
Método de Hartree-Fockdepa.fquim.unam.mx/amyd/archivero/trabajo5maestria_35287.pdf ·...
21
1 Método de Hartree-Fock Integrantes Moreno Narváez María Esther Orta Sotelo Cynthia Padilla Mayne Sofía Ruiz Alemán Cecilia
Transcript of Método de Hartree-Fockdepa.fquim.unam.mx/amyd/archivero/trabajo5maestria_35287.pdf ·...

1
Método de Hartree-Fock
Integrantes
Moreno Narváez María Esther
Orta Sotelo Cynthia
Padilla Mayne Sofía
Ruiz Alemán Cecilia

APROXIMACIÓN DE HARTREE-FOCK
• Da una mejor aproximación en cuanto a la función de onda que se obtiene a partir de un determinante de Slater.
• En ves de utilizar funciones de onda hidrogenoides, se utilizan funciones variacionales de prueba, por ejemplo 1 ϕ 2 ϕ , por una función antisimétrica de espín 2 Τ1 2 α 1 β 2 − β(1)α(2) . La función φ debe disminuir la integral variacional, además puede tener cualquier forma.
Las funciones variacionales son productos antisimetrizados de espines orbitales.
2

3
Douglas Hartree
Físico inglés
Vladimir Fock
Físico soviético
Entre los años 1927 y 1930 desarrollaron un procedimiento con el
que se encontraba a las mejoras posibles para orbitales atómicos.

Función de onda de Hartree-Fock:Función de onda variacional producto antisimetrizado de orbitales óptimos.
• Para cada estado de un sistema hay una única función de onda de Hartree-Fock.
• Las funciones φ satisfacen la siguiente ecuación:
𝐹 ϕ𝑖= ε𝑖ϕ𝑖
Donde:
𝐹 = operador Hartree-Fock.
Φ= orbital que es función de tres coordenadas espaciales.
ε = energía del sistema.
• Los cálculos numéricos para los orbitales de Hartree-Fock y los resultados obtenidos son valores de φ endistintos puntos del espacio.
4

• Rothan presentó a los orbitales de Hartree-Fock como combinaciones lineales de un conjunto de funciones base, estas forman un conjunto completo.
• Las funciones base que se utilizan para esto deben formar parte de un conjunto completo.
𝑔1, 𝑔2, 𝑔3… 𝑓 = σ𝑘 𝑔𝑘 𝑐𝑘
• Rothan calculó los coeficientes que dan los mejores resultados.
• El conjunto completo de funciones base para los cálculos de los orbitales de Hartree-Fock es un conjunto de orbitales tipo Slater.
• El factor radical es 𝐺𝑛 r = N𝑟𝑛−1𝑒 Τζ𝑟 𝑎0
N= constante de normalización
n = número cuántico principal
ζ = parámetro variacional
5
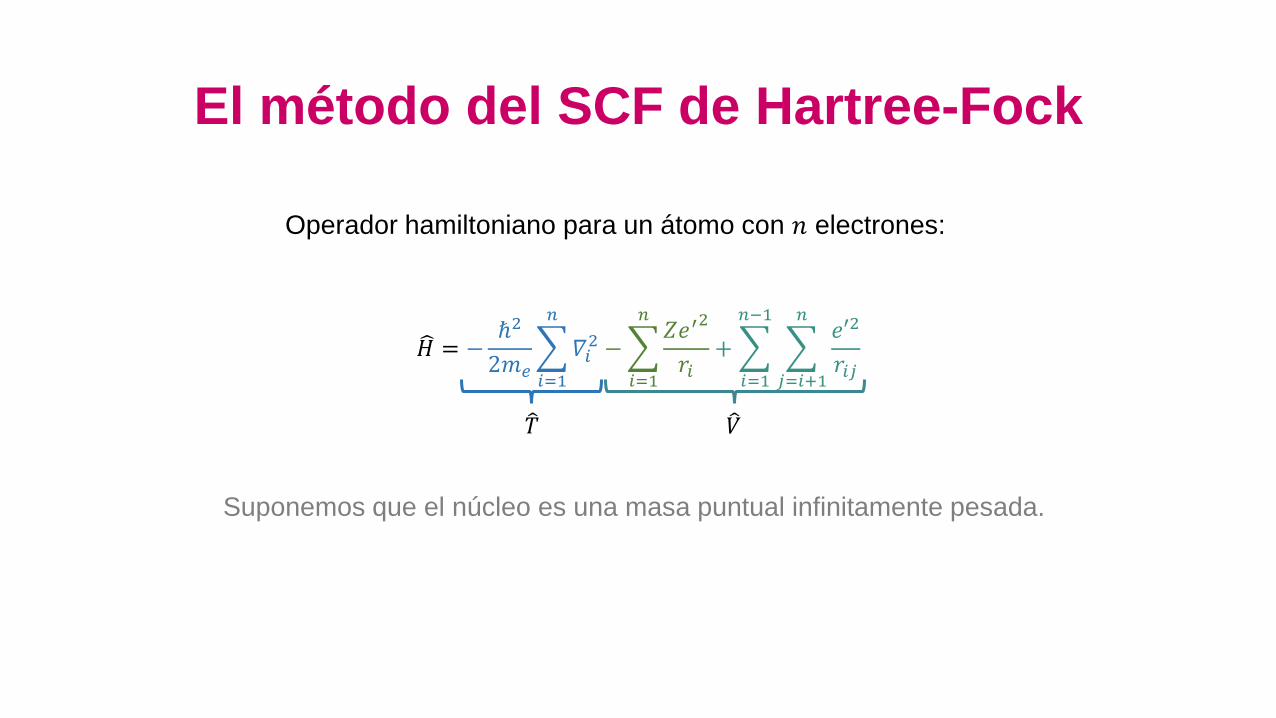
El método del SCF de Hartree-Fock
𝐻 = −ℏ2
2𝑚𝑒
𝑖=1
𝑛
𝛻𝑖2 −
𝑖=1
𝑛𝑍𝑒′
2
𝑟𝑖+
𝑖=1
𝑛−1
𝑗=𝑖+1
𝑛𝑒′2
𝑟𝑖𝑗
Operador hamiltoniano para un átomo con 𝑛 electrones:
𝑇 𝑉
Suponemos que el núcleo es una masa puntual infinitamente pesada.

La ecuación de Schrödinger polielectrónica para el átomo:
repulsión interelectrónica no separable
si despreciamos las repulsiones separable
mejor aproximación usando 𝑍∗
𝜓 0 = 𝑓1 𝑟1, 𝜃1, 𝜙1 𝑓2 𝑟2, 𝜃2, 𝜙2 …𝑓𝑛 𝑟𝑛, 𝜃𝑛, 𝜙𝑛
𝑓 = 𝑅𝑛𝑙 𝑟 𝑌𝑙𝑚 𝜃, 𝜙

❖ Usamos una función variacional que no esté restringida al orbital hidrogenoide:
y buscamos las funciones 𝑔𝑖 que ↓ la integral variacional 𝜙∗ 𝐻𝜙𝑑𝑣
𝜙∗𝜙𝑑𝑣
❖ Aproximamos los mejores OA posibles, haciendo uso de los armónicos esféricos:
𝜙 = 𝑔1 𝑟1, 𝜃1, 𝜙1 𝑔2 𝑟2, 𝜃2, 𝜙2 …𝑔𝑛 𝑟𝑛, 𝜃𝑛, 𝜙𝑛
𝑔𝑖 = ℎ𝑖 𝑟𝑖 𝑌𝑚𝑖
𝑙𝑖 𝜃𝑖 , 𝜙𝑖

cada función 𝑠𝑖 es una función normalizada de 𝑟 multiplicada por un armónico esférico
𝑉12 =𝑄′1𝑄′2𝑟12
=𝑄′1𝑄′24𝜋𝜀0𝑟12
𝜌2 = −𝑒 𝑠22
𝑄1 = −𝑒
𝜙0 = 𝑠1 𝑟1, 𝜃1, 𝜙1 𝑠2 𝑟2, 𝜃2, 𝜙2 …𝑠𝑛 𝑟𝑛, 𝜃𝑛, 𝜙𝑛
𝑉12 =𝑄14𝜋𝜀0
න𝜌2𝑟12
𝑑𝑣2 = 𝑒′2න𝑠2
2
𝑟12𝑑𝑣2
𝑉12 + 𝑉13 +⋯+ 𝑉1𝑛 =
𝑗=2
𝑛
𝑒′2න𝑠𝑗
2
𝑟1𝑗𝑑𝑣𝑗
𝑉1 𝑟1𝜃1, 𝜙1 =
𝑗=2
𝑛
𝑒′2න𝑠𝑗
2
𝑟1𝑗𝑑𝑣𝑗 −
𝑍𝑒′2
𝑟1
Electrón 1: los electrones 2, 3, …, 𝑛 como distribución
estática de carga eléctrica:
𝑄2 → distribución continua de carga → 𝜌2la carga infinitesimal en 𝑑𝑣2 es 𝜌2𝑑𝑣2
y la interaccion con 𝑄1 es:
sumando las interacciones

Aproximación del campo central:
promediamos sobre los ángulos 𝑉1 𝑟1𝜃1, 𝜙1obtenemos 𝑉1 𝑟1
Ecuación de Schrödinger monoelectrónica
…proceso iterativo…
El conjunto final de orbitales proporciona la función de onda del campo
autoconsistente de Hartree
−ℏ2
2𝑚𝑒𝛻12 + 𝑉1 𝑟1 𝑡1 1 = 𝜀1𝑡1 1

Para obtener la energía del átomo:
incorrecto
correcto
El conjunto de orbitales 𝑛, constituye una capa
Las capas 𝑛 = 1, 2, 3, … son las capas 𝐾, 𝐿,𝑀,…, respectivamente.
𝐸 =
𝑖=1
𝑛
𝜀𝑖 −
𝑖=1
𝑛−1
𝐽=𝑖+1
𝑛
ඵ𝑒′2 𝑔𝑖 𝑖
2 𝑔𝑗 𝑗2
𝑟𝑖𝑗𝑑𝑣𝑖𝑑𝑣𝑗 =
𝑖=1
𝑛
𝜀𝑖 −
𝑖
𝑗>𝑖
𝐽𝑖𝑗
𝜀1 + 𝜀2 +⋯+ 𝜀𝑛

Fock y Slater, en 1930; cálculo SCF que utiliza espín-orbitales antisimetrizados
(cálculo de Hartree-Fock):
La energía total del átomo contiene integrales de intercambio 𝐾𝑖𝑗
En 1951, Roothaan; orbitales de Hartree-Fock como combinaciones lineales de un
conjunto completo de funciones conocidas (funciones de base):
conjunto de funciones de base - orbitales de tipo Slater (STO):
𝐹𝑢𝑖 = 𝜀𝑖𝑢𝑖 𝑖 = 1, 2, … , 𝑛
𝑓 =
𝑖
𝑏𝑖𝑥𝑖 𝑔 =
𝑖
𝑐𝑖𝑥𝑖
2 Τ𝜁 𝑎0𝑛+ Τ1 2
2𝑛 ! Τ1 2𝑟𝑛−1𝑒−𝜁 Τ𝑟 𝑎0𝑌𝑙
𝑚 𝜃, 𝜙 𝜁 =𝑍 − 𝑠
𝑛exponente orbital
13
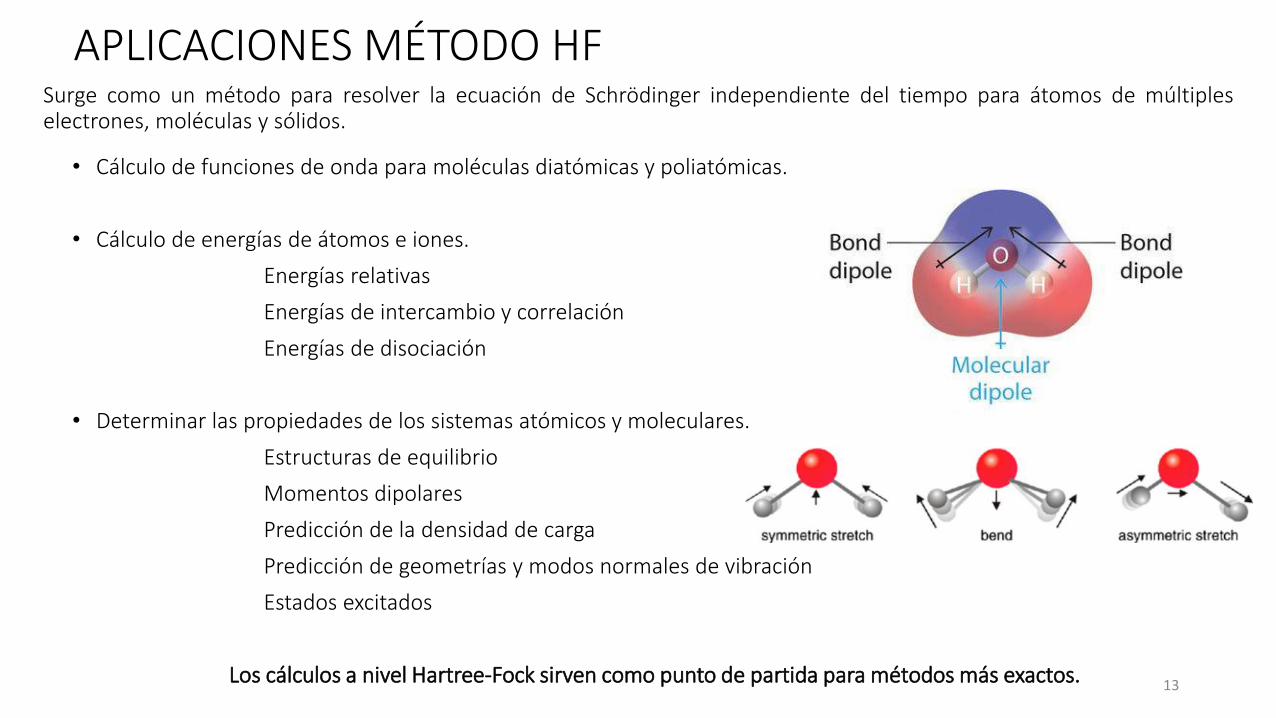
APLICACIONES MÉTODO HF
• Cálculo de funciones de onda para moléculas diatómicas y poliatómicas.
• Cálculo de energías de átomos e iones.
Energías relativas
Energías de intercambio y correlación
Energías de disociación
• Determinar las propiedades de los sistemas atómicos y moleculares.
Estructuras de equilibrio
Momentos dipolares
Predicción de la densidad de carga
Predicción de geometrías y modos normales de vibración
Estados excitados
Los cálculos a nivel Hartree-Fock sirven como punto de partida para métodos más exactos.
Surge como un método para resolver la ecuación de Schrödinger independiente del tiempo para átomos de múltipleselectrones, moléculas y sólidos.

14
MÉTODOS POST HARTREE-FOCK
Toman en cuenta la correlación electrónica
Pertenecen a tres categorías:
• Métodos de Interacción de Configuraciones (IC)
Representa la función de onda como con una combinación lineal de determinantes de Slater.
• Métodos perturbativos (p.ej. Møller-Plesset)
Resuelve la ecuación de Schrödinger mediante una aproximación matemática que consiste en calcular la energía decada orbital con un operador que sí tenga solución y posteriormente corregir su valor aplicando al sistema unoperador formado por la diferencia entre el operador problema y el conocido.
• Coupled Cluster (CC)
Toma el método HF y construye funciones de onda multielectrónicas usando el operador de cluster exponencial paradar cuenta de la correlación electrónica.
La función de onda a mejorar es exponencial
La función de onda a mejorar es lineal
La función de onda a mejorar es lineal

15
PROGRAMAS QUE UTILIZAN HARTREE-FOCK
General
• Visualización de informaciónsobre la estructura.
• Parámetros de geometría(ángulos, distancias, etc.)
Química Cuántica
• Densidad electrónica
• Orbitales moleculares
• Potenciales electrostáticos
• Momentos dipolares
• Modos vibracionales
• Espectro IR
MOLEKEL

16
MÉTODO HF UTILIZANDO HERRAMIENTAS COMPUTACIONALES
Obtención de propiedades moleculares:• Método
• Conjuntos base
Química medicinal• Relación actividad-estructura (SAR, siglas en inglés)
• Relación cuantitativa actividad-estructura (QSAR)
• Relación cuantitativa propiedad-estructura (QSPR)
Santos, C.B.R., Lobato, C.C., Braga, F.S., Morais, S.S.S., Santos, C.F., Fernandes, C.P., ... Carvalho, J.C.T. (2014). Application of Hartree-Fock Method for Modeling of Bioactive MoleculesUsing SAR and QSPR. CMB, 4, 1-24.
17
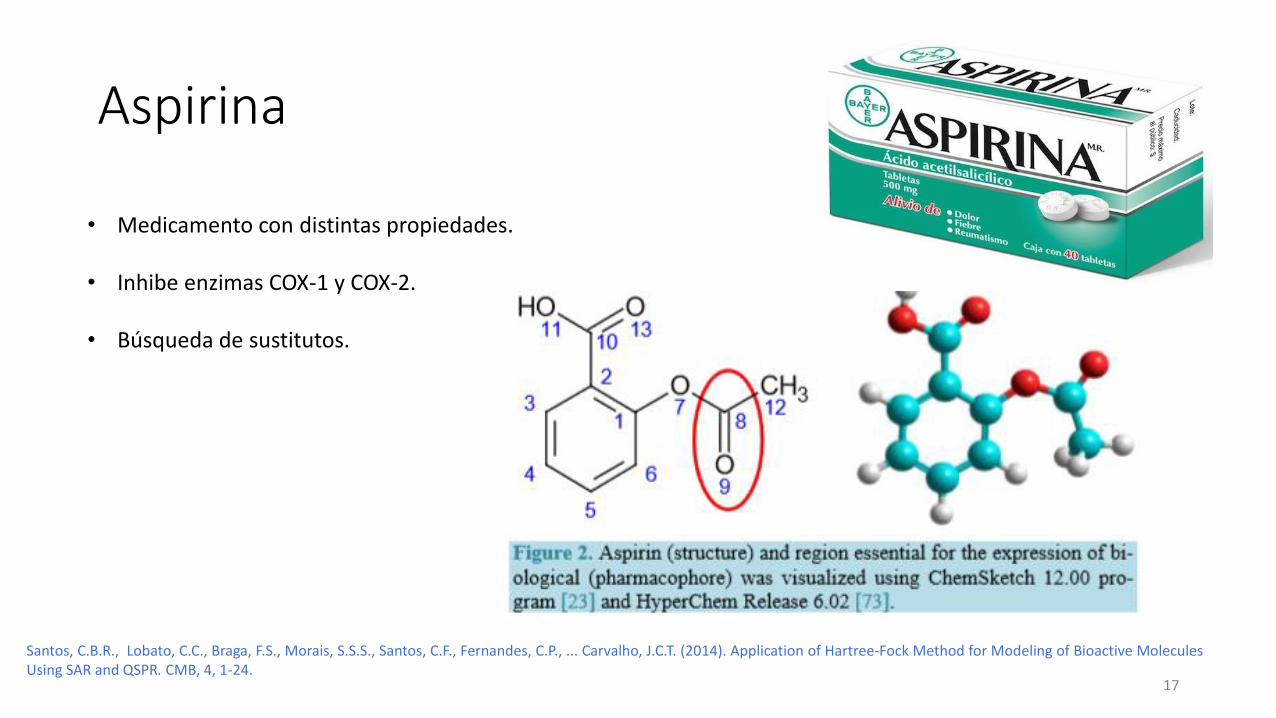
Aspirina
• Medicamento con distintas propiedades.
• Inhibe enzimas COX-1 y COX-2.
• Búsqueda de sustitutos.
Santos, C.B.R., Lobato, C.C., Braga, F.S., Morais, S.S.S., Santos, C.F., Fernandes, C.P., ... Carvalho, J.C.T. (2014). Application of Hartree-Fock Method for Modeling of Bioactive MoleculesUsing SAR and QSPR. CMB, 4, 1-24.
18
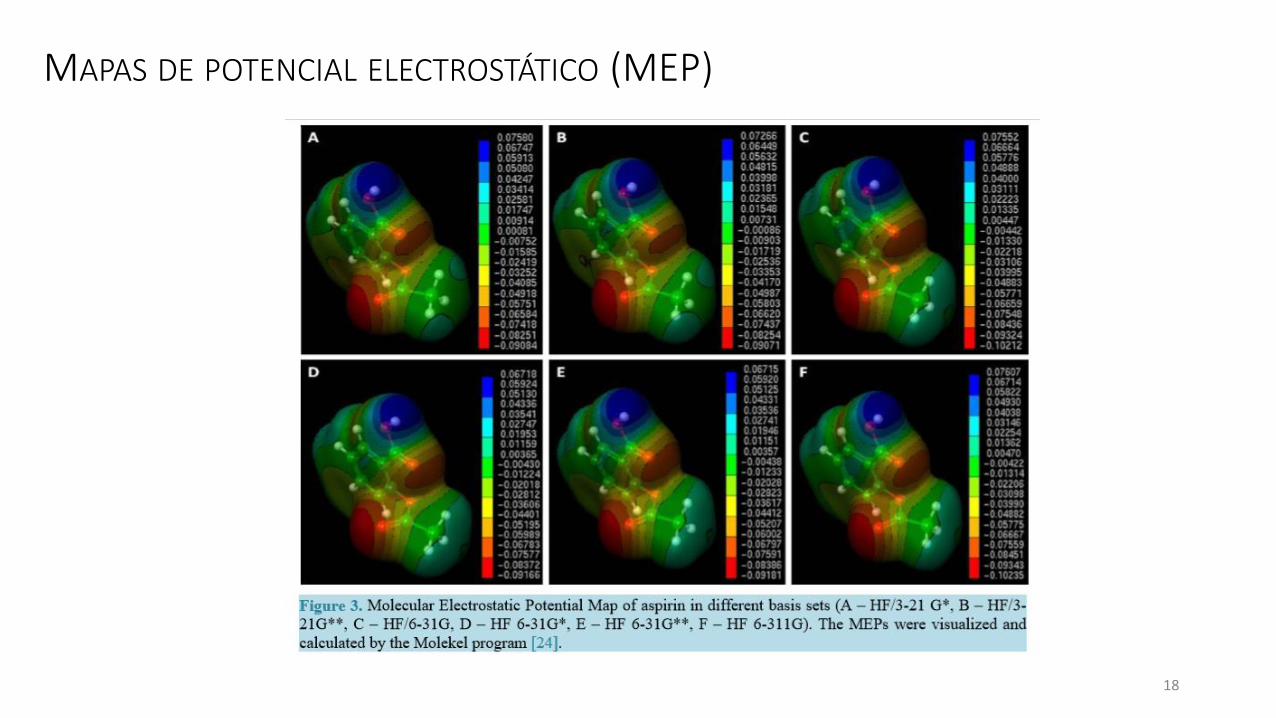
MAPAS DE POTENCIAL ELECTROSTÁTICO (MEP)

19

20

21
Bibliogafía
• Santos, C.B.R., Lobato, C.C., Braga, F.S., Morais, S.S.S., Santos, C.F., Fernandes, C.P., ... Carvalho, J.C.T. (2014). Application ofHartree-Fock Method for Modeling of Bioactive Molecules Using SAR and QSPR. CMB, 4, 1-24.
• Molekel – a visualization tool for quantum chemistry data U. Varetto, M. G. Giuffreda, Y. Jang Molekel – ACS meeting –August 2009 Swiss National Supercomputing Centre (CSCS).
• Bort Juan Andrés et. Al., Química teórica y computacional, Ciències Experimentals, Universidad Jaume, 2001, pag. 79
• Exploring chemistry with electronic structure methods, James B. Foresman, Æleen Frisch, Gaussian, Inc., 1996, Químicacuántica, Ira N. Levine, Pearson Educación, 2001


















