
Patología hematopoyética 2 y 3 · PDF fileLinfoma de Hodgkin •Se origina en...
107
Patología hematopoyética 2 y 3 UNIBE – Patología 2 III cuatrimestre - 2012
-
Upload
hoangduong -
Category
Documents
-
view
218 -
download
1
Transcript of Patología hematopoyética 2 y 3 · PDF fileLinfoma de Hodgkin •Se origina en...

Patología hematopoyética 2 y 3
UNIBE – Patología 2
III cuatrimestre - 2012

Temas
• Linfadenitis aguda y crónica.
• Linfomas.
• Anemia.
• Policitemia.
• Coagulación intravascular diseminada.

Linfadenitis aguda inespecífica• Ganglios hinchados y enrojecidos.
• Centros germinativos grandes con
numerosas figuras mitóticas.
• Macrófagos con detritus celulares o de
microorganismos fagocitados.
• Neutrófilos en los sinusoides o necrosis
con abscesos en casos severos.
• Hiperplasia de células endoteliales
sinusoidales.

Patrones de linfadenitis crónica
inespecífica
• Hiperplasia folicular.
• Hiperplasia parafolicular (interfolicular).
• Histiocitosis sinusal (hiperplasia reticular).
• Patrón mixto.
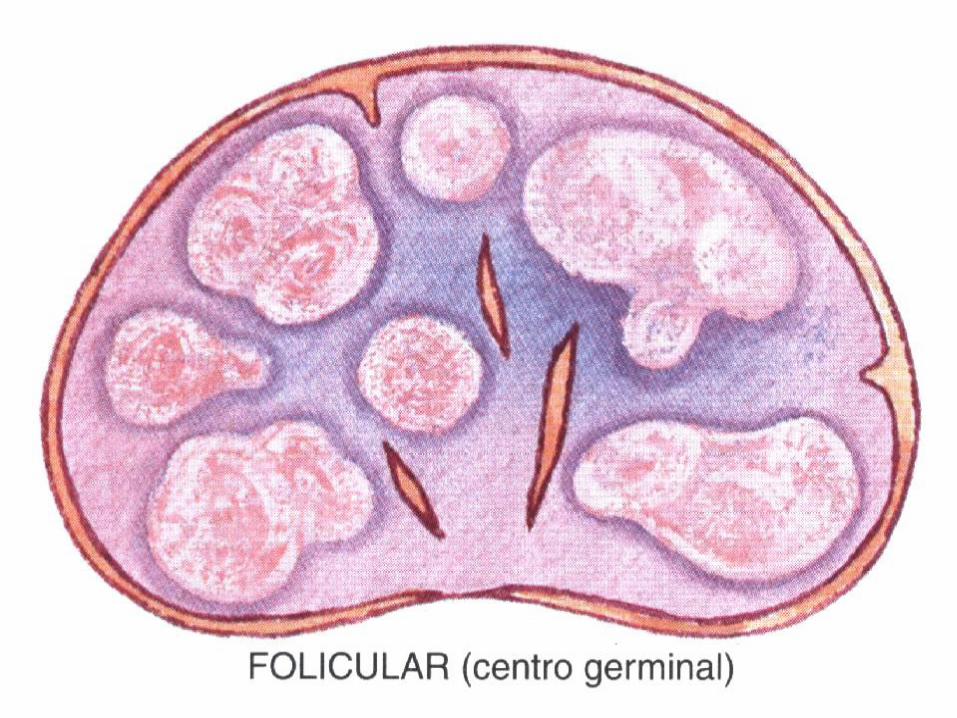
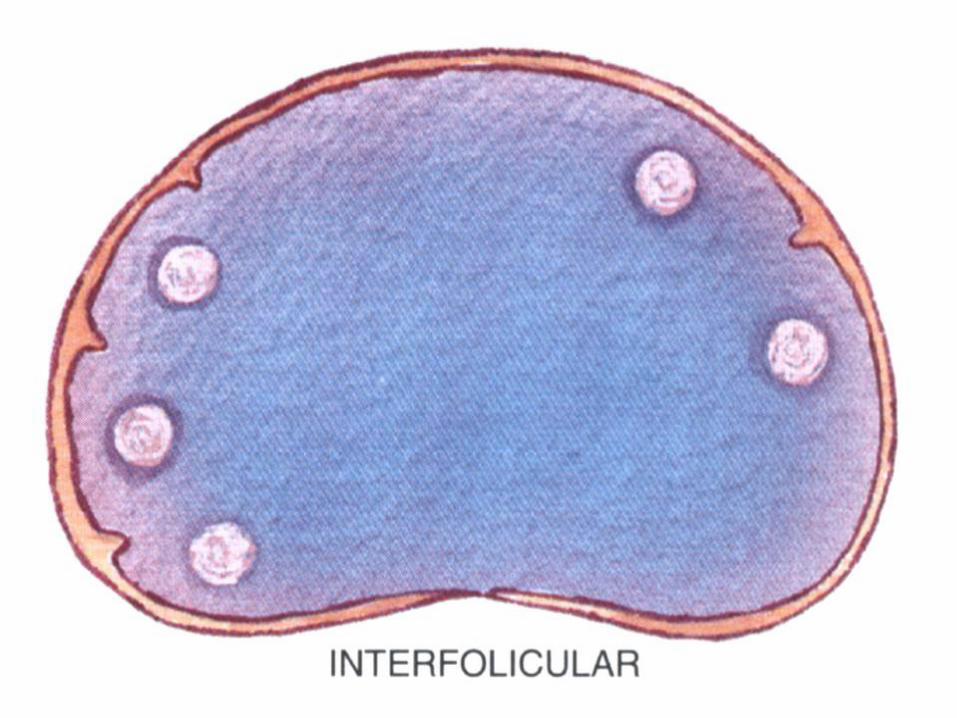

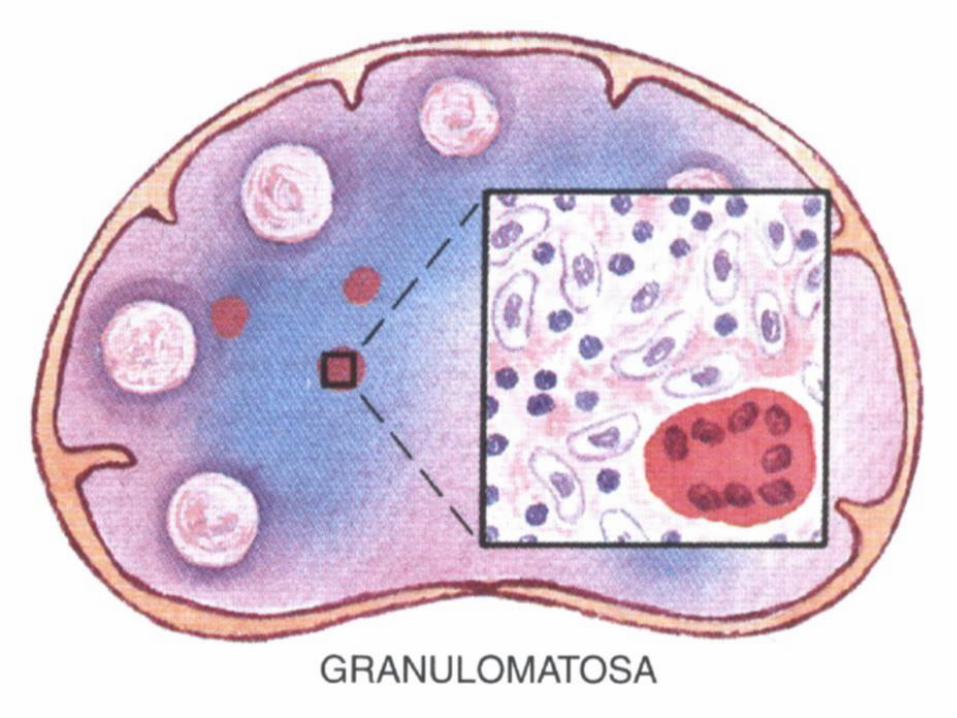

Clasificación de la OMS de neoplasia
linfoides
I Neoplasia de precursores de células B:
Linfoma/leucemia aguda de células B
linfoblásticas.

II. Neoplasias de células B periféricas:
Linfoma/leucemia crónica de linfocitos
pequeños.
Leucemia de células B prolinfocíticas.
Linfoma linfoplasmocítico.
Linfoma de las zona marginal esplénico y
nodal.
Linfoma de la zona marginal extranodal.

Linfoma de células del manto.
Linfoma folicular.
Linfoma de la zona marginal.
Leucemia de células peludas.
Plasmacitoma/mieloma de células
plasmáticas.
Linfoma difuso de células grandes B.
Linfoma de Burkitt.

III. Neoplasia de precursores de células T:
Linfoma/leucemia linfoblástico agudo de
células T.

IV. Neoplasias de células T periféricas y
de células NK:
Leucemia prolinfocítica de células T.
Leucemia linfocítica de células grandes
granulares.
Micosis fungoide/síndrome de Sézary.
Linfoma de célula T periféricas, no
especificado.
Linfoma anaplásico de células grandes.

Linfoma angioinmunoblástico T.
Linfoma de células T asociado a enteropatía.
Linfoma de células T semejante a paniculitis.
Linfoma hepatoesplénico de T γδ.
Leucemia/linfoma de células T adultas.
Linfoma de células NK/T extranodal.
Leucemia de células NK.

V. Linfoma de Hogkin:
Subtipos clásicos:
Esclerosis nodular.
Celularidad mixta.
Rico en linfocitos.
Depleción linfocítica.
Predominancia de linfocitos.

Linfoma linfoblástico
• Linfocitos inmaduros B o T.
• Las masas mas comunes son en el timo.
• Sin embargo la presentación más común
es como leucemia.

Linfoma linfoblástico en timo

Linfoma de linfocitos pequeños
• Infiltración por linfocitos pequeños con
agregados laxos de linfocitos más
grandes (centros de proliferación).
• Translocaciones cromosómicas raras.
• Mayoría asintomáticos, o pérdida de
peso, fatigabilidad, anorexia,
hepatomegalia y esplenomegalia.
• Supervivencia media 4 a 6 años.

Linfoma de linfocitos pequeños

Linfoma de linfocitos pequeños

Linfoma folicular
• Patrón nodular o nodular y difuso con 2
tipos de linfocitos: centrocitos y
centroblastos.
• Bcl2 expresado en el 90%, debido a
t(14;18).
• Se manifiestan con linfadenopatía
generalizada no dolorosa, con
supervivencia de 7 a 9 años.
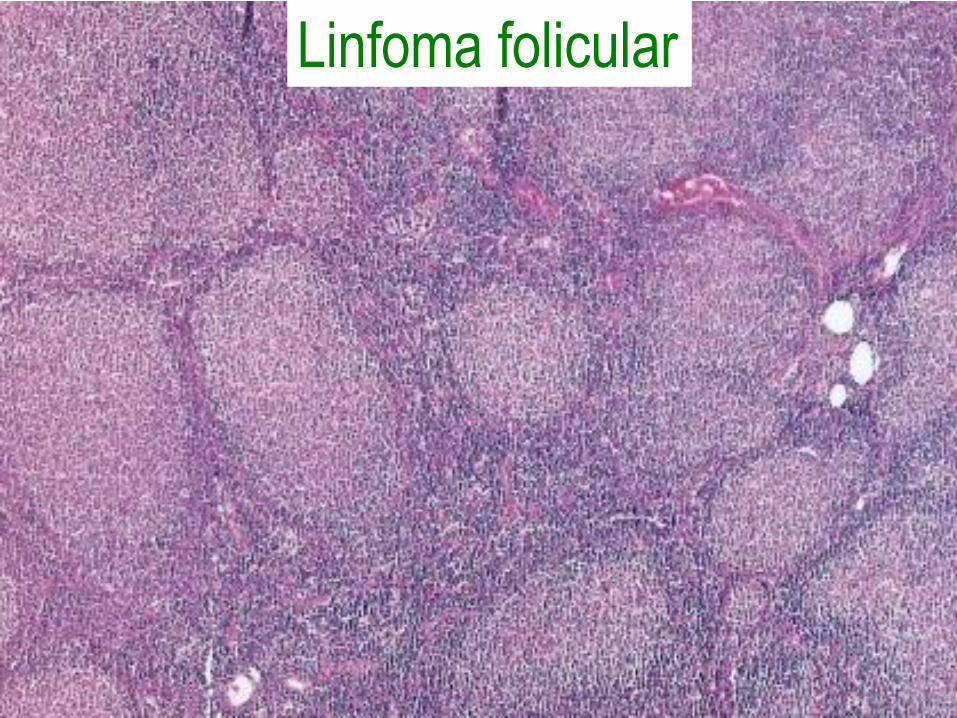
Linfoma folicular

Linfoma folicular con centroblastos

Linfoma folicularFolículo hiperplásico
Inmunotición con Bcl2

Linfoma de células grandes• Células grandes con patrón de
crecimiento difuso, 1 a 3 nucleolos y citoplasma abundante.
• Grupo heterogéneo, con disregulación de Bcl6 o Bcl2, o asociados a inmunodeficiencia (con virus EBV o HHV8).
• Masa nodal o extranodal de crecimiento rápido.
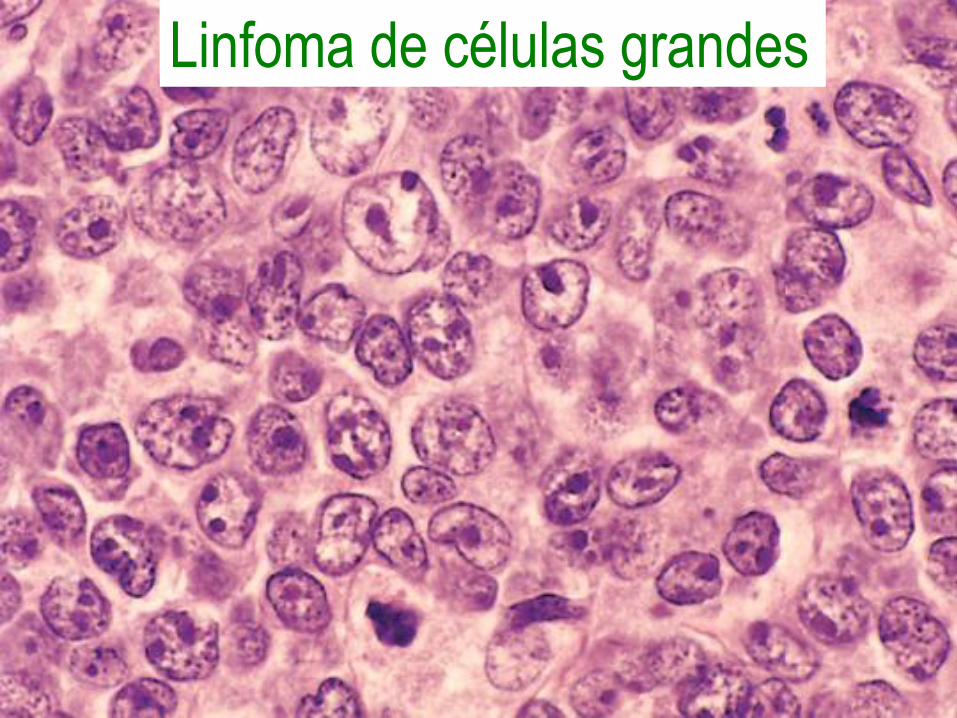
Linfoma de células grandes

Linfoma de Burkitt
• Infiltrado difuso de células de tamaño
intermedio, cromatina gruesa, varios
nucleolos, mucha mitosis y apoptosis.
• Translocación del gen c-MYC con
aumento de su expresión.
• Afecta niños y adultos jóvenes, agresivo
pero curable en la mayoría de los casos.

Linfoma deBurkitt
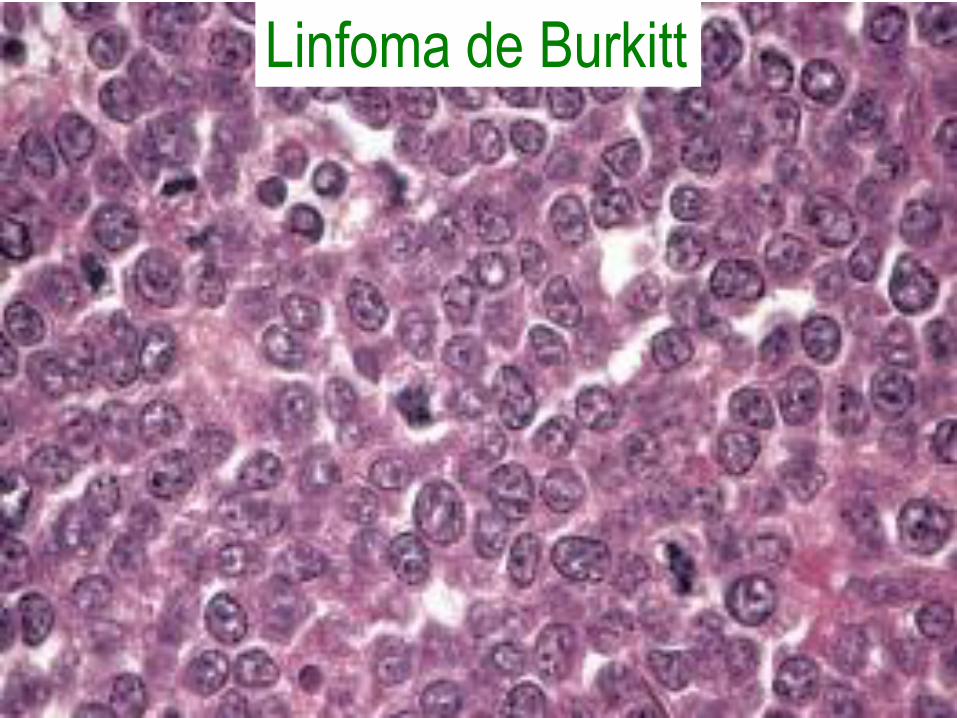
Linfoma de Burkitt

Linfoma de células del manto
• Linfocitos pequeños con núcleos de
contorno irregular que infiltran
difusamente o rodean centros germinales.
• Sobrexpresión de ciclina D1 por
translocación (11;14).
• Presentación más común con
linfadenopatías y la supervivencia es de 3
a 4 años.

Linfoma de células del manto

Linfoma de células T periférico
• Infiltrado difuso del ganglio con mezcla de
linfocitos T pleomórficos de tamaño
variable.
• Frecuente infiltrado prominente de células
reactivas como eosinófilos y macrófagos.
• Se manifiestan con linfadenopatía
generalizada, a veces con eosinofilia,
prurito y pérdida de peso.

Linfoma de células T periférico

Linfoma anaplásico de células grandes
(ALK+)
• Células grandes anaplásicas, algunas con
núcleos en herradura, con rearreglo en el
gen ALK en el cromosoma 2p23.
• En niños y adultos jóvenes, con
frecuencia en tejidos blandos y de buen
pronóstico con tratamiento.

Linfoma anaplásico de células grandes

Micosis fungoide/síndrome de Sezary
• Linfocitos T CD4+ neoplásicos, de
aspecto (cerebriforme), que infiltran
incialmente piel y pueden extenderse
luego a otros órganos.
• En el síndrome de Sezary hay
eritrodermia exfoliativa generalizada
asociada a leucemia.
• Supervivencia de 8 a 9 años.

Micosis fungoide

Linfoma de Hodgkin• Se origina en un ganglio o una cadena de
ganglios, raramente extranodal, diseminándose a los tejidos linfoides anatómicamente contiguos.
• Las células neoplásicas son llamadas de Reed-Sternberg y secretan factores que atraen linfocitos, macrófagos y granulocitos, los cuales forman más del 90% del tumor.
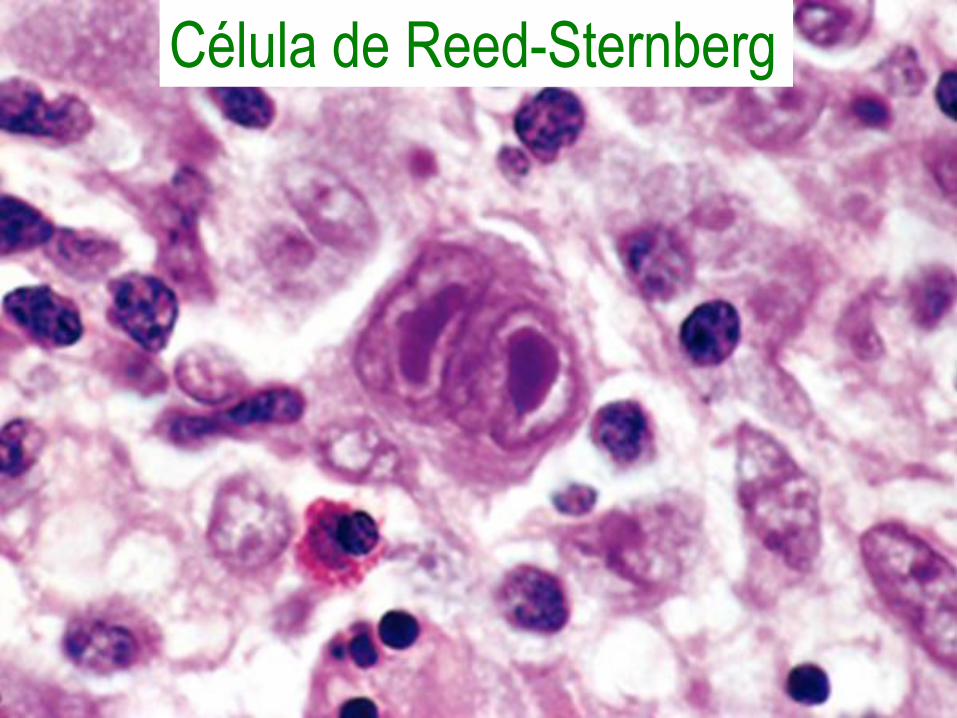
Célula de Reed-Sternberg
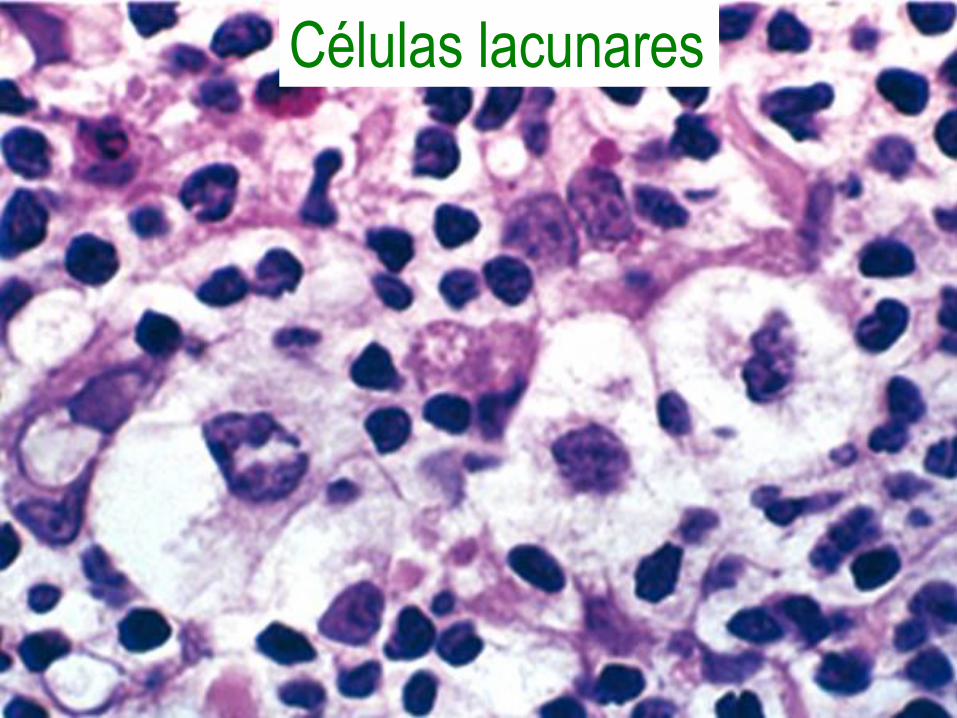
Células lacunares

Variante H&L

Linfoma de Hodgkin tipo esclerosis
nodular
• 65 a 70% de los casos.
• Con la variante lacular de células de
Reed-Sternberg.
• Con bandas de colágeno que dividen al
ganglio en nódulos circunscritos.
• Muy buen pronóstico.
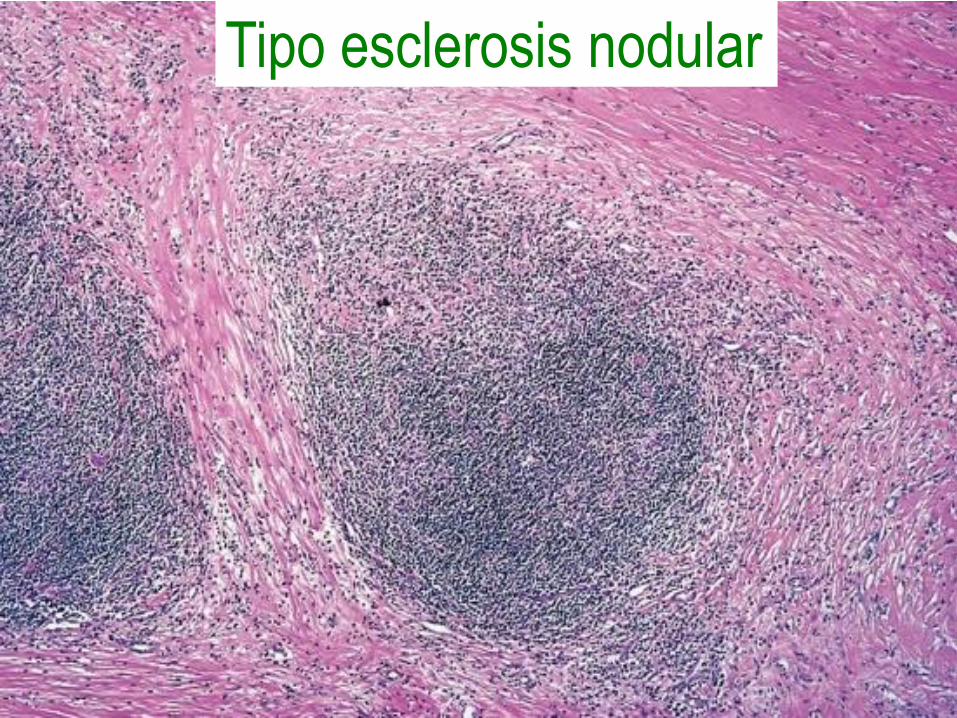
Tipo esclerosis nodular

Linfoma de Hodgkin de celularidad
mixta
• 20 a 25% de los casos.
• Infiltrado heterogénio de linfocitos T,
eosinófilos, plasmocitos, macrófagos,
células de Reed-Sternberg y variantes
mononucleares (con 70 % de infección
con EBV).

Tipo celularidad mixta

Linfoma de Hodgkin de la variedad
clásica rica en linfocitos
• Infiltrado difuso formado principalmente
por linfocitos reactivos.
• Células de Reed-Stemberg o variantes
mononucleares con inmunofenotipo
clásico (positivo por PAX5, CD15 y CD30,
negativo para otros marcadores B de
linfocitos B, de linfocitos T o CD45).

Variedad clásica rica en linfocitos

Linfoma de Hodgkin tipo depleción
linfocítica
• Menos del 5%.
• Pocos linfocitos y muchas células de
Reed-Stemberg con sus variantes.
• Con virus EBV en el 90%.
• Pronóstico menos favorable.

Tipo depleción linfocítica

Linfoma de Hodgkin del tipo nodular de
predominancia de linfocitos
• 5% de los casos.
• Infiltrado nodular de linfocitos mezclados
con macrófagos
• Células clásicas de Reed-Sternberg
difíciles de encontrar, pero hay muchas
de la variante H&L (postivas para CD20 y
Bcl6, negativas para CD15, CD30 y EBV).

Tipo con predominancia de linfocitos


Manifestaciones clínicas
• Linfadenopatía no dolorosa en la mayoría
de los casos.
• Con enfermedad avanzada hay fiebre,
sudores nocturnos y pérdida de peso.
• Hay anergia cutánea por depresión de la
inmunidad celular.

Anemia: definición
• Reducción de la masa total de eritrocitos
circulantes bajo los límites normales.
• El diagnóstico se hace por la disminución
del hematócrito o de la concentración de
hemoglobina.

Hombre Mujer
Hemoglobina
(g/dL)
13.6–17.2 12.0–15.0
Hematocríto 39–49 33–43
Conteo de
eritrocitos
(×106/μL)
4.3–5.9 3.5–5.0

Anemia

Anemia: clasificación
• Por tamaño: normocítica, macrocítica
o microcítica.
• Por contenido de hemoglobina:
normocrómica o hipocrómica.

Clasificación según mecanismo
• Pérdida de sangre.
• Hemólisis.
• Disminución en la eritropoyesis.

Por sangrado agudo• La pérdida de volumen intravascular, que
puede llevar a choque y muerte.
• Inicialmente los eritrocitos son de aspecto
normal y luego hay reticulocitosis.
Por sangrado crónico• Se excede la capacidad regenerativa de
la médula ósea o cuando se agotan los
depósitos de hierro.

Anemias hemolíticas
• Defectos hereditarios del eritrocito.
• Defectos genéticos adquiridos.
• Destrucción mediada por anticuerpos.
• Trauma mecánico.
• Infecciones de eritrocitos.
• Daño tóxico o químico.
• Anormalidades de lípidos de membrana.
• Secuestro de eritrocitos.

Eritropoyesis reducida•Defectos genéticos hereditarios.
•Deficiencias nutricionales.
•Deficiencia de eritropoyetina.
•Lesión inmunológica de progenitores.
•Secuestro de hierro por inflamación.
•Neoplasias hematopoyéticas primarias.
•Lesiones medulares espacio ocupantes.
•Infección de progenitores de eritrocitos.
•Mecanismos desconocidos.

Anemias hemolíticas
• Destrucción prematura de eritrocitos,
extravascular o intravascular.
• Niveles elevados de eritropoyetina con
aumento en la eritropoyesis.
• Acumulación de productos de la
degradación de la hemoglobina.

Morfología en anemia hemolítica
• Aumento de normoblastos en la médula.
• Reticulocitosis prominente.
• Hemosiderosis.
• Hematopoyesis extramedular.
• Colelitiasis.

Esferocitosis hereditaria

Deficiencia de glucosa-6-fosfato
deshidrogenasa
• Reducción en la protección de los
eritrocitos al daño oxidativo.
• Herencia ligada al cromosoma X de las
formas en que se degradan más rápido.
• Hemólisis episódica al presentarse stress
oxidativo (por infecciones, drogas o
algunos alimentos).

Anemia de células falciformes
• Mutación del codón 6 de la β-globina con sustitución de glutamato por valina.
• Polimerización con deoxigenación.
• Hemólisis crónica y oclusión microvascular.
• Empeora con deshidratación y acidez.

Morfología
• Eritrocitos falciformes, forma de hoja de
acebo, en diana, reticulocitosis, cuerpos de
Howell-Jolly, hiperplasia medular,
hematopoyesis extramedular,
autoesplenectomía, colelitiasis e
hiperbilirrubinemia.
• Infartos en huesos, cerebro, riñones,
hígado, retina, pulmón y úlceras cutáneas.
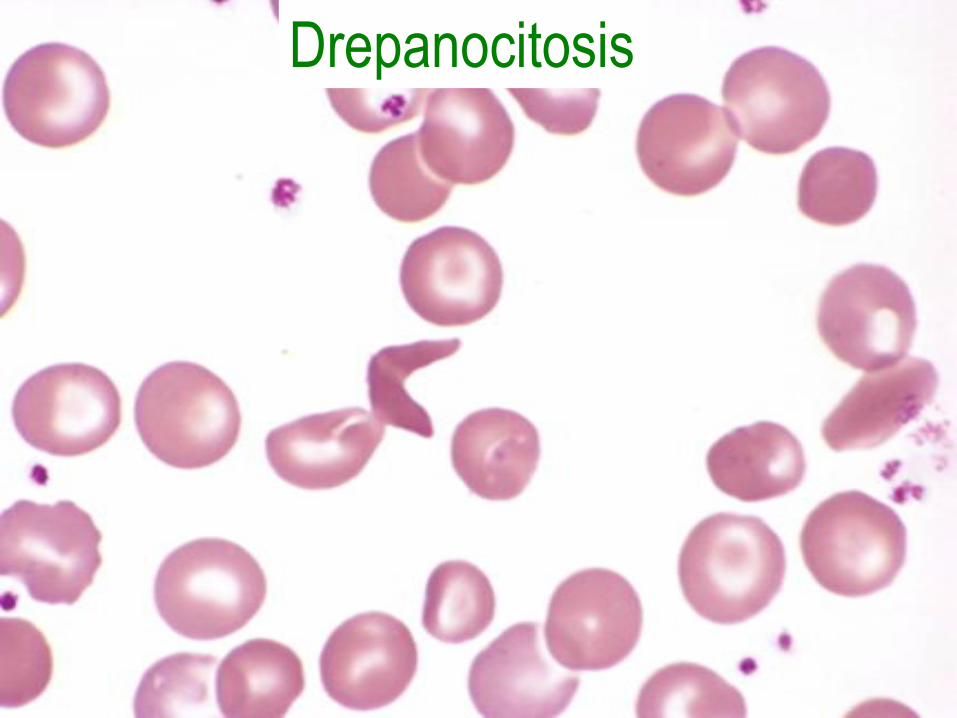
Drepanocitosis
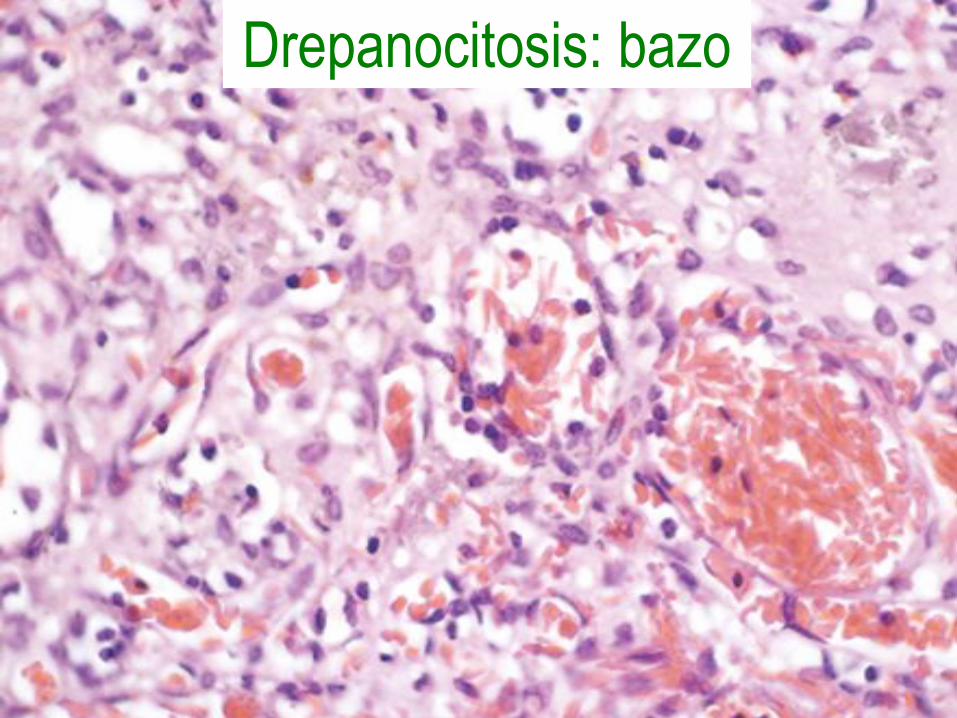
Drepanocitosis: bazo

Drepanocitosis: bazo

Drepanocitosis: crisis vasoclusiva en bazo

Síndromes talasémicos
• Causados por mutaciones heredadas que
disminuyen la síntesis la HbA (α2β2).
• La β-talasemia afecta la síntesis de
cadenas β y la α-talasemia afecta la
síntesis de cadenas α.

β-talasemia• Con la mutaciones β0, no hay β-globina y
con las β+ esta reducida.
• Disminución en cantidad de hemoglobina
normal y en la vida de los eritrocitos y sus
precursores.
• Hay eritropoyesis ineficaz porque las
cadenas α en exceso precipitan formando
inclusiones insolubles que dañan la
membrana.
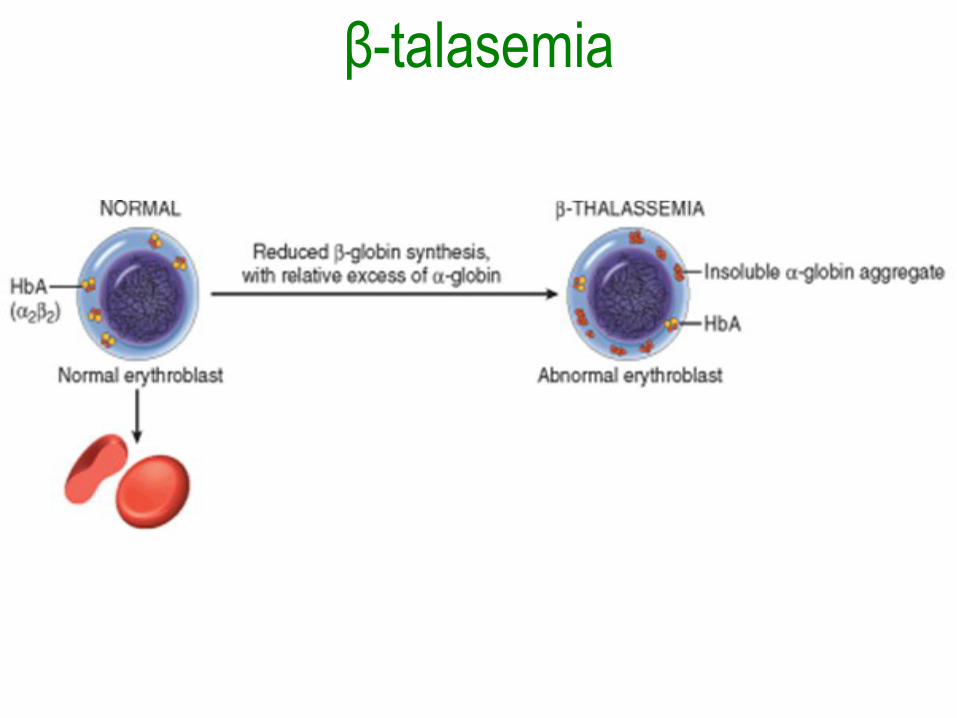
β-talasemia

β-talasemia
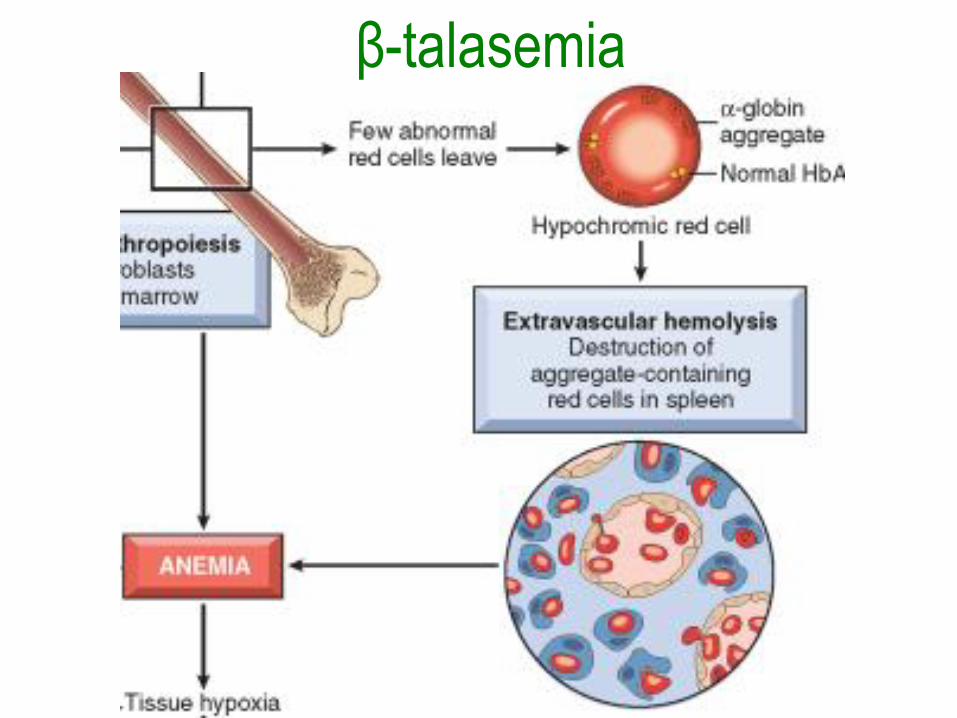
β-talasemia

Talasemia: hiperplasia de médula ósea

Talasemia

Hemoglobinuria paroxística nocturna
• Mutación adquirida en el gen del grupo A
del fosfatidilinositol glicano con deficiencia
en varias proteínas, en particular CD59
(inhibidor de C3 convertasa).
• En el 25% de los casos es paroxística y
nocturna y en el resto es una anemia
crónica.

Manifestaciones adicionales
• 40% de los pacientes sufren trombosis
venosa.
• Por disfunción plaquetaria por falta de
proteínas ligadas al fosfatidilinositol
glicano, así como a la absorción del NO a
la hemoglobina libre.


Anemia inmunohemolítica
• Debida a anticuerpos que se unen
a los eritrocitos produciendo su
destrucción prematura.

Anemia hemolítica por trauma de
eritrocitos
• Prótesis valvulares cardíacas.
• Coagulación intravascular diseminada,
púrpura trombótica trombocitopénica,
síndrome hemolítico urémico, hipertensión
maligna, LES y cáncer diseminado.
• Reducción del lumen por depósito de
fibrina y plaquetas.

Anemias megaloblásticas• Sangre con macro-ovalocitos, anisocitosis,
poiquilocitosis, bajo conteo de reticulocitos,
progenitores nucleados de eritrocitos,
macropolimorfonucleares y leucocitos
hipersegmentados.
• Médula ósea hipercelular con cambios
megaloblásticos en todas las etapas de la
eritropoyesis y metamielocitos gigantes.
• Apoptosis en la médula lleva a pancitopenia.

Anemia megaloblástica

Anemia perniciosa • Por gastritis autoinmune con fallo en
producción de factor intrínseco y
deficiencia en vitamina B12.
• El daño lo inician linfocitos T
aurorreactivos con producción secundaria
de anticuerpos.
• Médula espinal con desmielinización de
tractos dorsales y laterales (paraparesis,
ataxia sensorial y parestesias).

Anemia por deficiencia de folato
• Por aporte insuficiente, aumento de
necesidades o mala utilización.
• Aporte insuficiencia por dieta o fallo en la
absorción. Drogas, como fenitoína y
contraceptivos interfieren su absorción.
• Aumento en necesidades por embarazo,
infancia, hematopoyesis hiperactiva o
cáncer diseminado.

Anemia por deficiencia de hierro
• Causas: falta en la dieta, fallo en
absorción, aumento en necesidades o
pérdida de sangre.
• Las necesidades aumentan durante el
crecimiento y el embarazo.

Anemia ferropénica
• Aumento en progenitores eritroides,
desaparición de hierro en los macrófagos.
• Anemia microcítica hipocrómica con
poiquilocitosis.
• Disminución en enzimas con hierro:
koiloniquia, alopecia, atrofia de mucosa
lingual, atrofia gástrica, malabsorción
intestinal y pica.

Anemia ferropénica

Anemia de enfermedad crónica
• En enfermedades infecciosas,
inmunitarias y neoplasias.
• Reducción de hematopoyesis y utilización
del hierro.
• Mediadores inflamatorios (IL-6), estimulan
producción de hepcidina que inhibe la
ferroportina y la eritropoyetina.

Anemia aplásica adquirida•Idiopática: Defecto adquirido en células madre
o inmunológica (dos tercios de los casos).
•Por agentes químicos:
Relacionada con dosis: Agentes alquilantes,
antimetabolitos, benzeno, cloranfenicol,
arsenicales.
Idiosincrática: Cloranfenicol, fenilbutazona,
arsenicales orgánicos, metilfeniletilhidantoína,
carbamazepina, penicilamina y sales de oro.

Anemia aplásica adquirida
• Por agentes físicos:
Irradiación corporal total.
• Infecciones virales: Hepatitis,
citomegalovirus, virus Epstein-Barr y
herpes zoster.

Anemia aplásica hereditaria
• Anemia de Fanconi: mutación en uno de
15 genes diferentes relacionados con la
reparación del ADN.
• Defectos de la telomerasa.
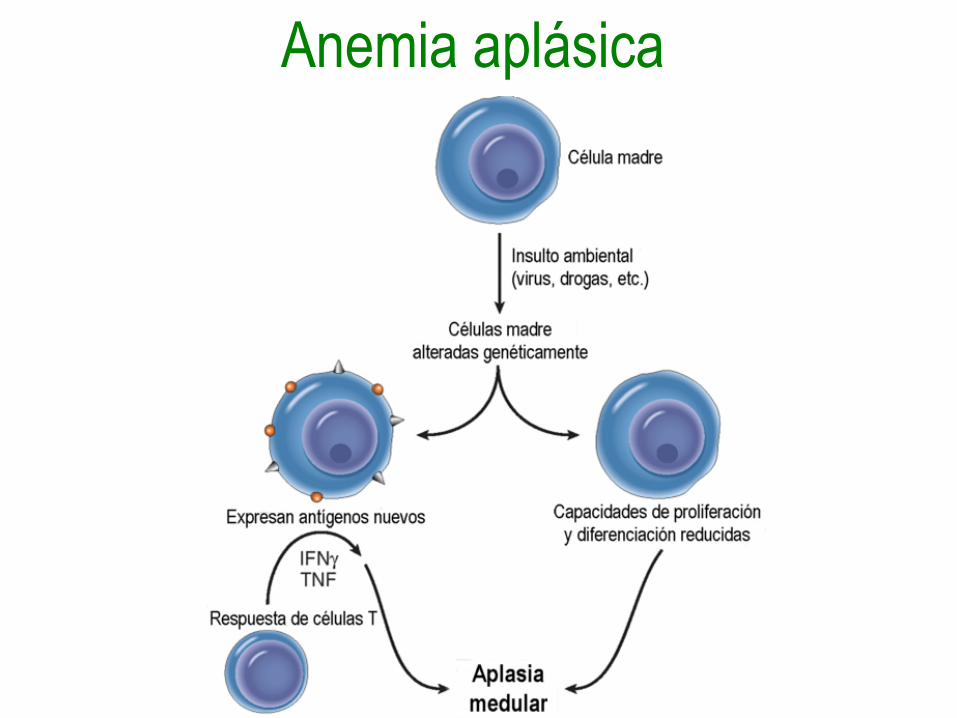
Anemia aplásica

Policitemia absoluta
• Primaria (baja eritropoyetina):
Policitemia vera.
Mutación de receptores de
eritropoyetina.

Policitemia absoluta
• Secundaria (elevada eritropoyetina):
Compensatoria.
Paraneoplásica.
Hemoglobinas mutantes con alta
afinidad al O2.
Mutaciones hereditarias que estabilizan
el factor inducido por la hipoxia 1α.

Coagulación intravascular diseminada
• Enfermedad trombohemorrágica aguda,
subaguda o crónica, caracterizada por
activación excesiva de la coagulación,
que conduce a la formación de trombos
en la microcirculación.

Presentación clínica
• Anemia hemolítica microangiopática.
• Disnea, cianosis y fallo respiratorio.
• Covulsiones y coma.
• Oliguria y fallo renal agudo.
• Fallo circulatorio súbito o progresivo
y choque.

Causas principales
• Complicaciones obstétricas.
• Neoplasias malignas.
• Sepsis.
• Trauma severo.

Fisiopatología
• Liberación del factor tisular o
sustancias tromboplastínicas en la
circulación.
• Daño diseminado de las células
endoteliales.

Fisiopatología
• Hay oclusión vascular con lesión
isquémica de los tejidos.
• Son consumidos los factores de
coagulación y se activa la plasmina con
fibrinólisis, llevando todo a hemorragias.

Morfología
• Trombos, microinfartos y hemorragias
en cerebro, corazón, pulmones, riñones,
adrenales, bazo e hígado.
• Puede producir el síndrome de
Waterhouse-Friderichsen con necrosis
de adrenales o el síndrome de Sheehan
con necrosis posparto de hipófisis.

Coagulación intravascular diseminada
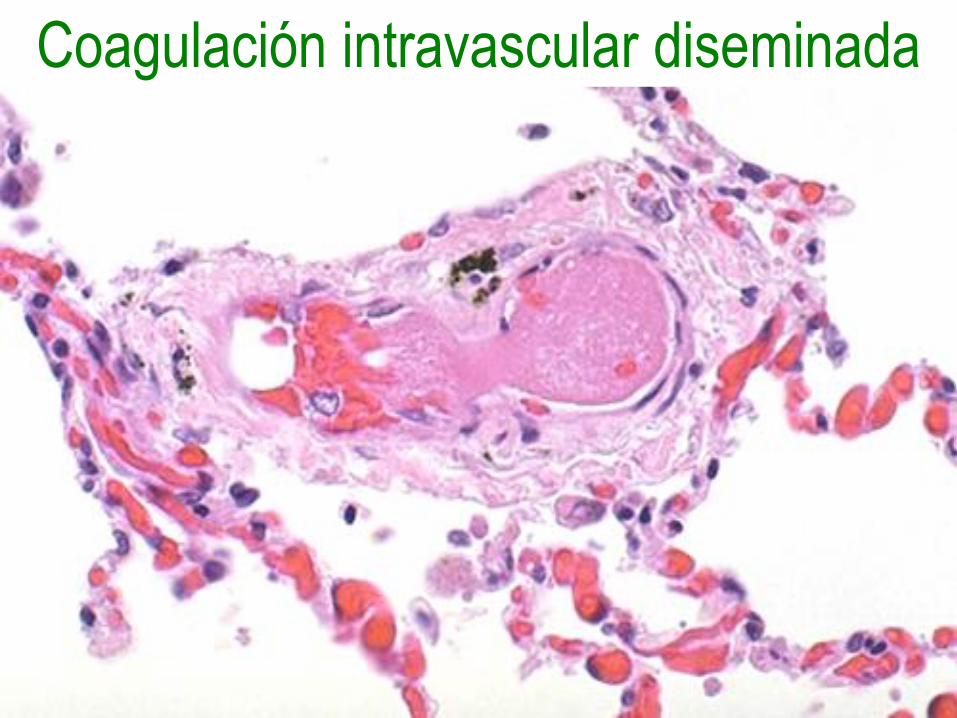
Coagulación intravascular diseminada
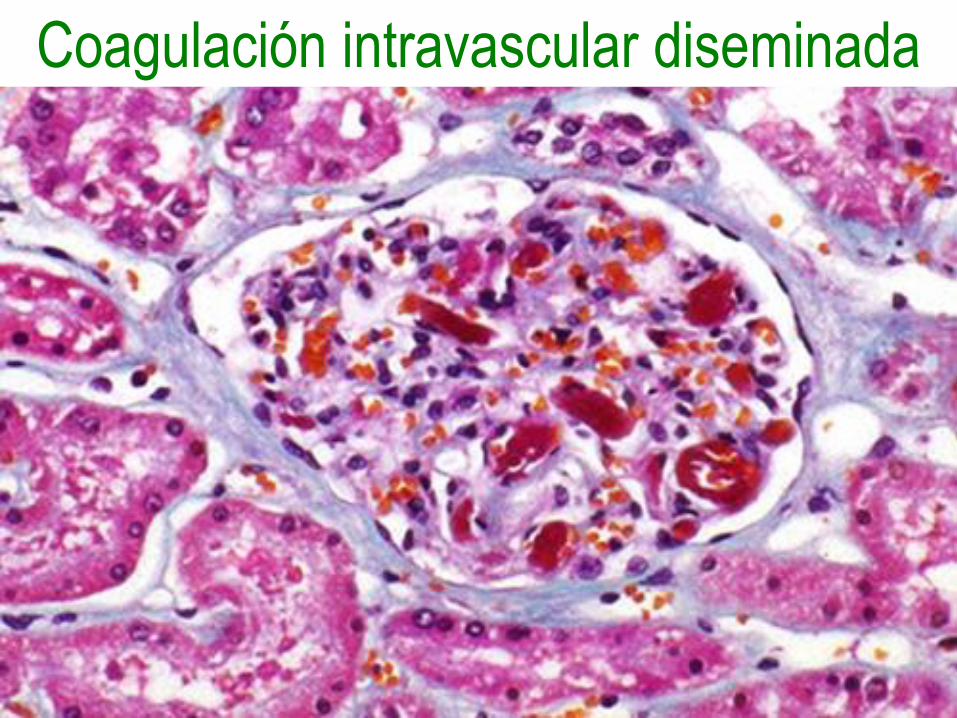
Coagulación intravascular diseminada

Fin
Gracias por su atención


















