento de las inspecciones.1. Las inspecciones podrán ser realizadas durante el curso del ensayo o bien después de su fina-
lización.2. Una vez realizada la inspección, sus resultados serán comunicados por la autoridad sanitaria
responsable al promotor, al investigador, y al Comité Ético de Investigación Clínica, en un plazo detreinta días.
3. Como consecuencia de la inspección, la autoridad sanitaria responsable procederá a la inte-rrupción cautelar del ensayo clínico, siempre que se produzca alguno de los supuestos previstos enel artículo 31 de este Real Decreto.
4. Para las anomalías detectadas en el curso de la inspección será de aplicación el Título IX sobrerégimen sancionador de la Ley 25/1990, del Medicamento. Cuando se estime que estas anomalíasafectan a la credibilidad de los datos obtenidos, la Dirección General de Farmacia y Productos Sani-tarios invalidará éstos.
Disposición adicional primera. Ensayos clínicos con productos sanitarios.Los ensayos clínicos con productos sanitarios se regirán por los principios recogidos en el Título I
sobre consideraciones generales y principios básicos y en el Título III de los Comités Éticos de Inves-tigación Clínica, siempre que les sean aplicables.
Disposición adicional segunda. Facultad de modificación.Se autoriza al Ministro de Sanidad y Consumo para modificar el contenido de los anexos del pre-
sente Real Decreto.
Disposición transitoria primera. Validez de actuaciones de los Comités.Durante el primer año de vigencia de este Real Decreto se considerarán válidas las actuacio-
nes de los Comités Éticos de Investigación Clínica que ya se hubieran constituido o, en su de-fecto, los de los Comités de Ensayos Clínicos existentes o constituidos con arreglo a la normati-va anterior.
Disposición transitoria segunda. Excepción del artículo 20.Lo dispuesto en el artículo 20 solamente será exigible a partir del año siguiente a la entrada en vi-
gor de este Real Decreto.
105
REAL DECRETO 561/1993

Disposición derogativa única. Derogación normativa.Queda derogado el Real Decreto 944/1978, de 14 de abril, por el que se regulan los ensayos clí-
nicos de productos farmacéuticos y preparados medicinales, modificado por el Real Decreto424/1988, de 29 de abril, y la Orden ministerial de 3 de agosto de 1982, por la que se desarrolla elcitado Real Decreto 944/1978.
Disposición final única. Entrada en vigorEl presente Real Decreto entrará en vigor a los tres meses de su publicación en el «Boletín Oficial
del Estado».
106
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

ANEXO 1
Estructuración y contenido de un protocolo de ensayo clínico con medicamentos
1. Resumen.Se presentará al principio del protocolo y de acuerdo con el siguiente contenido y formato:0. Tipo de solicitud. Se hará referencia a uno de los siguientes supuestos:
a) Primer ensayo clínico de un PEI autorizado o en trámite.b) Ensayo clínico posterior al primero autorizado con un PEI, indicando el código de éste.c) Primer ensayo clínico en una nueva indicación.d) Ensayo clínico en una nueva indicación con ensayo clínico previamente autorizado en
esa condición, indicando el número de ese ensayo.e) Ensayo clínico con un principio activo de una especialidad farmacéutica en nuevas con-
diciones de uso.f) Ensayo clínico con una especialidad farmacéutica en las condiciones de uso autoriza-
das.g) Modificación de ensayo autorizado.i) Solicitud de anulación.
1. Identificación del promotor.2. Título del ensayo clínico.3. Código del protocolo.4. Investigador principal. Dirección de su centro de trabajo.5. Centros en los que se prevé realizar el ensayo.6. Comités Éticos de Investigación Clínica que han aprobado el ensayo.7. Nombre y calificación de la persona responsable de la monitorización.8. Fármaco experimental y control: dosis, forma farmacéutica, vía de administración, grupo tera-
péutico.9. Fase del ensayo clínico.
10. Objetivo principal (eficacia, seguridad, farmacocinética, búsqueda de dosis, etcétera).11. Diseño (aleatorizado, controlado, doble ciego, ...).12. Enfermedad o trastorno en estudio.13. Variable principal de valoración.14. Población en estudio y número total de pacientes.15. Duración del tratamiento.16. Calendario y fecha prevista de finalización.
2. Índice.
3. Información general.A. Identificación del ensayo.1.º Código de protocolo: Clave de 15 caracteres como máximo, que será específica para cada en-
sayo, e idéntica para todas las versiones de un mismo protocolo. Será asignada por el promotor yquedará reflejada con claridad junto al título en la primera página del protocolo, e irá seguida de lafecha correspondiente a la versión de que se trate.
El código estará compuesto por letras y números disponibles en una máquina de escribir de tecla-do español. También se podrán incluir los signos ortográficos guión (-) y barra (/). Se hará clara dis-tinción entre ceros y oes, así como entre íes y unos. No se dejarán espacios en blanco entre carac-teres. Las letras se entenderán como mayúsculas a todos los efectos.
2.º Título.B. Tipo de ensayo clínico.Indicar:
a) Si se refiere a un PEI (identificando éste y si procede, el código del primer protocolo dedicho PEI).
b) Si se refiere a una nueva indicación con un producto contenido en una especialidad far-macéutica.
107
REAL DECRETO 561/1993

c) Si se refiere a una especialidad farmacéutica en otras condiciones de uso diferentes alas de su autorización.
d) Si se refiere a una especialidad farmacéutica en las condiciones de uso especificadas ensu autorización sanitaria.
C. Descripción de los productos en estudio (experimental y control).a) Denominación genérica, nombre comercial y países en que está comercializado cuando
proceda.b) Composición cuantitativa y cualitativa de los mismos, indicando los principios activos y
aquellos excipientes que sea obligado especificar en el material de acondicionamientode las especialidades farmacéuticas.
c) Forma farmacéutica.d) Características organolépticas cuando se utilice algún procedimiento de enmascara-
miento.e) Entidades elaboradoras de las muestras.
D. Datos relativos al promotor.Nombre, dirección, teléfono y telefax o télex, si lo hubiera. En el caso de que el promotor esté ubi-
cado fuera de España, nombre, dirección, teléfono y telefax o télex, si lo hubiera, del responsableautorizado en España.
E. Director técnico responsable de la elaboración/control de las muestras.F. Identificación del monitor.G. Datos de los investigadores del ensayo.Especificar el investigador principal y colaboradores, incluyendo su lugar de trabajo en cada cen-
tro.H. Centros en que se realizará el ensayo.Identificar el Comité Ético de Investigación Clínica que ha informado favorablemente la realización
del ensayo en cada centro. Indicar también los centros internacionales si procede.I.Duración prevista del ensayo.
4. Justificación y objetivos.a) Justificar la realización del ensayo en base a toda la información relevante y específica
de que se disponga. (Incluir tanto referencias bibliográficas como datos no publica-dos.)
b) En base a dicha justificación, concretar el (los) objetivo(s) del ensayo diferenciandocuando proceda el principal de los secundarios.
5. Tipo de ensayo clínico y diseño del mismo.a) Fase de desarrollo (indicar si es un estudio piloto).b) Descripción detallada del proceso de aleatorización (procedimiento, consideraciones
prácticas...).c) Tipo de control (placebo u otros) y diseño (cruzado, paralelo...).d) Técnicas de enmascaramiento. Medidas que se adoptarán para el mantenimiento del
carácter ciego del estudio, situaciones en que pueda romperse y forma de proceder enestos casos, etcétera.
e) Períodos de preinclusión o lavado, seguimiento, etcétera.
6. Selección de los sujetos.a) Criterios de inclusión y exclusión.b) Criterios diagnósticos para las patologías en estudio (si es posible reconocidos a nivel in-
ternacional).c) Número de sujetos previstos (total y por centros si procede) y justificación del mismo.
Indicar el método de cálculo para determinar el tamaño de la muestra y los datos emple-ados para ello.
d) Criterios de retirada y análisis previstos de las retiradas y los abandonos.e) Tratamiento de las pérdidas prerrandomización.f) Duración aproximada del período de reclutamiento en función del número de pacientes
disponibles.
108
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

7. Descripción del tratamiento.a) Descripción de las dosis, intervalo, vía y forma de administración y duración del (de los)
tratamientos del ensayo.b) Criterios para la modificación de pautas a lo largo del ensayo (tanto en los estudios de
búsqueda de dosis, como en los de tolerancia o en casos de reacciones adversas o toxi-cidad…).
c) Tratamientos concomitantes permitidos y prohibidos.d) Especificación de «mediación de rescate» en los casos en que proceda.e) Normas especiales de manejo de los fármacos en estudio.f) En caso de tratamientos no permitidos, especificar el período de tiempo mínimo transcu-
rrido desde su suspensión hasta que el sujeto pueda ser incluido en el estudio.g) Medidas para valorar el cumplimiento.
8. Desarrollo del ensayo y evaluación de la respuesta.a) Especificar la variable principal de evaluación (preferentemente objetiva y la más rele-
vante desde el punto de vista clínico) y aquellas otras que se consideren secundarias.b) Desarrollo del ensayo en el que se indicará el número y tiempo de las visitas durante el
mismo, especificando las pruebas o exploraciones que se realizarán para la valoraciónde la respuesta.
c) Descripción de los métodos (radiológicos, de laboratorio, etc.), utilizados para la valoraciónde la respuesta y control de calidad de los mismos. Pueden ir incluidos en un anexo.
9. Acontecimientos adversos.a) Indicar la información mínima que se deberá especificar para los acontecimientos ad-
versos que ocurran a un sujeto durante el ensayo (descripción, gravedad, duración, se-cuencia temporal, método de detección, tratamiento administrado, en su caso, causasalternativas o factores predisponentes, ...).
b) Indicar criterios de imputabilidad que se van a utilizar.c) Indicar los procedimientos para la notificación inmediata de los acontecimientos adver-
sos graves o inesperados.d) Se incluirá un modelo de hoja de notificación de acontecimientos adversos a las autori-
dades sanitarias (anexo 8).
10. Aspectos éticos.a) Consideraciones generales: aceptación de las normas nacionales e internacionales al
respecto (versión actual de la declaración de Helsinki,etcétera).b) Información que será proporcionada a los sujetos y tipo de consentimiento que será soli-
citado en el ensayo (anexo 6).c) Especificar quiénes tendrán acceso a los datos de los voluntarios en aras a garantizar su
confidencialidad.d) Contenidos del presupuesto del ensayo (compensación para los sujetos del ensayo, in-
vestigadores, etcétera), que deban ser comunicados al Comité Ético de Investigación Clí-nica correspondiente.
e) Garantía de la existencia de una póliza de seguro o indemnización suscrita y característi-cas de la misma. (Puede incluirse en un anexo.)
11. Consideraciones prácticas.a) Especificar las responsabilidades de todos los participantes en el ensayo.b) Especificar las condiciones de archivo de datos, su manejo y procesamiento, correcciones…c) Identificación de muestras para investigación clínica y responsables de su suministro y
conservación. Etiquetado de las mismas.d) Condiciones de publicación.
12. Análisis estadístico.a) Especificar las pruebas estadísticas que se prevé utilizar en el análisis de los resultados,
especialmente en lo que a la variable de valoración principal se refiere.
109
REAL DECRETO 561/1993

b) Indicar si está prevista la realización de análisis intermedios, especificando cuáles seríanlos criterios que determinarían la finalización del ensayo.
c) Indicar dónde se realizará dicho análisis.
Anexo I. Cuaderno de recogida de datos.Específico para cada ensayo clínico.
Anexo II. Manual del investigador.Versión actualizada de la información preclínica y clínica relevante para el ensayo sobre los pro-
ductos en estudio.
Anexo III. Procedimientos normalizados de trabajo.a) Deberán contemplarse, al menos, los procedimientos normalizados de trabajo referentes
a los apartados, incluidos en el artículo 45 de este Real Decreto.b) Programa de auditorías internas si existieran. Este apartado será imprescindible en
aquellos ensayos cuyos resultados pretendan ser utilizados como soporte para registrode una especialidad farmacéutica o modificaciones de las condiciones de una autoriza-ción previa.
Anexo IV. Memoria analítica de las muestras a utilizar.Deberá presentarse siempre a la Dirección General de Farmacia y Productos Sanitarios, excepto
cuando los productos sean especialidad farmacéutica en nuestro país o tengan la calificación dePEI.
110
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

ANEXO 2A
Solicitud de ensayo clínico con medicamentos
Don (nombre y apellidos del promotor) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(relación con la entidad promotora, nombre y dirección social del promotor) . . . . . . . . . . . . . . . . . .
EXPONE:Que desea llevar a cabo el estudio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(título) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Que el ensayo clínico será realizado en el Servicio de . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .del Hospital/Centro . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .por . . . . . . . . . . . . . . . . . . . . . . . (nombre y apellidos) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .como investigador principal.
Que el estudio se realizará tal y como se ha planteado respetando la normativa legal aplicable pa-ra los ensayos clínicos que se realicen en España y siguiendo las normas éticas internacionalmenteaceptadas (Helsinki/Tokio y otras).
Por lo expuesto,
SOLICITA:Le sea autorizada la realización de este ensayo por el procedimiento (1):
■■ Simplificado ■■ Ensayo clínico posterior al primero autorizado con un PEI (indicar código).■■ Ensayo clínico en una nueva indicación con ensayo clínico previamente autorizado en esa con-
dición (indicar el número de este último).■■ Ensayo clínico con un principio activo de una especialidad farmacéutica en nuevas condicio-
nes de uso.■■ Ensayo clínico con una especialidad farmacéutica en las condiciones de uso autorizadas.■■ Ensayo de bioequivalencia con genéricos.
■■ Autorización previa■■ Primer ensayo clínico de un PEI autorizado o en trámite.■■ Primer ensayo clínico en una nueva indicación.■■ Otros.
para lo cual se adjunta la siguiente documentación:
Protocolo del ensayo.Compromiso del investigador.Informe del CEIC sobre la realización del ensayo.Conformidad del Director del Centro.
Firmado:
El promotor.Don/doña . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
El investigador principal.Don/doña . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
En . . . . . . . . . . . . . . . . . . . . . .a . . . . . .de . . . . . . . . . . . . . . . . . . .de . . . . . . . . .
Ilmo. Sr. Director general de Farmacia y Productos Sanitarios.
(1) Señálese lo que proceda
111
REAL DECRETO 561/1993

ANEXO 2B
Modificación/anulación de ensayo clínico autorizado con medicamentos
Don (nombre y apellidos del promotor) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(relación con la entidad promotora, nombre y dirección social del promotor) . . . . . . . . . . . . . . . . . .
EXPONE:Que desea solicitar le sea autorizada la modificación/anulación (1) del estudio: . . . . . . . . . . . . . .
(título y código) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Que dicha modificación/anulación (1) se solicita por . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(justificar el motivo de la petición) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Que la modificación solicitada se refiere a . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . (especificar) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
SOLICITA:Le sea autorizada la modificación/anulación (1) anteriormente referida, para lo cual se adjunta la
siguiente documentación:
Resumen del protocolo según el formato reglamentario.Aprobación del CEIC (2)
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Fecha y firma del promotor Fecha y firma del investigador principal
Ilmo. Sr. Director general de Farmacia y Productos Sanitarios.
(1) Táchese lo que no proceda(2) Sólo es necesario cuando se soliciten modificaciones relevantes.
112
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

ANEXO 3
Compromiso del investigador
Don . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Hace constar:
Que conoce y acepta participar como investigador principal en el ensayo clínico código de proto-colo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .titulado . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Que se compromete a que cada sujeto sea tratado y controlado siguiendo lo establecido en el pro-tocolo autorizado por el Comité Ético de Investigación Clínica y por la Dirección General de Farmaciay Productos Sanitarios.
Que respetará las normas éticas aplicables a este tipo de estudios.
Que dicho ensayo se llevará a cabo contando con la colaboración de . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
como investigadores colaboradores.
En . . . . . . . . . . . . . . . . . . . . . .a . . . . . .de . . . . . . . . . . . . . . . . . . .de . . . . . . . . .
Firmado: Firmado:
Don/doña . . . . . . . . . . . . . . . . . . . . . . . . Don/doña . . . . . . . . . . . . . . . . . . . . . . . . . .Investigador principal. Investigadores colaboradores (si procede).
113
REAL DECRETO 561/1993

ANEXO 4
Informe del Comité Ético de Investigación Clínica
Don/doña . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Secretario del Comité Ético de Investigación Clínica de . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(título) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
CERTIFICAQue este Comité ha evaluado la propuesta del promotor . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
para que se realice el ensayo clínico código de protocolo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .titulado . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .con los medicamentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .y considera que:
Se cumplen los requisitos necesarios de idoneidad del protocolo en relación con los objetivos delestudio y están justificados los riesgos y molestias previsibles para el sujeto.
La capacidad del investigador y los medios disponibles son apropiados para llevar a cabo el estu-dio.
Son adecuados tanto el procedimiento para obtener el consentimiento informado como la com-pensación prevista para los sujetos por daños que pudieran derivarse de su participación en el en-sayo.
El alcance de las compensaciones económicas previstas no interfiere con el respeto a los postula-dos éticos.
Y que este Comité acepta que dicho ensayo clínico sea realizado en el Centro . . . . . . . . . . . . . . .,por . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
como investigador principal.
Lo que firmo en . . . . . . . . . . . . . . . . . . .a . . . .de . . . . . . . . . . . . . . . . . . . . . .de . . . . . . .
Firmado:Don/doña . . . . . . . . . . . . . . . . . . . . . . . .
114
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

ANEXO 5
Conformidad de la Dirección del Centro
Don/doña . . . . . . . . . . . . . . . . . . . . . . . . .Director del Hospital/Centro . . . . . . . . . . . . . . . . . . . .y vista la autorización del Comité Ético de Investigación Clínica,
CERTIFICAQue conoce la propuesta realizada por el promotor
para que sea realizado en este Centro el ensayo clínico código de protocolo . . . . . . . . . . . . . . . . . .titulado . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .con los medicamentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .y que será realizado por . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .como investigador principal.
Que está de acuerdo con el contrato firmado entre el Centro y el promotor en el que se especifi-can todos los aspectos económicos de este ensayo clínico.
Que acepta la realización de dicho ensayo clínico en este Centro.
Lo que firma en . . . . . . . . . . . . . . . . . . .a . . . .de . . . . . . . . . . . . . . . . . . . . . .de . . . . . . .
Firmado:Don/doña . . . . . . . . . . . . . . . . . . . . . . . .
115
REAL DECRETO 561/1993

ANEXO 6
Consentimiento informado
1. Hoja de información para el posible participante.
Es el documento escrito, específico para cada ensayo clínico, que se entregará al posible partici-pante antes de que éste otorgue su consentimiento para ser incluido en el mismo.
Contendrá información referente a los siguientes aspectos del ensayo clínico:1.º Objetivo.2.º Metodología empleada.3.º Tratamiento que puede serle administrado, haciendo referencia al placebo, si procede.4.º Beneficios esperados para él o la sociedad.5.º Incomodidades y riesgos derivados del estudio (número de visitas, pruebas complementarias
a que se someterá…).6.º Posibles acontecimientos adversos.7.º Tratamientos alternativos disponibles.8.º Carácter voluntario de su participación, así como posibilidad de retirarse del estudio en cual-
quier momento, sin que por ello se altere la relación médico-enfermo ni se produzca perjui-cio en su tratamiento.
9.º Personas que tendrán acceso a los datos del voluntario y forma en que se mantendrá la con-fidencialidad.
10.º Modo de compensación económica y tratamiento en caso de daño o lesión por su participa-ción en el ensayo, tal como consta en la Ley del Medicamento.
11.º Investigador responsable del ensayo y de informar al sujeto y contestar a sus dudas y pre-guntas, y modo de contactar con él en caso de urgencia.
2. Modelo de consentimiento por escrito.
Título del ensayo: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Yo, . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(nombre y apellidos) . . . . . . . . . . . . . . . . . . . . . . . . . .
He leído la hoja de información que se me ha entregado.He podido hacer preguntas sobre el estudio.He recibido suficiente información sobre el estudio.He hablado con: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(nombre del investigador)
Comprendo que mi participación es voluntaria.Comprendo que puedo retirarme del estudio:1.º Cuando quiera.2.º Sin tener que dar explicaciones.3.º Sin que esto repercuta en mis cuidados médicos.
Presto libremente mi conformidad para participar en el estudio.
Fecha Firma del participante
116
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

3. Modelo de consentimiento oral ante testigos.
Título del ensayo: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Yo, . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(nombre y apellidos) . . . . . . . . . . . . . . . . . . . . . . . . . .declaro bajo mi responsabilidad que: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(nombre del participante en el ensayo) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Ha recibido la hoja de información sobre el estudio.Ha podido hacer preguntas sobre el estudio.Ha recibido suficiente información sobre el estudio.Ha sido informado por: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(nombre del investigador)Comprende que su participación es voluntaria.Comprende que puede retirarse del estudio:1.º Cuando quiera.2.º Sin tener que dar explicaciones.3.º Sin que esto repercuta en sus cuidados médicos.
Y ha expresado libremente su conformidad para participar en el estudio.
Fecha Firma del testigo
4. Modelo de consentimiento del representante.
Título del ensayo: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .Yo, . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(nombre y apellidos) . . . . . . . . . . . . . . . . . . . . . . . . . .en calidad de . . . . . . . . . . . . . . . . . . . . . .(relación con el participante) . . . . . . . . . . . . . . . . . . . .de . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(nombre del participante) . . . . . . . . . . . . . . . . . . . . . . .
He leído la hoja de información que se me ha entregado.He podido hacer preguntas sobre el estudio.He recibido respuestas satisfactorias a mis preguntas.He recibido suficiente información sobre el estudio.
He hablado con: . . . . . . . . . . . . . . . . . . . .(nombre del investigador) . . . . . . . . . . . . . . . . . . . . . . .
Comprendo que la participación es voluntaria.Comprendo que puede retirarse del estudio:1.º Cuando quiera.2.º Sin tener que dar explicaciones.3.º Sin que esto repercuta en sus cuidados médicos.
En mi presencia se ha dado a . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(nombre del participante) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .toda la información pertinente adaptada a su nivel de entendimiento y está de acuerdo en participar.Y presto mi conformidad con que . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(nombre del participante) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .participe en este estudio.
Fecha Firma del representante
117
REAL DECRETO 561/1993

ANEXO 7
Estructuración y documentación de un producto en fase de investigación clínica
A. Expediente.I. Resumen.1. Impreso de solicitud.
a) Identificación del promotor.b) Compromiso de realizar el plan de investigación tal y como se ha planteado, y siguiendo
las normas éticas reconocidas a nivel internacional.c) Compromiso de respetar la normativa legal aplicable para los ensayos clínicos que se
realicen en España.d) Firma del promotor o representante autorizado.1.º Tipo de solicitud. Se hará referencia a alguno de los siguientes supuestos:
Solicitud de PEI.Solicitud de ampliación del plan de investigación.Ampliación de información.Solicitud de renovación.Solicitud de anulación.
2.º Promotor (nombre, dirección, teléfono y telefax o télex). Cuando el promotor no esté ubicadoen nuestro país se indicarán además los datos del representante legalmente autorizado en Es-paña.
3.º Identificación del PEI en el caso de que haya sido solicitado o autorizado previamente. Núme-ro asignado por la Dirección General de Farmacia y Productos Sanitarios.
4.º Nombre de los principios activos del producto (DCI, DOE, código de identificación del producto…).5.º Forma(s) farmacéutica(s) y vía(s) de administración.6.º Indicación/es solicitada/s.7.º Fase clínica de los ensayos previstos (I, II o III).8.º Nombre y calificación de la persona responsable de la monitorización.9.º Nombre y calificación de la persona responsable de evaluar la seguridad del producto.2. Resumen de las características del producto.3. Informes de experto.
II. Documentación química farmacéutica y biológica.III. Documentación toxicológica y farmacológica.IV. Documentación clínica.La documentación se estructura de acuerdo con la nota explicativa para solicitantes de autoriza-
ciones de comercialización de medicamentos de uso humano en la CEE y contendrá toda la infor-mación disponible en el momento de su presentación.B. Plan de investigación clínica.
1. Descripción del plan de investigación para al menos los dos años siguientes a la concesión delproducto en fase de investigación clínica. Razón de ser de los ensayos propuestos, haciendoreferencia a los objetivos buscados; indicación o indicaciones que se estudiarán; ensayos clíni-cos que se realizarán en España y en total; calendario aproximado; diseño general que se se-guirá en cada ensayo planteado, número aproximado de pacientes que serán sometidos a tra-tamiento; riesgos y reacciones adversas previsibles.
2. Calificaciones y requisitos que se exigirán a los investigadores que van a realizar los ensayos.3. Identificación de la persona responsable de la monitorización de los ensayos que se plantean.4. Protocolos de los ensayos clínicos a realizar en España, de acuerdo con el formato y contenido
del anexo 1.Estos protocolos serán incorporados a la documentación a medida que se disponga de ellos o se
presenten para su autorización.5. Versión actualizada del manual del investigador.Normas generales de presentación.Se deberá incluir un índice de toda la documentación presentada.La documentación se presentará en el idioma oficial del Estado, pudiendo presentarse en inglés
las partes, II, III y IV del expediente y el manual del investigador.
118
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

ANEXO 8
119
REAL DECRETO 561/1993
NOTIFICACIÓN DE ACONTECIMIENTOSADVERSOS
PRODUCTO EN FASE DE INVESTIGACIÓN CLÍNICA
I. INFORMACIÓN SOBRE EL ACONTECIMIENTO ADVERSO
II. INFORMACIÓN DEL PRODUCTO EN INVESTIGACIÓN
III. HISTORIA CLÍNICA Y MEDICAMENTOS CONCOMITANTES
IV. INFORMACIÓN SOBRE PROMOTOR E INVESTIGADOR
PROTOCOLO N.º
PACIENTE N.º
N.º NOTIFICACIÓN(Laboratorio)
N.º NOTIFICACIÓN
1. INICIALES DEL PACIENTE
1a. PAÍS 2. FECHA DE NACIMIENTO 2ª. EDAD 3. SEXO 3a PESO 4-6. INICIODÍA MES AÑO DÍA MES AÑO
8-13. CONSECUENCIAS
FALLECIMIENTO
LA VIDA DELPACIENTE HAESTADO ENPELIGRO
HOSPITALIZACIÓNO PROLONGACIÓNDE LAHOSPITALIZACIÓN
INCAPACIDADPERMANENTE OSIGNIFICATIVA
PERSISTENCIA DELACONTECIMIENTOADVERSO
RECUPERACIÓN
7. DESCRIPCIÓN DEL ACONTECIMIENTO ADVERSO (incluyendo resultados relevantes de exploración o de laboratorio)
14. NOMBRE
15. DOSIS DIARIA
17. ENFERMEDAD EN ESTUDIO
18. FECHAS DEL TRATAMIENTO (DESDE/HASTA)
16. VÍA DE ADMINISTRACIÓN
19. DURACIÓN DEL TRATAMIENTO
20. ¿REMITIÓ ELACONTECIMIENTO ALSUSPENDER LAMEDICACIÓN?
21. ¿REAPARECIÓ ELACONTECIMIENTO ALADMINISTRAR DE NUEVOLA MEDICACIÓN?
No procedeNoSí
No procedeNoSí
22. MEDICAMENTOS CONCOMITANTES Y FECHA DE ADMINISTRACIÓN
23. DATOS IMPORTANTES DE LA HISTORIA CLÍNICA (Ej.: diagnósticos, alergias, embarazo, enfermedades concomitantes, etc.)
24a. NOMBRE Y DIRECCIÓN DEL PROMOTOR 24b. NOMBRE Y DIRECCIÓN DEL INVESTIGADOR
24d. CÓDIGO DE LABORATORIO (N.º DGFPS)
25a. TIPO DE INFORME INICIAL SEGUIMIENTO
24c. TÉCNICO DEL PROMOTOR QUE INFORMANOMBRE:TFNO:FIRMA:
24e. FECHA DEL INFORME 24f. FECHA DE ENTRADA DGFPS 25b. SE ADJUNTA INFORME COMPLEMENTARIO

INSTRUCCIONES GENERALES
1.Este formulario se utilizará solamente para comunicar los acontecimientos adversos mortales,graves o inesperados que ocurran durante el desarrollo del ensayo clínico.
2.Los acontecimientos adversos mortales o que entrañen riesgo vital (aquellos que de no habermediado una intervención terapéutica inmediata hubieran supuesto la muerte del paciente) secomunicarán en el plazo máximo de 72 horas; si no se dispusiera de toda la información, éstapodrá completarse en el plazo de 15 días. Los demás acontecimientos adversos graves o ines-perados se comunicarán en el plazo de 15 días.
3.Cuando el espacio disponible sea insuficiente, se añadirá una hoja de información adicional,correctamente identificada con el nombre del promotor y el número asignado a la notificación.En esta información adicional podrá hacerse constar la evaluación de la causalidad realizadapor el técnico que informa.
INSTRUCCIONES PARA RELLENAR CORRECTAMENTE ESTA HOJA
Ø. El número de protocolo es el código alfanumérico específico asignado por el promotor. El número de notificación del promotor es el que éste utiliza para su archivo. Cuando se tratede información de seguimiento se utilizará el mismo número o bien, si se modifica, se indicaráel número de la notificación inicial. Se dejará sin rellenar el espacio “Nº de notificación” queaparece sombreado.
1.Las iniciales del paciente (o del profesional sanitario que detectó la R.A.) seguirán las siguien-tes normas:
Las dos primeras posiciones por la izquierda para las iniciales del primer y segundo nombre ylas dos últimas para las del primer y segundo apellido.
Poner un Ø en la primera posición si sólo se tiene un nombre, que se situará en la segundaposición. En caso de apellidos compuestos utilizar sólo las iniciales del primer componente delapellido compuesto. Ejemplo:
JLPG . . . . . . .José Luis Pérez GonzálezØJPG . . . . . .José Pérez GonzálezJLPR . . . . . . .José Luis Pérez-González y RodríguezØJPR . . . . . .José Pérez-González y Rodríguez-Gómez
Al codificar apellidos, se prescindirá de preposiciones y artículos.
2. La edad se pondrá en años, meses, semanas o días según convenga.
7.Se describirá el acontecimiento adverso en forma completa, indicando la fecha de finalizacióndel mismo e incluyendo los resultados de las exploraciones complementarias o pruebas de la-boratorio que se consideren de interés. A esta notificación podrán acompañarse cuantos infor-mes se estimen convenientes para la adecuada interpretación del cuadro clínico.
14.Los productos en fase de investigación se identificarán a ser posible por su nombre genérico(CDI) o, en su defecto, por el código de investigación del producto. Se considera producto deinvestigación tanto el producto específicamente investigado como el control.
15.En caso de que la administración no sea diaria se intentará describirla con alguna de las si-guientes posibilidades: cíclica, semanal, mensual, anual o número de veces que se ha utiliza-do (poniendo en este caso la dosis de cada toma, no la total)
17.Se hará constar el proceso patológico del paciente al que va destinado el producto en investi-gación o bien “voluntario sano” en caso de tratarse de tal.
19.Se hará constar la duración del tratamiento hasta el inicio del acontecimiento adverso.
120
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

Circular N.º 12/93
DEPENDENCIA: Dirección General de Farmacia y Productos Sanitarios
CONTENIDO: Intervención del Ministerio Fiscal cuando los sujetos de un ensayo clínico sean menores de edad o incapaces.
AMBITO DE AMPLIACION: Industria Farmacéutica. Consejo General de Colegios de Médicos. Comités Éticos de Investigación Clínica. Promotores de Ensayos Clínicos en general. Comunidades Autónomas.
El Real Decreto 561/1993, de 16 de abril, por el que se establecen los requisitos para la realiza-ción de ensayos clínicos con medicamentos, publicado en el Boletín Oficial del Estado el 13 de ma-yo de 1993, en el artículo 12 punto 5, dispone:
“En los casos de sujetos menores de edad e incapaces, el consentimiento lo otorgará siempre porescrito su representante legal (anexo 6 apartado 4), tras haber recibido y comprendido la informa-ción mencionada. Cuando las condiciones del sujeto lo permitan y, en todo caso, cuando el menortenga 12 o más años, deberá prestar además su consentimiento (anexo 6, apartado 2) para partici-par en el ensayo, después de haberle dado toda la información pertinente adaptada a su nivel deentendimiento. El consentimiento del representante legal y del menor, en su caso, será puesto enconocimiento del Ministerio Fiscal previamente a la realización del ensayo”.
Según se establece en el artículo 16.4C de dicho Real Decreto, debe ser el investigador quien ob-tenga el consentimiento informado del representante legal y, si procede, del menor o incapaz, antesde su inclusión en el estudio. Por tanto, será él quien firme, en la misma fecha en que sea otorgado,el documento de notificación al Ministerio Fiscal (ANEXO I).
Por su parte, el promotor del ensayo, que como se establece en el artículo 14.1 del mismo RealDecreto “es la persona física o jurídica que tiene interés en su realización, firma las solicitudes deautorización dirigidas al Comité Ético de Investigación Clínica o a la Dirección General de Farmacia yProductos Sanitarios, y se responsabiliza de él, incluyendo su organización, comienzo y financia-ción”, debe ser el encargado de trasladar al Ministerio Fiscal el consentimiento al que se refiere elartículo 12.5., asegurando en todo caso la confidencialidad de la identidad del sujeto.
Para ello, deberá enviar el modelo de comunicación que se adjunta como ANEXO I de esta Circu-lar, dirigido al Fiscal Jefe de la Audiencia Provincial donde tenga su domicilio el menor o incapacita-do (ANEXO II), acompañando una copia del consentimiento otorgado por el representante legal, y siprocede, por una copia del consentimiento otorgado por el menor o por el incapaz.
Madrid, a 28 de Julio de 1992La Directora General de Farmacia y Productos Sanitarios.Fdo: Regina Revilla.
121

ANEXO I
En cumplimiento de lo establecido en el artículo 12, apartado 5 del Real Decreto 561/1993, de 16de Abril (B.O.E.. 13.5.93), por el que se establecen los requisitos para la realización de ensayos clí-nicos con medicamentos:
“En los casos de sujetos menores de edad e incapaces, el consentimiento lo otorgará siempre porescrito su representante legal (anexo 6 apartado 4), tras haber recibido y comprendido la informa-ción mencionada. Cuando las condiciones del sujeto lo permitan, y en todo caso, cuando el menortenga 12 o más años, deberá prestar además su consentimiento (anexo 6, apartado 2) para partici-par en el ensayo, después de haberle dado la información pertinente adaptada a su nivel de enten-dimiento. El consentimiento del representante legal y del menor, en su caso, será puesto en conoci-miento del Ministerio Fiscal previamente a la realización del ensayo”.
Le informamos que en (Hospital o Centro) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .se está realizando el ensayo clínico Nº de protocolo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .autorizado por el Ministerio de Sanidad y Consumo, en el cual se ha incluido a D. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .menor de edad o incapaz, y remitidos a ese Ministerio Fiscal copia de los siguientes documentos:
Consentimiento otorgado por el representante legal del menor o incapaz (anexo 6 apartado 4 delReal Decreto 61/1993)
Consentimiento otorgado por el menor de edad cuando tenga 12 o más años (anexo 6 apartado 2)
Consentimiento otorgado por el incapaz o por el menor de 12 años en caso de que su capacidadde entendimiento lo permita (anexo 6 apartado 2)
Fecha . . . . . . . . . . . . . . Firma del Investigador
Ilmo Sr. Fiscal-Jefe de la Audiencia Provincialde . . . . . . . . . . . . . . . . .
X
122
GLOSARIO DE INVESTIGACIÓN CLÍNICA Y EPIDEMIOLÓGICA

ANEXO II
DIRECCIONES DE LAS AUDIENCIAS PROVINCIALES
La documentación se remitirá a:Ilmo. Sr. Fiscal JefeAudiencia Provincial de . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .(la provincia que corresponda).
En las siguientes provincias se añadirá además la dirección concreta como sigue:
C/Ferraz 4128008 MADRID
Pau Clarís 158-16008071 Barcelona
Pza. de Alfonso el Magnánimo, glorietaPalacio de Justicia46071 Valencia
Prado de San Sebastián, s/n41071 Sevilla
Coso, 150071 Zaragoza
123
CIRCULAR N.º 12/93

Circular N.º 18/96
DEPENDENCIA: Dirección General de Farmacia y Productos Sanitarios.
CONTENIDO: Procedimientos de tramitación de solicitudes de autorización de ensayos clínicos(R.D. 561/1993).
DESTINATARIO: Industria Farmacéutica.
El artículo 24.1 del R.D. 561/1993, exige para la realización de ensayos clínicos en el territorio na-cional, la autorización de la Dirección General de Farmacia y Productos Sanitarios previo informe delcorrespondiente Comité Ético de Investigación Clínica (CEIC) acreditado.
La evaluación de la documentación del ensayo que prevé el Real Decreto no puede iniciarse has-ta no contar con el informe preceptivo del CEIC y la conformidad de la Dirección del Centro en quese realizará el ensayo lo que supone con cierta frecuencia una demora en el procedimiento de tra-mitación.
Por ello con el fin de agilizar los procedimientos de autorización de ensayos clínicos se hace nece-sario mejorar el sistema actual de forma que, garantizando el cumplimiento de lo establecido en elReal Decreto, se facilite la obtención de la correspondiente autorización.
En base a lo anteriormente expuesto se da la siguiente instrucción:La documentación que se presenta al CEIC podrá ser presentada al mismo tiempo en la Dirección
General de Farmacia y Productos Sanitarios con el din de facilitar el estudio y evaluación del proto-colo correspondiente por parte de este Centro Directivo. No obstante no se podrá contar con la auto-rización para la realización de los ensayos clínicos en los términos que prevé el Real Decreto561/1993, hasta no recibir la documentación completa.
Madrid, 25 de septiembre de 1996La Directora GeneralFdo. Ana Mª Naveira Naveira
124

1. El hospital de día y su repercusión en tera-péutica (1985).
2. Problemas que se plantean en el trata-miento de infecciones graves por S. au-reus (1986).
3. Contribución del biólogo a la farmacologíaen España (1987).
4. Un glosario para farmacólogos (1987).5. Aspectos biológicos de los síndromes de-
presivos (1988).6. Bases del tratamiento de las intoxicaciones
agudas (1988).7. Investigación básica y medicina clínica
(1988).8. Tratamiento de datos en farmacología
(1989).9. Perspectivas terapéuticas en la esclerosis
múltiple (1989).10. Biotecnología de aplicación farmacéutica
(1991).11. Metodología del ensayo clínico (1991).
12. Periodismo científico. Un simposio interna-cional (1991).
13. El ensayo clínico como tarea cooperativa(1992).
14. Terapéutica y calidad de vida (1993).15. Investigación sobre cáncer en España: de
la biología molecular a la clínica (1994).16. El tratamiento del dolor: del laboratorio a la
clínica (1994).17. Farmacología de los canales iónicos
(1995).18. Bases de datos en farmacología y terapéu-
tica (1996).19. Fármacos y conducción de vehículos (1996).20. Traducción y lenguaje en medicina
(1997).21. Medicina y medios de comunicación. Tra-
ducción al español de una serie publicadaen la revista The Lancet (1997).
22. Problemas y controversias en torno al en-sayo clínico (1998).
Monografías Dr. Antonio Esteve publicadas
125


















