
“Modelos celulares para la confirmación de la ... · PDF fileensayos de...
43
“Modelos celulares para la confirmación de la patogenicidad de nuevas mutaciones en el mtDNA y su aplicación al estudio de posibles compuestos terapéuticos. Terapia génica para las enfermedades mitocondriales”. Julio Montoya Departamento de Bioquímica, Biología Molecular y Celular CIBER de Enfermedades Raras (CIBERER) Universidad de Zaragoza
Transcript of “Modelos celulares para la confirmación de la ... · PDF fileensayos de...

“Modelos celulares para la confirmación de la patogenicidad de nuevas mutaciones en el mtDNA y
su aplicación al estudio de posibles compuestos terapéuticos. Terapia génica para las enfermedades
mitocondriales”.
Julio Montoya Departamento de Bioquímica, Biología Molecular y Celular
CIBER de Enfermedades Raras (CIBERER)
Universidad de Zaragoza


Enfermedades Genéticas Mitocondriales
Grupo de trastornos originados por defectos en el sistema de fosforilación oxidativa que
conlleva una deficiencia en la producción de ATP.
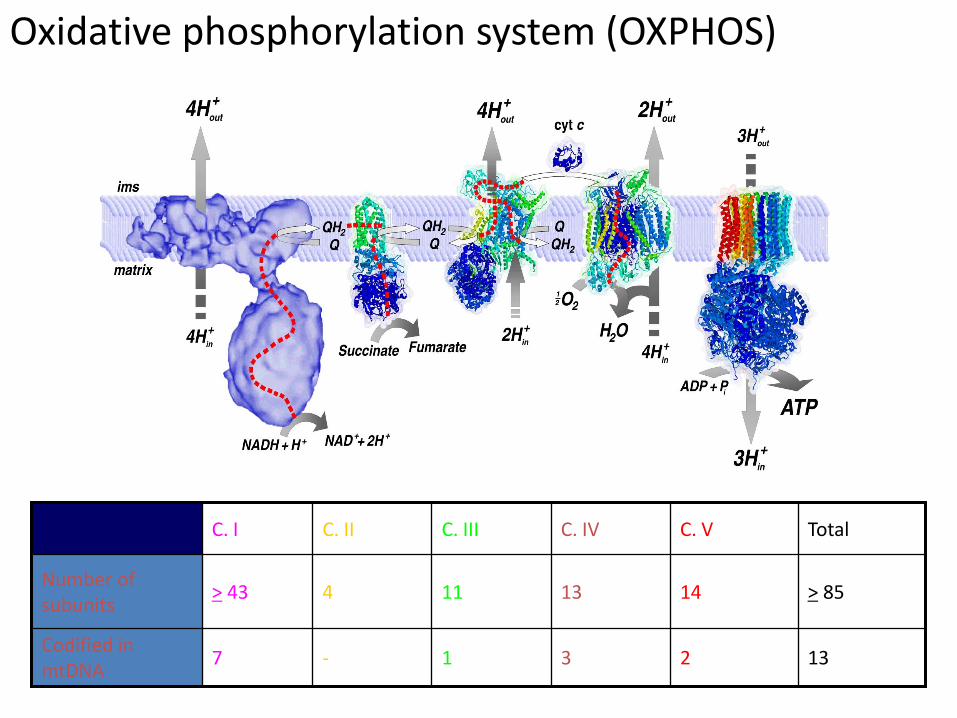
Oxidative phosphorylation system (OXPHOS)
2
14
C. V
13 3 1 - 7 Codified in mtDNA
> 85 13 11 4 > 43 Number of subunits
Total C. IV C. III C. II C. I

Herencia de las enfermedades genéticas
mitocondriales
•Mendeliana.-
Para genes mitocondriales codificados en el DNA nuclear
•Materna Para genes mitocondriales codificados en el DNA mitocondrial

Mapa genético del
mtDNA humano
16.569 pb
1.000-10.000 copias/cel.
37 genes
Información compacta

Genética Mitocondrial
• Herencia Materna
• Segregación mitótica
• Efecto Umbral
• Alta Velocidad de Mutación
•Carencia de recombinación

Mutaciones nuevas:
Secuenciación del mtDNA completo

MUTACIONES NUEVAS EN EL mtDNA
•Se conocen más de 150 mutaciones puntuales en el mtDNA asociadas a
enfermedades.
•A pesar de ello, se siguen encontrando mutaciones nuevas
•Para poder declararlas como mutaciones patológicas, éstas deben de
cumplir una serie de criterios

Criterios de Patogenicidad
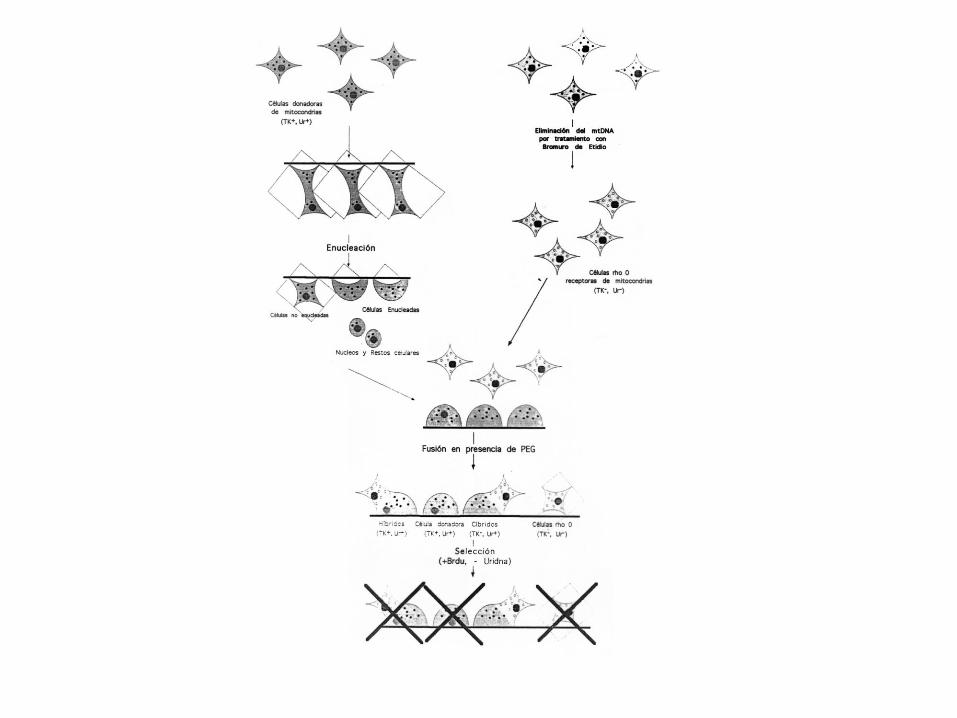
5.1.- La mutación debe de estar presente en pacientes y ausente en individuos control. 5.2.- La mutación debe de encontrarse en fondos genéticos diferentes. 5.3.- Existencia de correlación entre el porcentaje de la mutación y el fenotipo. 5.4.- La mutación debe de ser la mejor candidata a ser patogénica. 5.5.- La mutación debe afectar a nucleótidos muy conservados evolutivamente. 5.6.- La mutación debe afectar a dominios funcionales importantes de las proteínas. 5.7.- La transferencia de un mtDNA con una mutación a líneas celulares sin mtDNA, debe de ir acompañada de una transferencia del defecto molecular y/o celular: Construcción de cibridos transmitocondriales.

A New Pathologic Mitochondrial DNA Mutation in
the Cytochrome Oxidase Subunit I (MT-CO1)
María D. Herrero-Martín, Mercedes Pineda, Paz Briones, Ester López-Gallardo,
Magdalena Carreras, Mercedes Benac, Miguel Angel Idoate, María A. Vilaseca,
Rafael Artuch, Manuel J. López-Pérez, Eduardo Ruiz-Pesini, and Julio Montoya
HUMAN MUTATION 29:E103-E111, 2008

mtDNA point mutations
More than 100 mtDNA point mutations in have been described associated to different
phenotypes
Only a few of then have been frequentlyly associated to specific phenotypes (3243-MELAS; 8344-MERRF; 8993-Leigh and NARP; 3460, 11778, 14484-LHON, etc)
16-18% of the have a mutation in their mtDNA
New mutations increases this number to 20%. The others remain without a clear
molecular diagnostic.
Do this patients have nuclear mutations in genes that codify for components of the
OXPHOS system?
Do they suffer a real mitochondrial disease?
Are we missing patients, mainly children, with a very mild phenotype?

13 years old girl
Personal clinical history
Non-consanguineous parents: high blood pressure and
hypercholesterolemia
Normal pregnancy
Normal development during first years
Retarded speech development and moderate mental retardation (5 yo)
Loss of appetite
Recurrent diarrhoea and abdominal pain
Hypercholesterolemia
High transaminase levels
Retarded weight and height
Weakness
Slower at running than the children of the same age
Moderate mental retardation: special program of integration.

Examination at 13 yo
Normocephalia
Low weight
Growth retardation
Pes cavus
Moderate mental retardation
Anorexia
Generalized muscle atrophy
Weakness with fatigability to climb stairs
EMG: myopathic pattern

Mitochondrial disease?
Myopathic pattern
Pyruvate
Laboratory
parameters Haemogram
Organic
acids
Aminoacids
ECG
Hepatic
ultrasound
scan
Plasmatic
carnitine
EMG RMI-S
Audiometric
test
Hyperintensity
caudate nucleus
Cortex atrophy Lactate peak in
fronto-temporal-
occipital regions
CPK LDH
CSF
Blood
Lactate
Lactate
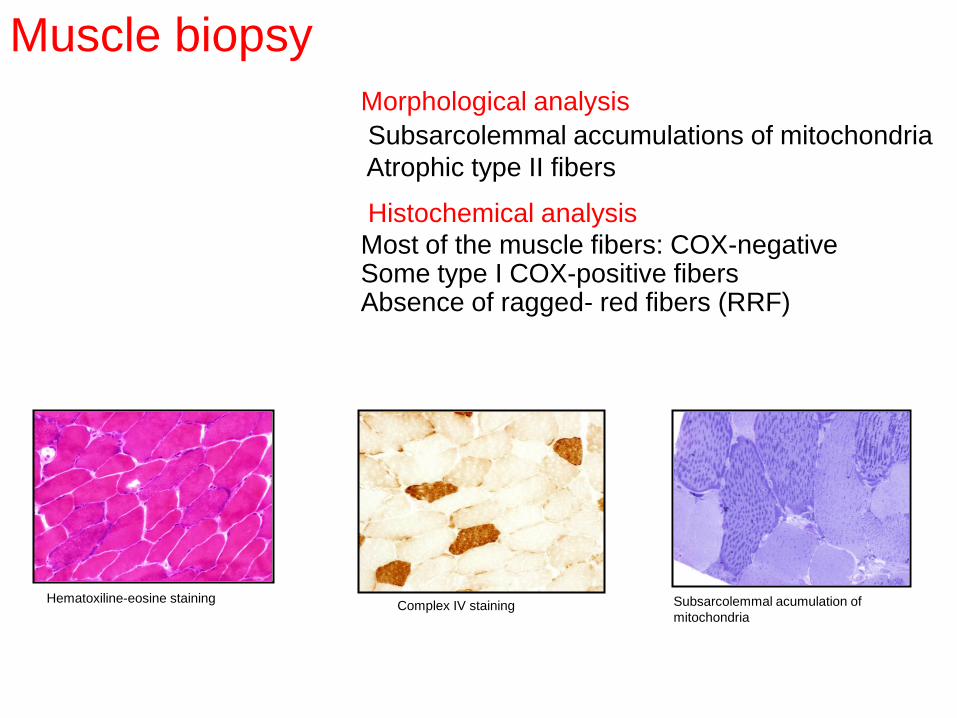
Hematoxiline-eosine staining Complex IV staining Subsarcolemmal acumulation of
mitochondria
Muscle biopsy
Morphological analysis
Subsarcolemmal accumulations of mitochondria
Atrophic type II fibers
Histochemical analysis
Most of the muscle fibers: COX-negative Some type I COX-positive fibers Absence of ragged- red fibers (RRF)

Specific activity
(nmol/min/mg prot)
mU/UCS
Patient Control
range
Patient Control
range
NADH:cyt c oxidoreductase
(Complex I+III)
12 (m) 12-56 (m) 41 (38.3%) (m) 107-560 (m)
Succinate:cyt c oxidoreductase
(Complex II+III)
6 (85.7%) (m)
6.5 (f)
7-24 (m)
2.4-17 (f)
21 (28.0%) (m)
0.24 (f)
75-149 (m)
0.14-1.0 (f)
Succinate:DCPIP oxidoreductase
(Complex II)
9 (m) 4-10 (m) 31 (93.9%) (m) 33-69 (m)
SDH (+PMS) 10 (m) 10-25 (m) 34 (59.6%) (m) 57-239 (m)
Decilubiquinol:cyt c oxidoreductase
(Complex III)
81 (m) 55-259 (m) 279 (45.7%) (m) 610-1760 (m)
Cytochrome oxidase
(Complex IV)
26 (44.1%) (m)
33 (f)
59-170 (m)
16-48 (f)
90 (15.3%) (m)
1.22 (84.1%) (f)
590-1300 (m)
1.45-3.13 (f)
Citrate synthase (CS) 290 (m)
27 (f)
71-200 (m)
11-29 (f)
Isolated C-IV deficiency Multienzymatic deficiency
Respiratory chain activity

Clinical symptoms: ETC disease
ETC:
-ETC: multienzymatic deficiency:
- mtDNA depletion, deletion
- Mitochondrial tRNA mutations
-CIV deficiency:
- Mutations of CIV genes encoded in mtDNA
- Mutations of CIV structural or assembly genes encoded in
nDNA
Muscle biopsy: No RRF and COX negative fibers:
- Mutations of CIV assembly genes encoded in nDNA
Where to check for mutations?
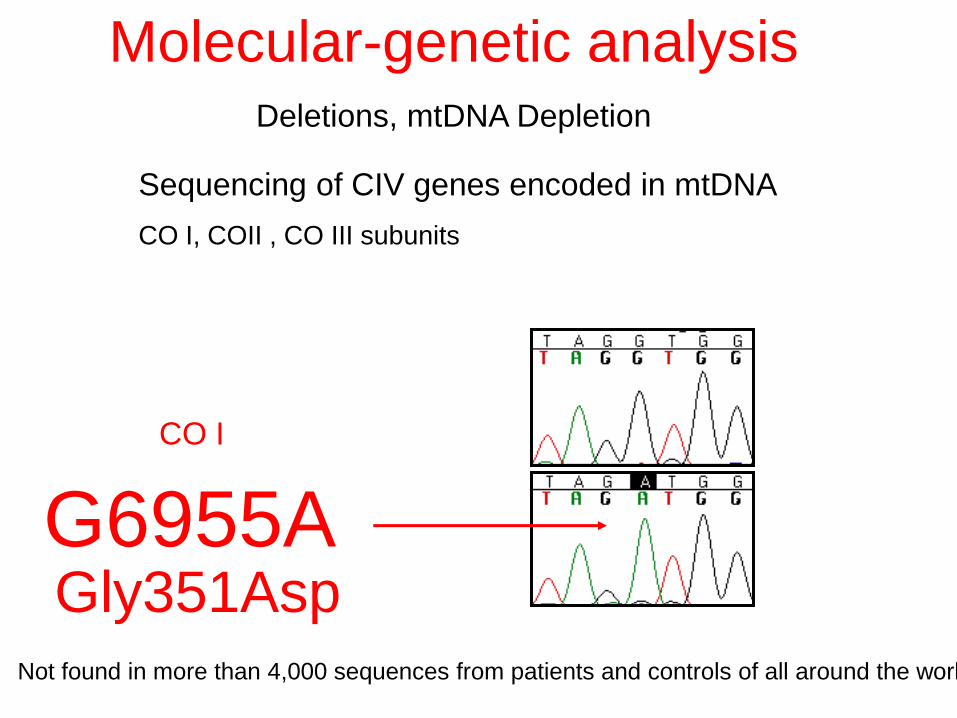
Sequencing of CIV genes encoded in mtDNA
CO I, COII , CO III subunits
G6955A
Molecular-genetic analysis Deletions, mtDNA Depletion
Gly351Asp Not found in more than 4,000 sequences from patients and controls of all around the world
CO I

Whole mtDNA sequence
Haplogroup H11
MT-GENE GENETIC VARIANTS
MT-RNR m.750A>G (MT-RNR1), m.961T>G (MT-RNR1), m.1438A>G (MT-RNR1)
MT-T rCRS
MT-ND m.4769A>G (S-p.MT-ND2), m.13759G>A (NS-p.MT-ND5:Ala475Thr)
MT-CYB m.15326A>G (NS-p.MT-CYB:Thr194Ala)
MT-CO m.6955G>A (NS-p.MT-CO1:Gly351Asp)
MT-ATP m.8448T>C (NS-p.MT-ATP8:Met28Thr), m.8860A>G (NS-p.MT-
ATP6:Thr112Ala)
MT-DLOOP m.195T>C (MT-DLOOP), m.263A>G (MT-DLOOP), m.309Ci (MT-DLOOP),
m.315Ci (MT-DLOOP), m.16265A>C (MT-DLOOP), m.16293A>G (MT-
DLOOP), m.16311T>C (MT-DLOOP)
Molecular-genetic analysis
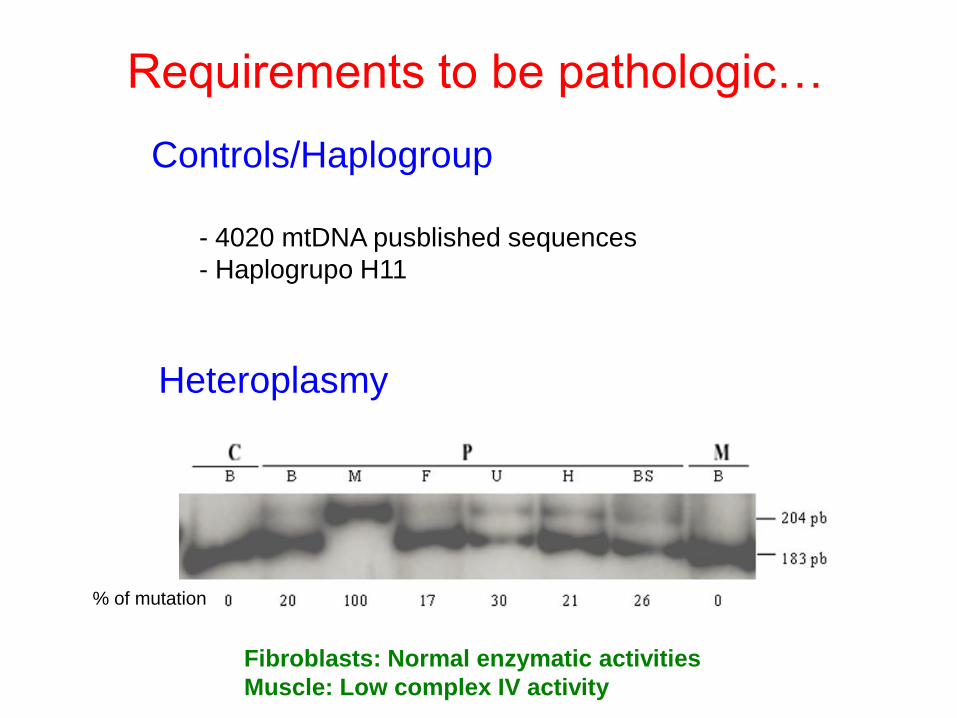
Heteroplasmy
Controls/Haplogroup
- 4020 mtDNA pusblished sequences
- Haplogrupo H11
Requirements to be pathologic…
% of mutation
Fibroblasts: Normal enzymatic activities
Muscle: Low complex IV activity
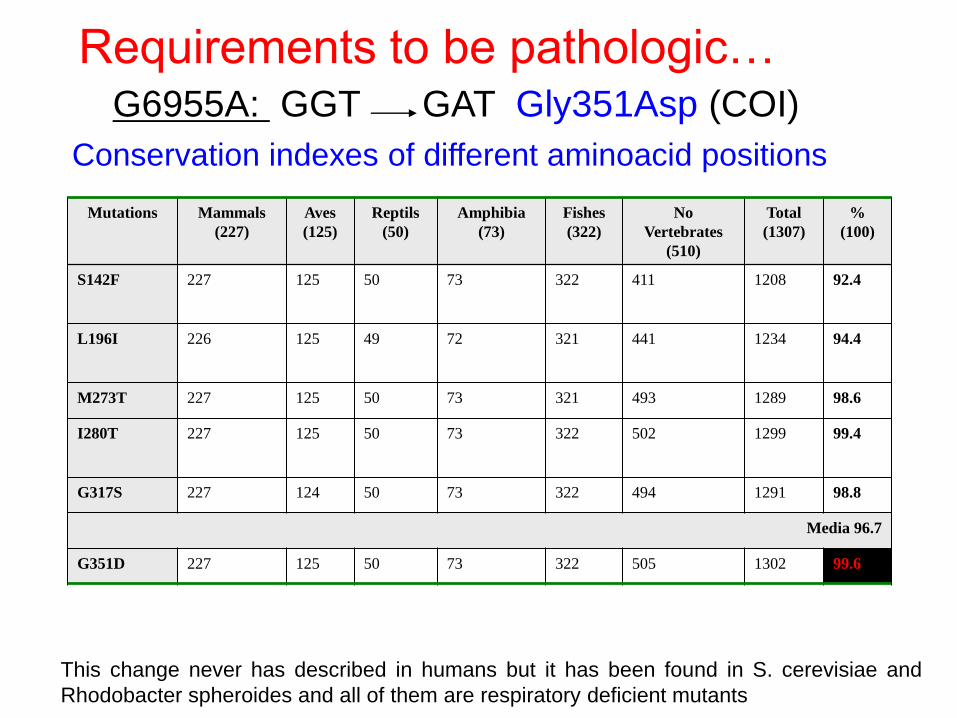
Conservation indexes of different aminoacid positions
Mutations Mammals
(227)
Aves
(125)
Reptils
(50)
Amphibia
(73)
Fishes
(322)
No
Vertebrates
(510)
Total
(1307)
%
(100)
S142F 227 125 50 73 322 411 1208 92.4
L196I
226 125 49 72 321 441 1234 94.4
M273T 227 125 50 73 321 493 1289 98.6
I280T
227 125 50 73 322 502 1299 99.4
G317S 227 124 50 73 322 494 1291 98.8
Media 96.7
G351D 227 125 50 73 322 505 1302 99.6
Requirements to be pathologic…
This change never has described in humans but it has been found in S. cerevisiae and
Rhodobacter spheroides and all of them are respiratory deficient mutants
G6955A: GGT GAT Gly351Asp (COI)
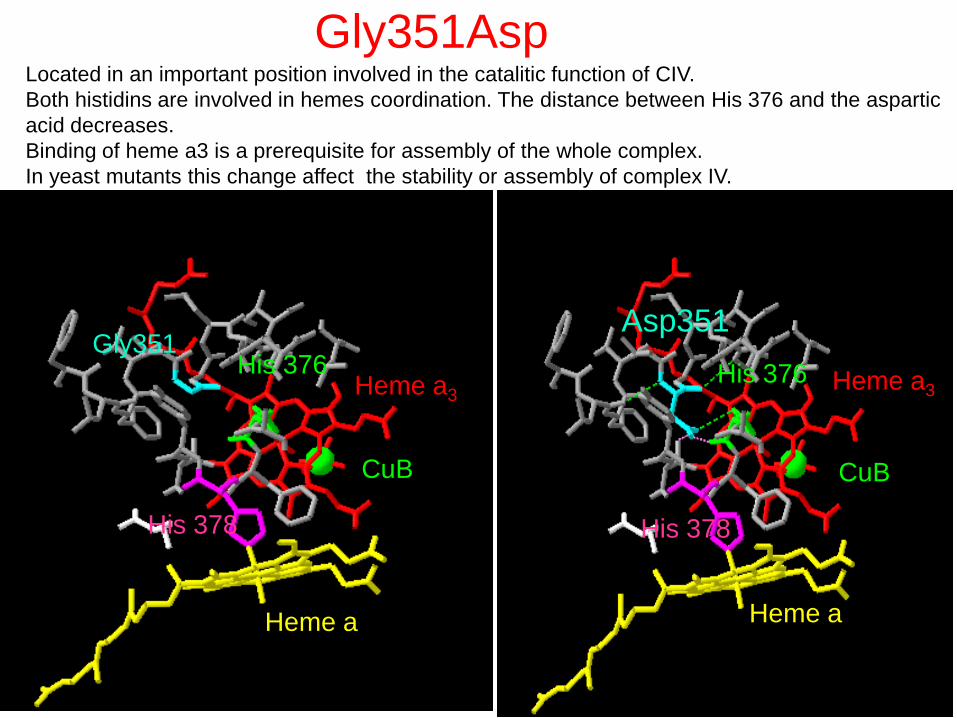
Gly351
His 378
His 376
Heme a
Heme a3
CuB
Asp351
CuB
Heme a3
His 378
Heme a
His 376
Gly351Asp Located in an important position involved in the catalitic function of CIV.
Both histidins are involved in hemes coordination. The distance between His 376 and the aspartic
acid decreases.
Binding of heme a3 is a prerequisite for assembly of the whole complex.
In yeast mutants this change affect the stability or assembly of complex IV.
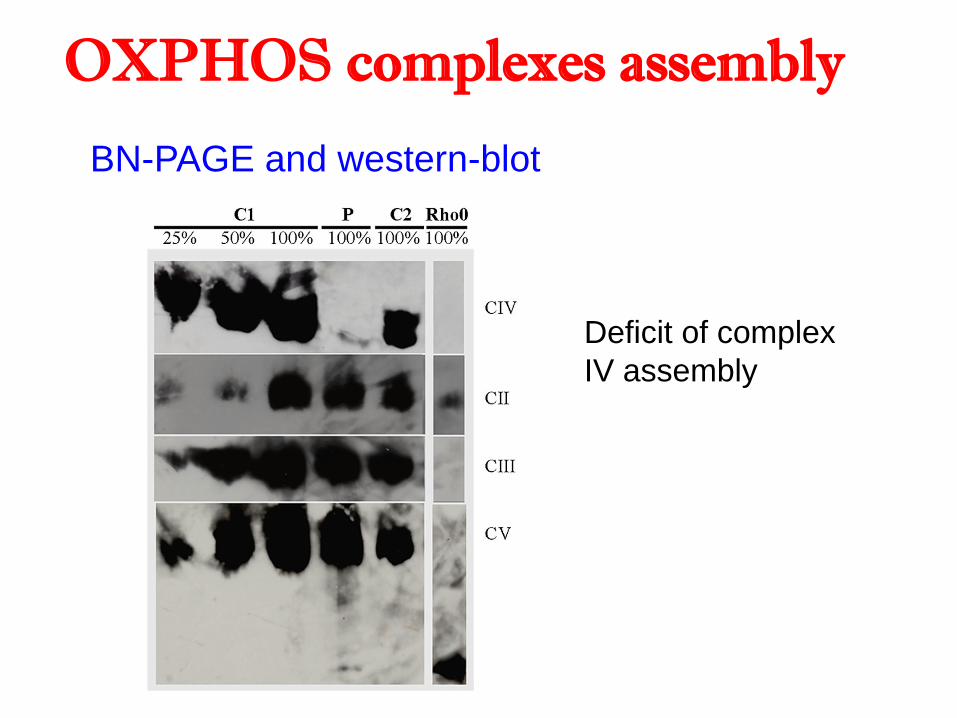
Múscul
o
BN-PAGE and western-blot
Deficit of complex
IV assembly
OXPHOS complexes assembly
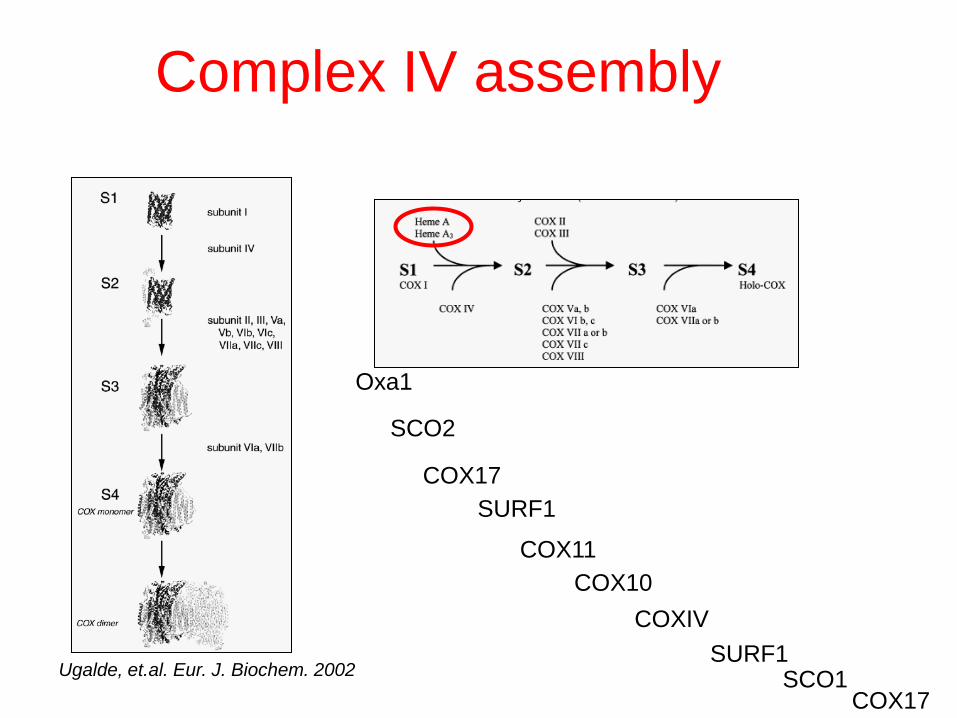
Ugalde, et.al. Eur. J. Biochem. 2002 SURF1
Oxa1
COX11
SCO1
COX17
SCO2
COX10
COXIV
COX17
SURF1
Complex IV assembly
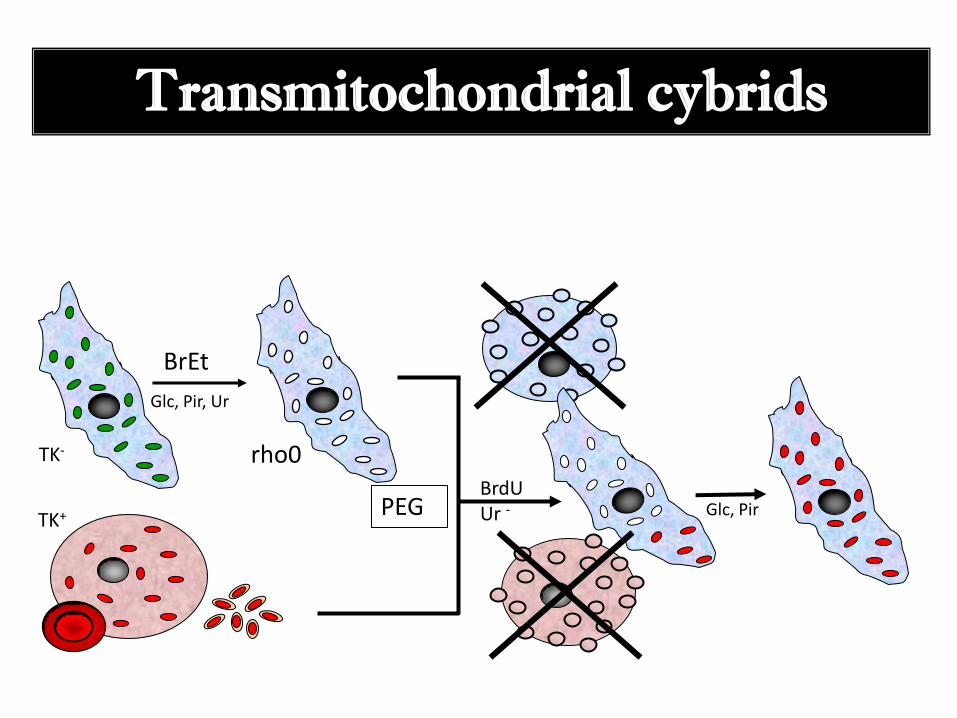
Transmitochondrial cybrids
TK-
BrEt
Glc, Pir, Ur
TK+
rho0
PEG BrdU Ur - Glc, Pir

Producción de líneas celulares cíbridas transmitocondriales
-Crecimiento de las células y tratamiento con BrEt para la eliminación del
mtDNA: Formación de células rho cero.
- Obtención de una muestra de sangre del paciente y preparación de
plaquetas (éstas tienen mtDNA pero no núcleo).
- Fusión de líneas celulares rho cero y plaquetas.
- Selección de las células que llevan las mitocondrias del paciente.
- Realización de análisis bioquímicos y genéticos.

Transmitochondrial cybrids
TK-
BrEt
Glc, Pir, Ur
TK+
rho0
PEG BrdU Ur - Glc, Pir



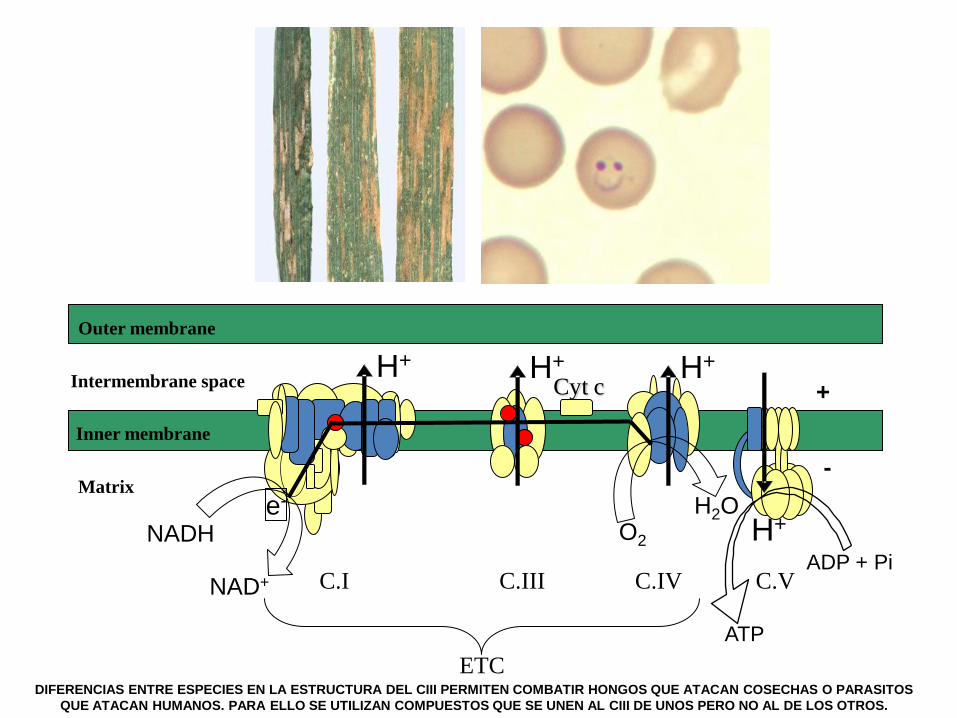
The OXPHOS system as a therapeutic target in personalized medicine
Nutrients supply electrons that reduce coenzyme Q10 of the mitochondrial
respiratory chain of the oxidative phosphorylation system. Some coenzyme Q10-
binding sites from the electron transport chain are, in part, formed by polypeptides
encoded in the mtDNA. Mitochondrial DNA is highly polymorphic and many of these
polymorphisms, that modify amino acids of the coenzyme Q10-binding sites, define
mitochondrial DNA haplogroups. This fact suggests that alternative coenzyme Q10-
binding sites from diverse mitochondrial DNA haplogroups might differentially
interact with coenzyme Q10 or its analogs and modify the oxidative phosphorylation
phenotype. In this scenario, this molecular pathway could become a therapeutic
target for custom-made treatments.
Methods. We synthesized, modeled and tested several coenzyme Q10 analogs in a
collection of transmitochondrial cell lines harboring different mitochondrial DNA
haplogroups.
Results. These compounds bind the p.MT-CYB Qi-site and modify several
OXPHOS parameters without decreasing cell viability. The effect of one of these
compounds depends on the mtDNA genetic background.
Conclusions. Our results confirm that mtDNA polymorphisms can modify the
interaction with these compounds and alter bioenergetic parameters. Therefore,
personalized treatment using the OXPHOS system as a therapeutic target is
possible.
Cyt c
Matrix
Intermembrane space
Outer membrane
+
-
C.I C.III C.IV C.V
ETC
Inner membrane
e- H2O
ATP
NADH
H+
O2
NAD+
H+ H+
H+
ADP + Pi
DIFERENCIAS ENTRE ESPECIES EN LA ESTRUCTURA DEL CIII PERMITEN COMBATIR HONGOS QUE ATACAN COSECHAS O PARASITOS
QUE ATACAN HUMANOS. PARA ELLO SE UTILIZAN COMPUESTOS QUE SE UNEN AL CIII DE UNOS PERO NO AL DE LOS OTROS.
182
N
C
a
F1
F2
H1
H2
ab
cd1
cd2
ef
E A B C D G
239 294
120
15
..
.
..
.
.
.
. .
..
.H16R
F18L
A229T
250 G251S
G251D
S35P
G34S
A122T
S151P
G166E T158A
190
A193T
D171N
Y278C
G290D
L236I
40
.G339E
.R318P
.
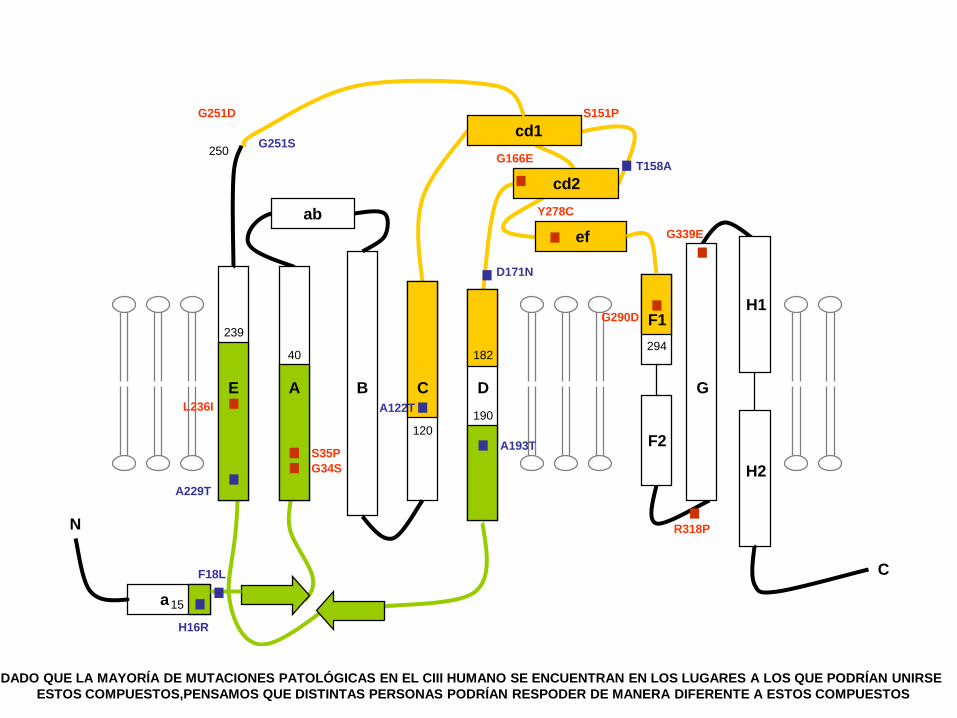
DADO QUE LA MAYORÍA DE MUTACIONES PATOLÓGICAS EN EL CIII HUMANO SE ENCUENTRAN EN LOS LUGARES A LOS QUE PODRÍAN UNIRSE
ESTOS COMPUESTOS,PENSAMOS QUE DISTINTAS PERSONAS PODRÍAN RESPODER DE MANERA DIFERENTE A ESTOS COMPUESTOS

EL DOCTOR ALVAREZ BUILLA DE LA UNIVERSIDAD DE ALCALA DE HENARES
SINTETIZÓ UNOS COMPUESTOS SIMILARES AL SUSTRATO NORMAL DEL CIII

1 nM – 1 mM
IC50
μM
[Q]plasmática = 0.5 μM
PROBAMOS DIFERENTES CONCENTRACIONES PARA VER QUE TENÍAN EFECTO SOBRE LAS CÉLULAS (AFECTABAN LA
SUPERVIVENCIA) Y LIMITAMOS LA CONCENTRACIÓN A AQUELLAS QUE DEJABAN VIVAS LA MAYORÍA DE LAS CÉLULAS
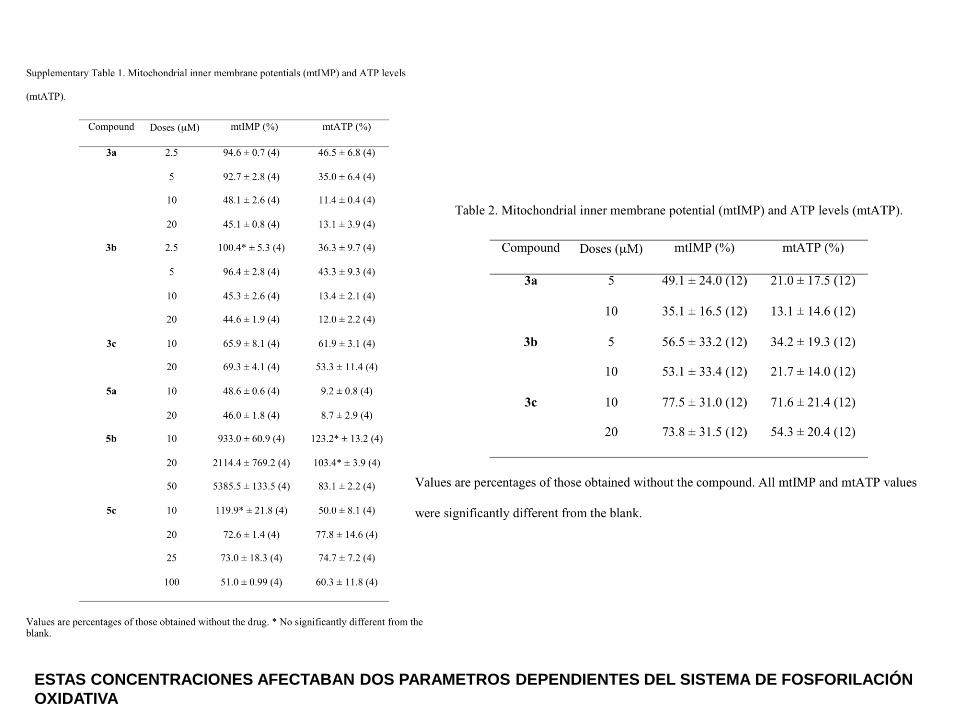
ESTAS CONCENTRACIONES AFECTABAN DOS PARAMETROS DEPENDIENTES DEL SISTEMA DE FOSFORILACIÓN
OXIDATIVA
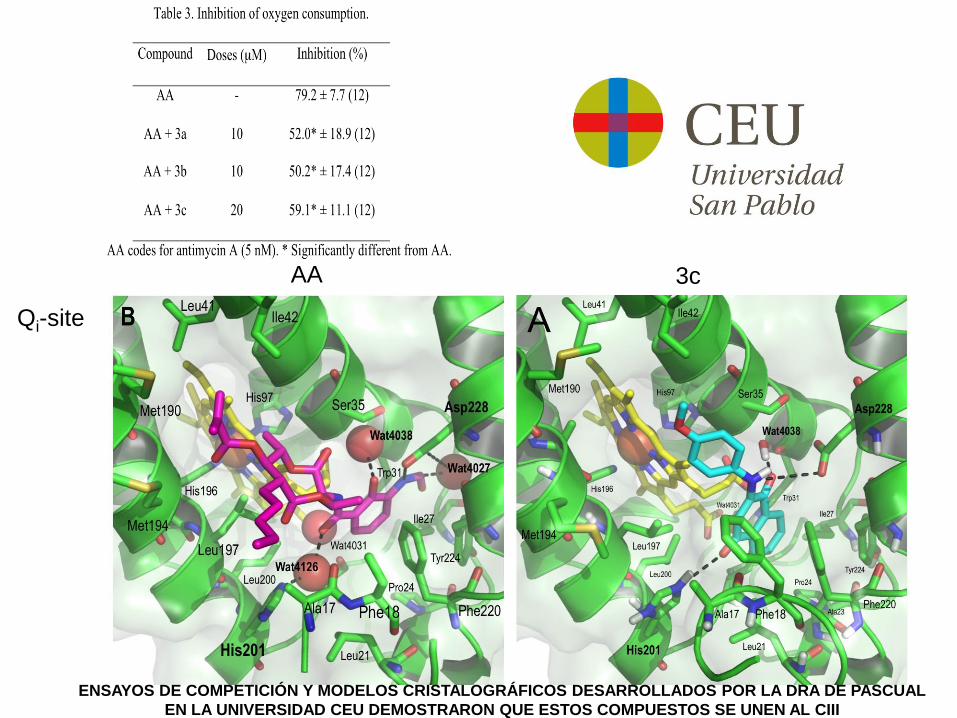
Qi-site
AA 3c
ENSAYOS DE COMPETICIÓN Y MODELOS CRISTALOGRÁFICOS DESARROLLADOS POR LA DRA DE PASCUAL
EN LA UNIVERSIDAD CEU DEMOSTRARON QUE ESTOS COMPUESTOS SE UNEN AL CIII

TK-
BrEt
Glc, Pir, Ur
TK+
rho0
PEG BrdU Ur -
Glc, Pir
TK-
BrEt
Glc, Pir, Ur
rho0
TK+ PEG
BrdU Ur -
Glc, Pir
HgH
HgJ2
Cybrids
CONSTRUIMOS CÍBRIDOS DE DISTINTOS HAPLOGRUPO
QUE DIFIEREN EN LA ESTRUCTURA DEL LUGAR DE UNIÓN DE
ESTOS COMPUESTOS
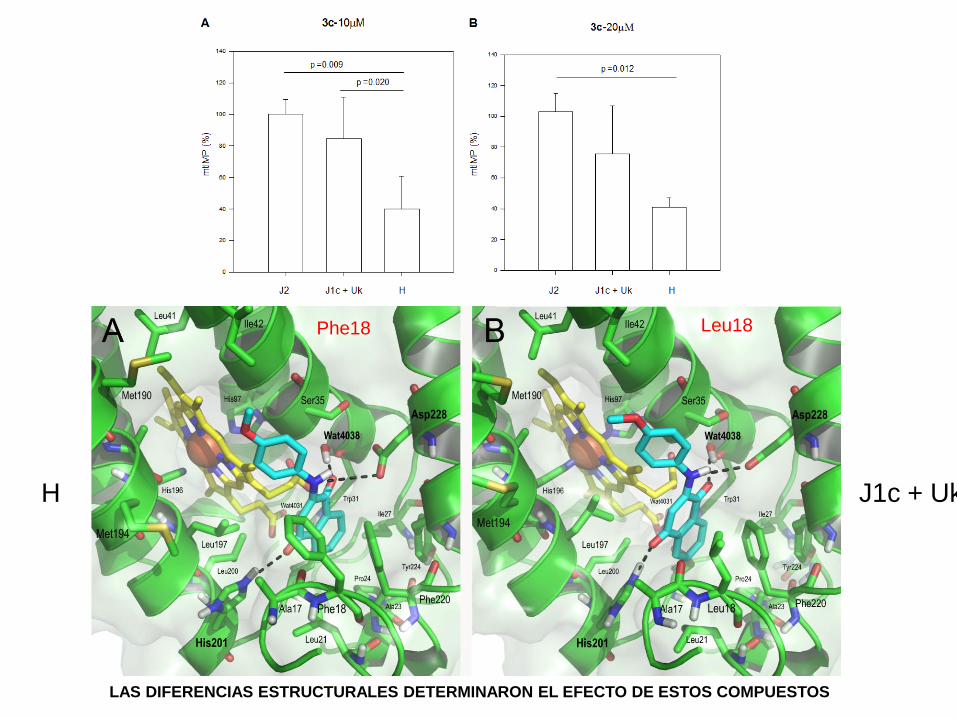
Phe18 Leu18
H J1c + Uk
LAS DIFERENCIAS ESTRUCTURALES DETERMINARON EL EFECTO DE ESTOS COMPUESTOS
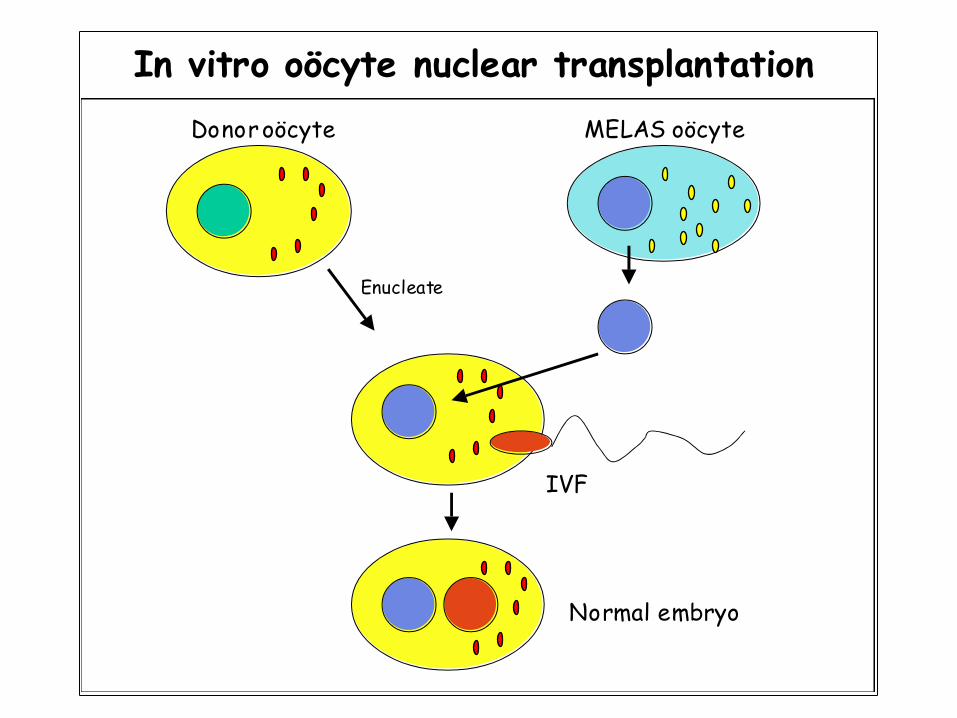
In vitro oöcyte nuclear transplantation
Enucleate
Donor oöcyte
IVF
Normal embryo
MELAS oöcyte


Ester López Gallardo
Julio Montoya Eduardo Ruiz-Pesini
David Pacheu
Loli Herrero
Manuel J. López-Pérez
Sonia
Emperador
Sonia Emperador


















