Presentación de PowerPoint - bioinfo2.ugr.es · tejidos en cualquier región del genoma....
57
Grupo de Genómica Computacional & Bioinformática Universidad de Granada
Transcript of Presentación de PowerPoint - bioinfo2.ugr.es · tejidos en cualquier región del genoma....

Grupo de Genómica Computacional & Bioinformática Universidad de Granada
INTRODUCCIÓN
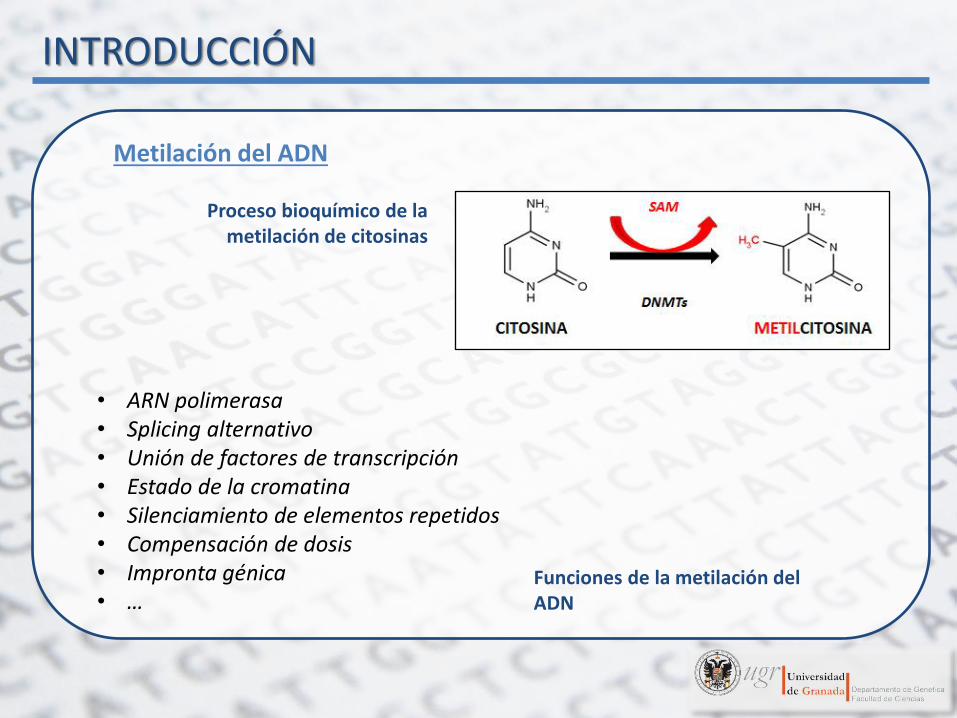
Proceso bioquímico de la metilación de citosinas
Metilación del ADN
• ARN polimerasa • Splicing alternativo • Unión de factores de transcripción • Estado de la cromatina • Silenciamiento de elementos repetidos • Compensación de dosis • Impronta génica • …
Funciones de la metilación del ADN

INTRODUCCIÓN
Distribución de la metilación en contextos
Células madre (H1)
Fibroblastos de pulmón (IMR90)
Lister et al. 2009 (Nature)
INTRODUCCIÓN
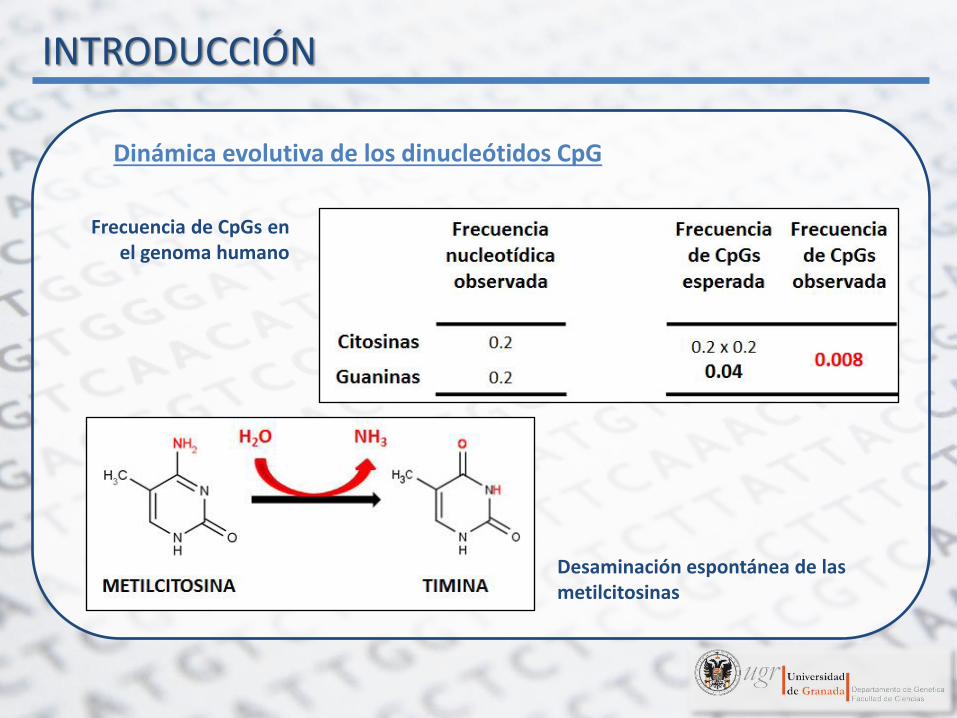
Dinámica evolutiva de los dinucleótidos CpG
Frecuencia de CpGs en el genoma humano
Desaminación espontánea de las metilcitosinas
INTRODUCCIÓN
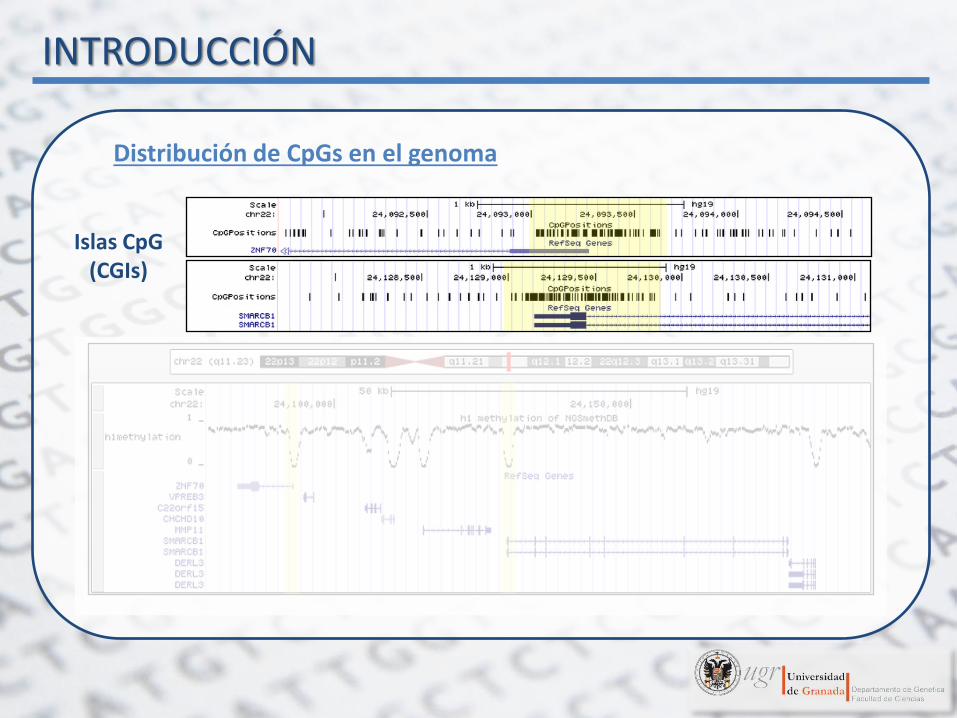
Distribución de CpGs en el genoma
Islas CpG (CGIs)

INTRODUCCIÓN
Distribución de CpGs en el genoma
Islas CpG (CGIs)

INTRODUCCIÓN
Metilación diferencial
H1
IMR90
Región diferencialmente metilada
(DMR)

OBJETIVOS
1. Desarrollo de la metodología necesaria para procesar y cuantificar los niveles de metilación, a partir de datos obtenidos mediante protocolos de secuenciación masiva.
2.Desarrollo de una base de datos relacional que permita el almacenamiento y la comparación de los datos de metilación en citosinas individuales.
3.Optimización del algoritmo CpGcluster, con objeto de mejorar sus predicciones.
4.Comparación del algoritmo mejorado con los métodos clásicos de identificación de islas CpGs.
5. Identificar y caracterizar regiones diferencialmente metiladas con una elevada densidad de CpGs.

IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS

IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
BLOQUE I • Inferencia de la metilación

IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
BLOQUE II • Mejora de CpGcluster • Comparación entre
predicciones de CGIs

IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
BLOQUE III • Almacenamiento de datos • Minería de datos • Análisis de regiones
diferencialmente metiladas

IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
BLOQUE I

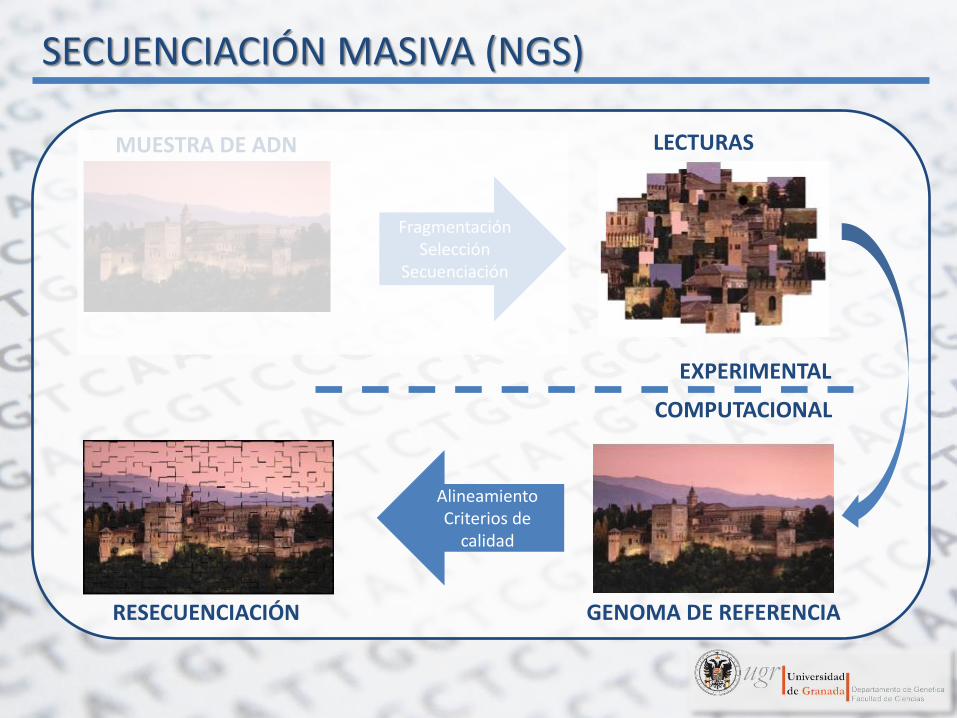
SECUENCIACIÓN MASIVA (NGS)
EXPERIMENTAL
COMPUTACIONAL
Fragmentación Selección
Secuenciación
Alineamiento Criterios de
calidad
MUESTRA DE ADN LECTURAS
GENOMA DE REFERENCIA RESECUENCIACIÓN
SECUENCIACIÓN MASIVA (NGS)
EXPERIMENTAL
COMPUTACIONAL
Fragmentación Selección
Secuenciación
Alineamiento Criterios de
calidad
MUESTRA DE ADN LECTURAS
GENOMA DE REFERENCIA RESECUENCIACIÓN

SECUENCIACIÓN MASIVA (NGS)
EXPERIMENTAL
COMPUTACIONAL
Troceado Selección
Criterios de calidad
MUESTRA DE ADN LECTURAS
GENOMA DE REFERENCIA RESECUENCIACIÓN
2) Variación
DIFERENCIAS
1) Errores de secuenciación y alineamiento

TRATAMIENTO CON BISULFITO
Fragmento de ADN
BSW BSC
BSW Referencia
CITOSINA METILADA BISULFITO
CM C
CITOSINA NO METILADA BISULFITO
C T
C C CITOSINA METILADA
T C CITOSINA NO METILADA

Hackenberg, M., G. Barturen and J. L. Oliver (2012). DNA methylation Profiling from High-Throughput Sequencing Data. DNA Methylation, InTech - Open
Access Publisher, ISBN 979-953-307-453-4.
Alineamiento de lecturas tratadas con bisulfito
Preprocesado de lecturas Desambiguación de multilecturas
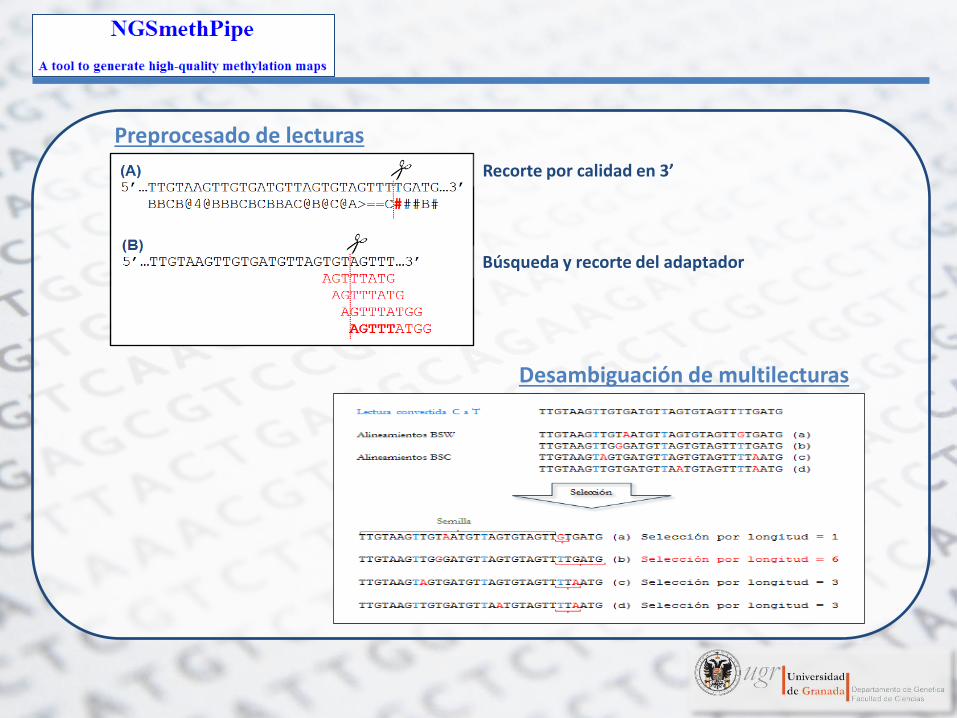
Preprocesado de lecturas
Recorte por calidad en 3’
Búsqueda y recorte del adaptador
Desambiguación de multilecturas
Barturen, G., A. Rueda, J. L. Oliver and M. Hackenberg (2013). "MethylExtract: High-Quality methylation maps and SNV calling from whole genome bisulfite sequencing data."
F1000Res 2(217): 217.
Inferencia de los niveles de metilación
Detección de variantes de secuencia Control de la calidad de secuenciación

Detección de variantes de secuencia
Detección de variantes Estimación errónea de la metilación
TCGA TTGA MUESTRA
BISULFITO
TTGA
TCGA
CITOSINA NO METILADA
TIMINA
REFERENCIA
¿CITOSINA NO METILADA?

Detección de variantes Estimación errónea de la metilación
TCGA TTGA MUESTRA
BISULFITO
TTGA
TCGA
CITOSINA NO METILADA
TIMINA
REFERENCIA
Guanina en la hebra complementaria
Detección de variantes de secuencia

Detección de variantes Estimación errónea de la metilación
TCGA TTGA MUESTRA
BISULFITO
TTGA
TCGA
CITOSINA NO METILADA
TIMINA
REFERENCIA
Adenina en la hebra complementaria
Detección de variantes de secuencia
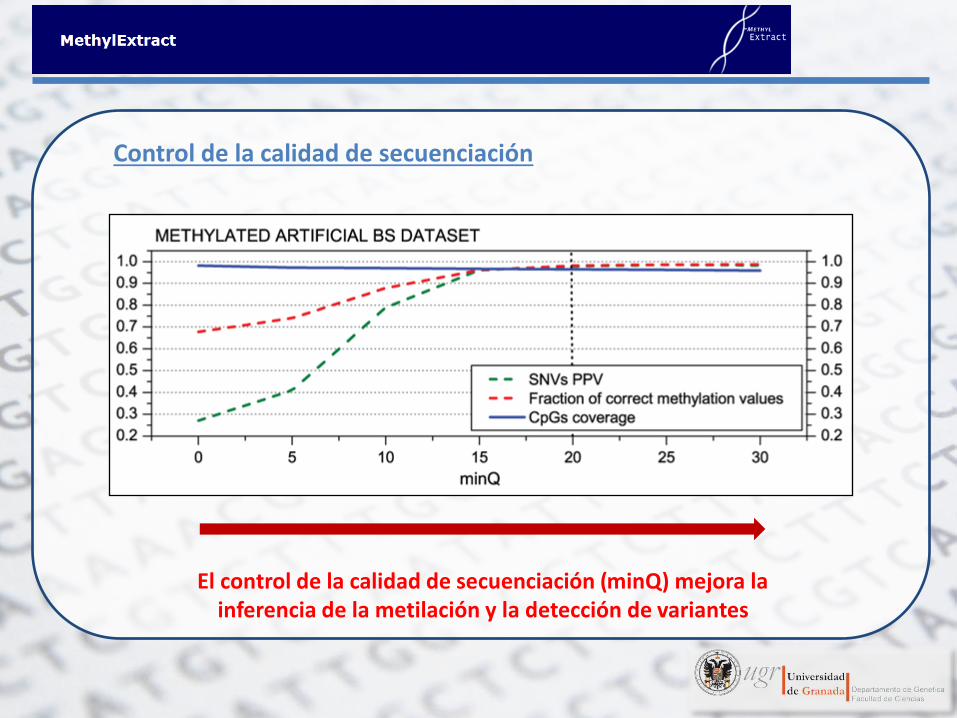
Control de la calidad de secuenciación
El control de la calidad de secuenciación (minQ) mejora la inferencia de la metilación y la detección de variantes

IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
BLOQUE II

Hackenberg, M., P. Carpena, P. Bernaola-Galvan, G. Barturen, A. M. Alganza and J. L. Oliver (2011). "WordCluster: detecting clusters of DNA words and genomic elements."
Algorithms Mol Biol 6: 2.
Mejora del algoritmo CpGcluster
Cálculo de la intersección genómica

Cálculo de la intersección genómica
Hackenberg et al. 2006 (BMC Bioinformatics)
Distribución de las distancias entre CpGs adyacentes

Comparación entre predicciones
Mayor solapamiento

Comparación entre predicciones
Menor Solapamiento

Hackenberg, M., G. Barturen, P. Carpena, P. L. Luque-Escamilla, C. Previti and J. L. Oliver (2010). "Prediction of CpG-island function: CpG clustering vs. sliding-window methods."
BMC Genomics 11: 327.
Comparación entre algoritmos de predicción de CGIs

Algoritmos de ventana Algoritmos de adición Algoritmos de agrupación (Clustering)
Algoritmos de predicción de CGIs

Especificidad funcional
Takai & Jones
CpGcluster
UCSC
TSSs
TFBSs
Las CGIs predichas por CpGcluster se asocian con mayor especificidad a elementos funcionales del genoma

Dominios de metilación homogéneos
Las CGIs predichas por CpGcluster muestran niveles de metilación más
homogéneos
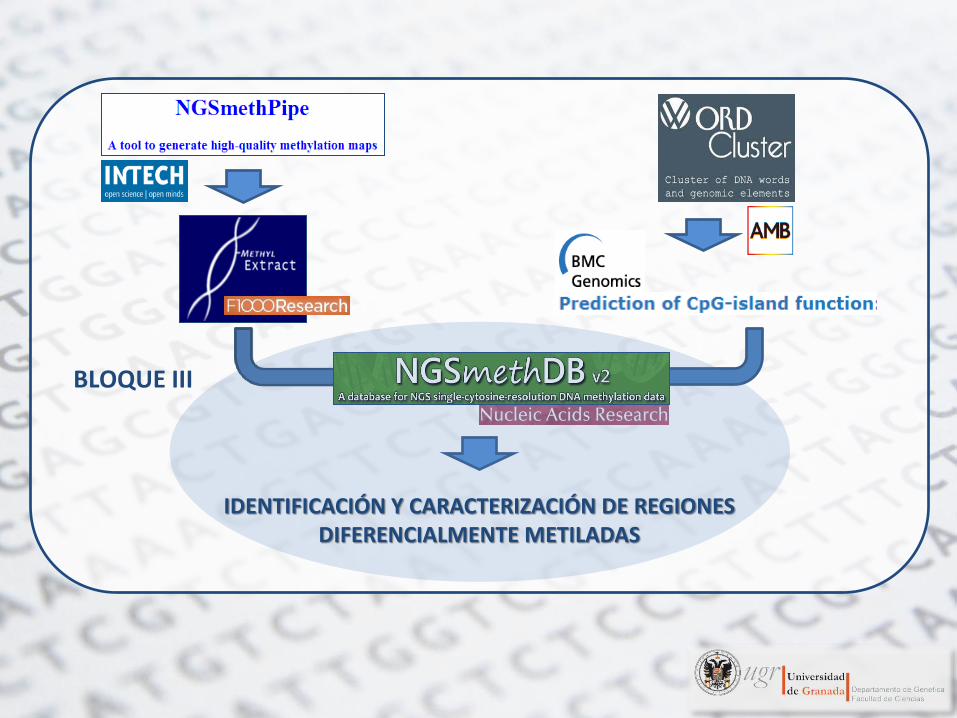
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
BLOQUE III

Hackenberg, M., G. Barturen and J. L. Oliver (2011). "NGSmethDB: a database for next-generation sequencing single-cytosine-resolution DNA methylation data." Nucleic Acids Res 39(Database issue): D75-79.
Geisen, S., G. Barturen, A. M. Alganza, M. Hackenberg and J. L. Oliver (2014). "NGSmethDB: an
updated genome resource for high quality, single-cytosine resolution methylomes." Nucleic Acids Res
42(1): D53-59.
Almacenamiento de niveles de metilación para múltiples tejidos Desarrollo de herramientas para visualizarlos y compararlos

Diseño y contenido de NGSmethDB
40 TB FASTQ
Procesados de manera uniforme
Humano – Chimpancé – Macaco – Ratón - Arabidopsis
Tomate (Niveles de metilación y variaciones de secuencia)
114 tejidos y/o condiciones diferentes
Minería de datos

IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
COMPARACIÓN DE MÉTODOS ESTADÍSTICOS
CLASIFICACIÓN DE ISLAS
ANÁLISIS FUNCIONAL
DISTRIBUCIÓN EN REGIONES GENÓMICAS
NIVELES DE METILACIÓN EN LAS CGIs
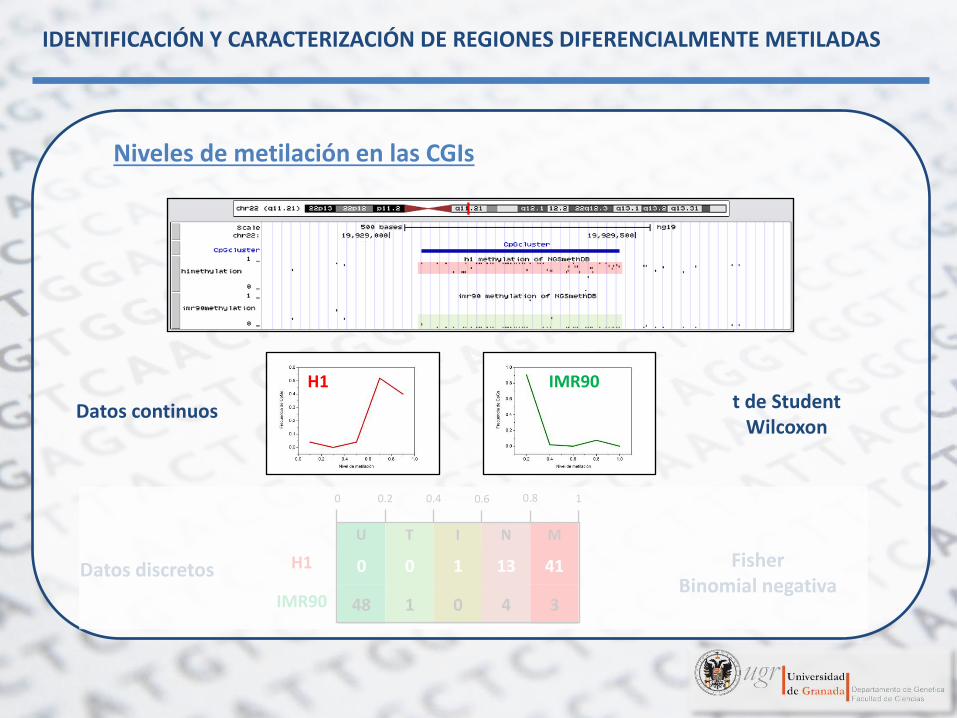
Niveles de metilación en las CGIs
H1 IMR90
U T N I M
0 0.2 0.4 0.6 0.8 1
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
H1 0 0 1 13 41
IMR90 48 1 0 4 3
Datos continuos
Datos discretos
t de Student Wilcoxon
Fisher Binomial negativa

Niveles de metilación en las CGIs
H1 IMR90
U T N I M
0 0.2 0.4 0.6 0.8 1
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
H1 0 0 1 13 41
IMR90 48 1 0 4 3
Datos continuos
Datos discretos
t de Student Wilcoxon
Fisher Binomial negativa

Comparación de métodos estadísticos
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
Diferencias de metilación entre
tejidos
Falsos positivos
Discretos Continuos Combinados
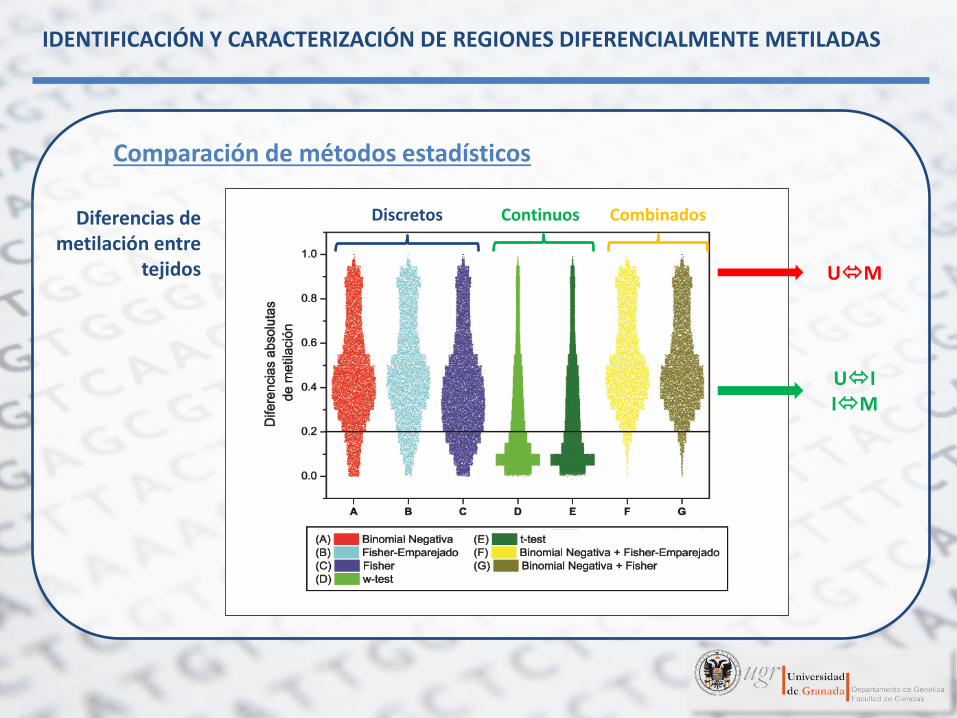
Comparación de métodos estadísticos
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
Diferencias de metilación entre
tejidos
Discretos Continuos Combinados
UM
UI IM

Comparación de métodos estadísticos
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
Diferencias de metilación entre
tejidos
Binomial Negativa +
Fisher

Clasificación de CGIs
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
Clases de CGIs en función de su metilación en múltiples tejidos
• Células madre (H1 y H9) • Células mononucleares sanguíneas • Células B • Células CD133 + • Células madre hematopoyéticas • Fibroblastos (pulmón y prepucio) • Córtex pre-frontal • Células epiteliales mamarias • Espermatozoides
UIs: en todos los tejidos U o T MIs: en todos los tejidos M o N DMIs: diferencias estadísticas en un par de tejidos NA: resto

Clasificación de CGIs
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
Tipos de DMIs
DMIs-M (mayoritariamente
metiladas)
DMIs-U (mayoritariamente no
metiladas)

Distribución en regiones genómicas
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
Enriquecimiento en elementos
génicos
Aleatorización valores p ≤ 0.01
𝑬𝒏𝒓𝒊𝒒𝒖𝒆𝒄𝒊𝒎𝒊𝒆𝒏𝒕𝒐 (𝒓) = 𝑭𝒓𝒂𝒄𝒄𝒊ó𝒏 𝒅𝒆 𝑪𝑮𝑰𝒔 𝒅𝒆𝒏𝒕𝒓𝒐 𝒅𝒆𝒍 𝒆𝒍𝒆𝒎𝒆𝒏𝒕𝒐
𝑭𝒓𝒂𝒄𝒄𝒊ó𝒏 𝒅𝒆 𝑪𝑮𝑰𝒔 𝒇𝒖𝒆𝒓𝒂 𝒅𝒆𝒍 𝒆𝒍𝒆𝒎𝒆𝒏𝒕𝒐
r>1 enriquecimiento r<1 empobrecimiento

Distribución en regiones genómicas
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
Enriquecimiento en elementos
génicos
Mayor enriquecimiento de UIs y DMIs-U en promotores
Aleatorización valores p ≤ 0.01

Distribución en regiones genómicas
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
Enriquecimiento en elementos
génicos
Elevado enriquecimiento de todas las clases en exones
Aleatorización valores p ≤ 0.01
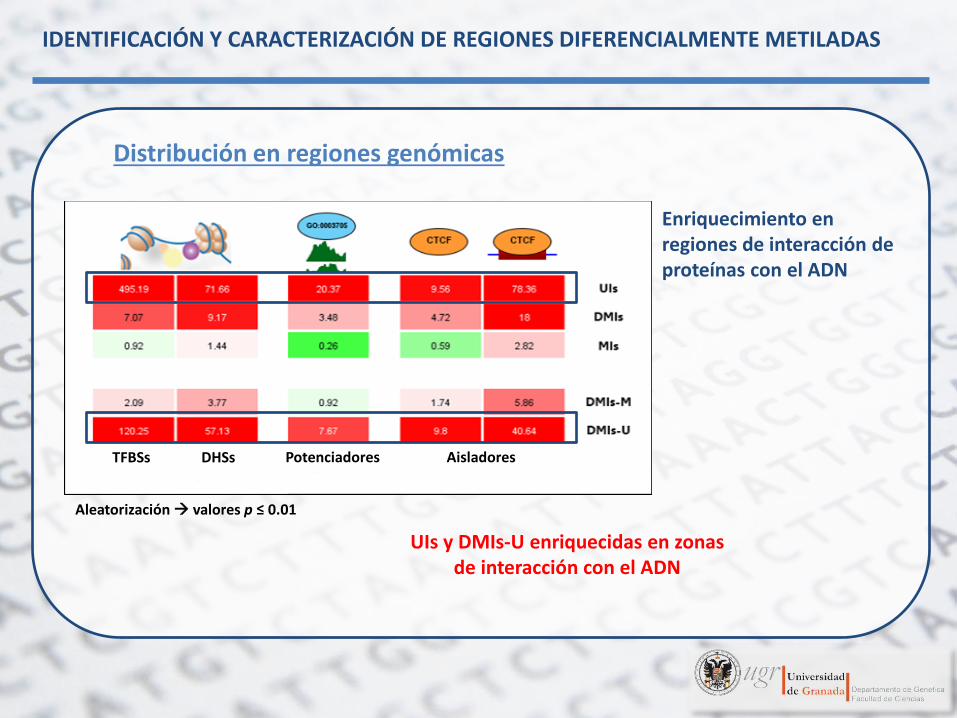
Distribución en regiones genómicas
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
Enriquecimiento en regiones de interacción de proteínas con el ADN
UIs y DMIs-U enriquecidas en zonas de interacción con el ADN
Aleatorización valores p ≤ 0.01
TFBSs DHSs Potenciadores Aisladores
Distribución en regiones genómicas
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
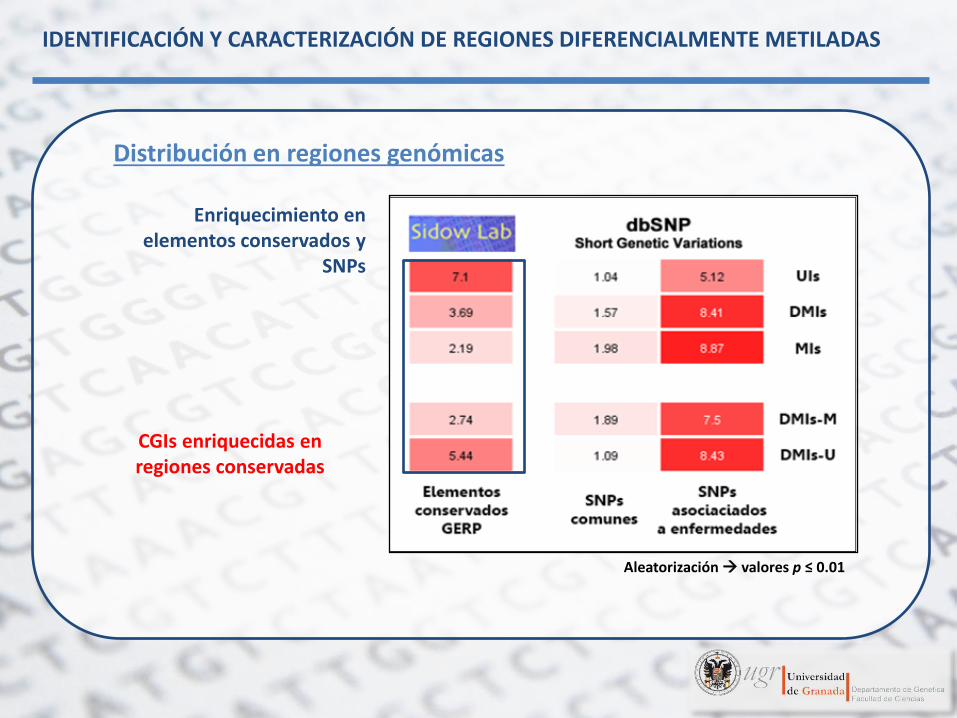
Enriquecimiento en elementos conservados y
SNPs
CGIs enriquecidas en regiones conservadas
Aleatorización valores p ≤ 0.01

Distribución en regiones genómicas
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
Enriquecimiento en elementos conservados y
SNPs
CGIs enriquecidas en SNPs asociados a
enfermedades
Aleatorización valores p ≤ 0.01

Análisis funcional (términos de ontologías)
IDENTIFICACIÓN Y CARACTERIZACIÓN DE REGIONES DIFERENCIALMENTE METILADAS
FUNCIONES ASOCIADAS A LOS TIPOS DE CGIs
• UIs Funciones básicas celulares (genes domésticos)
• DMIs-U Procesos de desarrollo y diferenciación
• DMIs-M Procesos involucrados en la reproducción
• MIs Actividad de receptores olfativos

CONCLUSIONES
1. En esta Tesis Doctoral se ha desarrollado un procedimiento para identificar regiones genómicas diferencialmente metiladas, que puedan servir como marcadores epigenéticos. Esto ha supuesto el desarrollo y la implementación de: 1) dos herramientas bioinformáticas para la obtención de metilomas de alta calidad en genoma completo; 2) una base de datos para el almacenamiento, visualización y gestión de los metilomas generados; 3) un algoritmo capaz de identificar islas CpG estadísticamente significativas; y 4) un método estadístico fiable para la identificación de islas diferencialmente metiladas, a partir del análisis comparado de múltiples metilomas.
2. NGSmethPipe, el primer programa desarrollado en esta Tesis, permite pre-procesar y alinear las lecturas cortas procedentes de protocolos de secuenciación masiva de ADN tratado con bisulfito. Los controles de calidad implementados consiguen una alta proporción de lecturas correctamente alineadas, sin aumentar en exceso los alineamientos erróneos. Esto, unido a la paralelización de todo el proceso, convierten a NGSmethPipe en una herramienta eficaz para alinear este tipo de librerías.

CONCLUSIONES
3. Por su parte, MethylExtract permite inferir simultáneamente los niveles de metilación y las variantes de un solo nucleótido a partir de una misma librería. Asimismo, este programa implementa estrictos controles para minimizar los errores de secuenciación y los fallos del tratamiento con bisulfito. Esto lo convierte en una herramienta fiable y versátil para la obtención de metilomas de alta resolución en genoma completo.
4. NGSmethDB es una base de datos relacional de metilomas completos, todos ellos procesados mediante el mismo protocolo, lo que permite comparar metilomas procedentes de diferentes estudios. Las herramientas de minería de datos implementadas y el navegador genómico de última generación que incorpora, han sido diseñados para realizar consultas complejas sobre múltiples tejidos en cualquier región del genoma. Actualmente, la base de datos contiene información para 6 especies y 114 tejidos y/o condiciones diferentes, lo que ha supuesto procesar aproximadamente 40 terabytes de librerías de secuenciación masiva. Estas características convierten a NGSmethDB en una herramienta muy potente para el análisis de metilomas de alta resolución.

CONCLUSIONES
5. Sobre la base de CpGcluster, se ha desarrollado WordCluster, un nuevo algoritmo capaz de identificar islas CpG estadísticamente significativas y con una alta densidad de CpGs. WordCluster presenta importantes ventajas con respecto a los métodos convencionales, destacando entre ellas la delimitación de dominios de metilación más cortos pero también más homogéneos, así como una asociación más específica con elementos reguladores y regiones evolutivamente conservadas del genoma.
6. El análisis de la metilación en las islas predichas por WordCluster ha permitido establecer que la combinación de la prueba exacta de Fisher y la binomial negativa es el método estadístico más apropiado para identificar islas diferencialmente metiladas.

CONCLUSIONES
7. Atendiendo al estado de metilación de los tejidos analizados, el 66% de las islas diferencialmente metiladas pueden ser de dos tipos: las que están generalmente metiladas pero aparecen no-metiladas en algún tejido (islas DMI-M), y aquellas que permanecen generalmente no-metiladas pero pueden metilarse en algún tejido (islas DMI-U).
8. El análisis funcional mediante el enriquecimiento en términos de ontologías ha permitido establecer que las islas de tipo DMI-M se asocian con funciones tejido-específicas, mientras que las islas de tipo DMI-U están implicadas en procesos de desarrollo y diferenciación.

CONCLUSIONES
9. Los estudios de enriquecimiento de las islas diferencialmente metiladas en distintos elementos genómicos han revelado importantes diferencias con respecto a las regiones diferencialmente metiladas, identificadas recientemente por otros autores. Entre estas diferencias destacan el elevado enriquecimiento de las islas diferencialmente metiladas en promotores y exones y su empobrecimiento en intrones, así como la significativa menor proporción de las islas de tipo DMI-M asociadas con sitios de unión a factores de transcripción. A su vez, es de destacar que las islas de tipo DMI-U solapan en mucho mayor grado con los sitios de unión a factores de transcripción que las islas de tipo DMI-M.
10.Las importantes características biológicas encontradas en las islas diferencialmente metiladas sugieren que WordCluster puede ser el algoritmo adecuado para preseleccionar las regiones a analizar en futuros estudios de metilación diferencial.

MUCHAS GRACIAS
Grupo de Genómica Computacional & Bioinformática
José L. Oliver Michael Hackenberg
Ángel Martín Alganza Ricardo Lebrón Francisco Dios Cristina Gómez
Ernesto Aparicio
Antonio Rueda (Plataforma Andaluza de Genómica y Bioinformática)
Stefanie Geisen Maarten Hamberg
(Saarland University)