
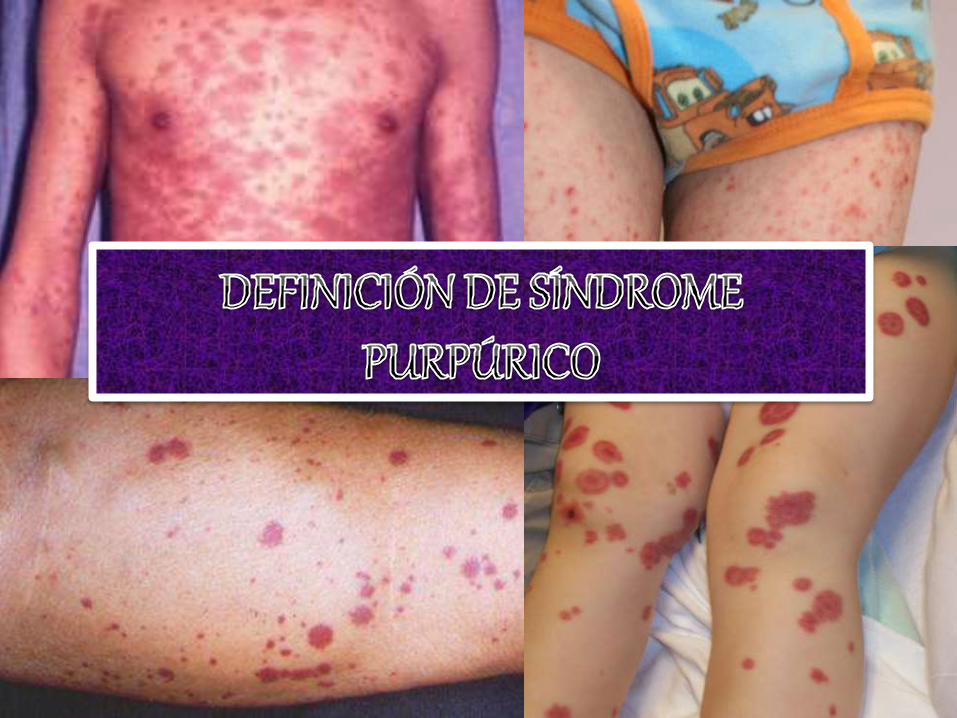
Purpuras jc guay
69
-
Upload
juan-guay -
Category
Health & Medicine
-
view
22 -
download
1
Transcript of Purpuras jc guay


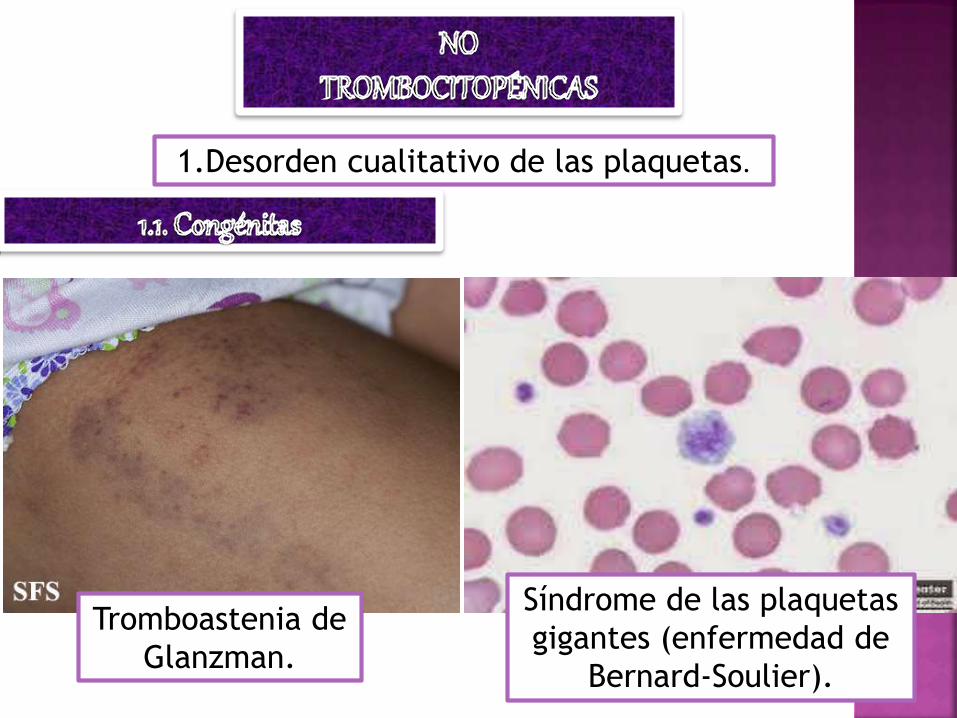
1.Desorden cualitativo de las plaquetas.
Tromboastenia de
Glanzman.
Síndrome de las plaquetas
gigantes (enfermedad de
Bernard-Soulier).
Insuficiencia renal
Aguda o crónica.
Enfermedad
hepática.
Consumo de
aspirina.
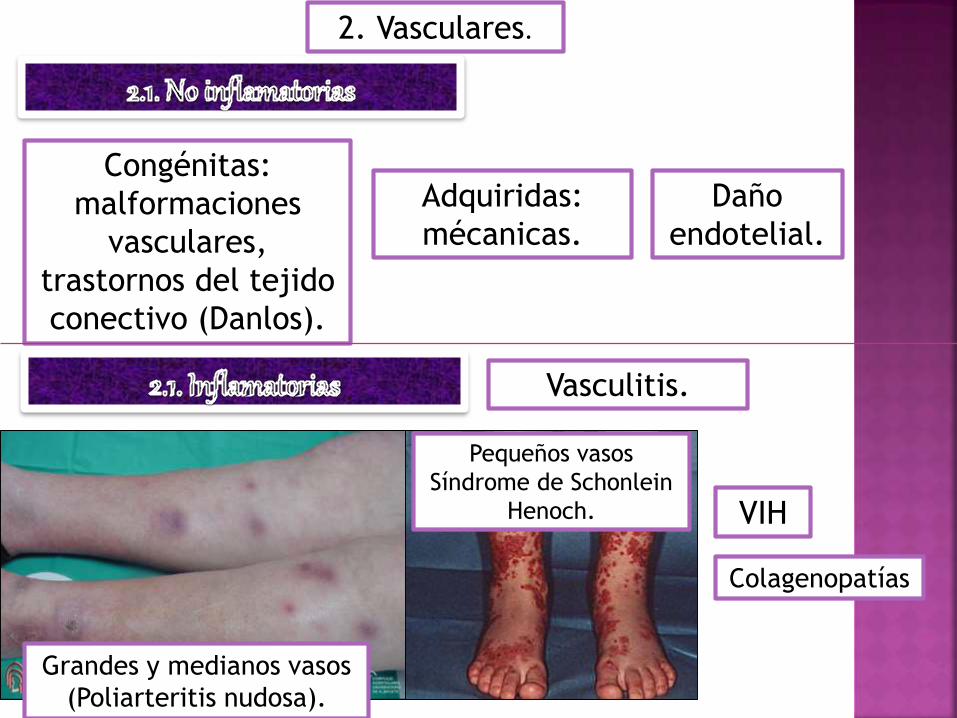
2. Vasculares.
Congénitas:
malformaciones
vasculares,
trastornos del tejido
conectivo (Danlos).
Adquiridas:
mécanicas.
Daño
endotelial.
Vasculitis.
Grandes y medianos vasos
(Poliarteritis nudosa).
Pequeños vasos
Síndrome de Schonlein
Henoch. VIH
Colagenopatías
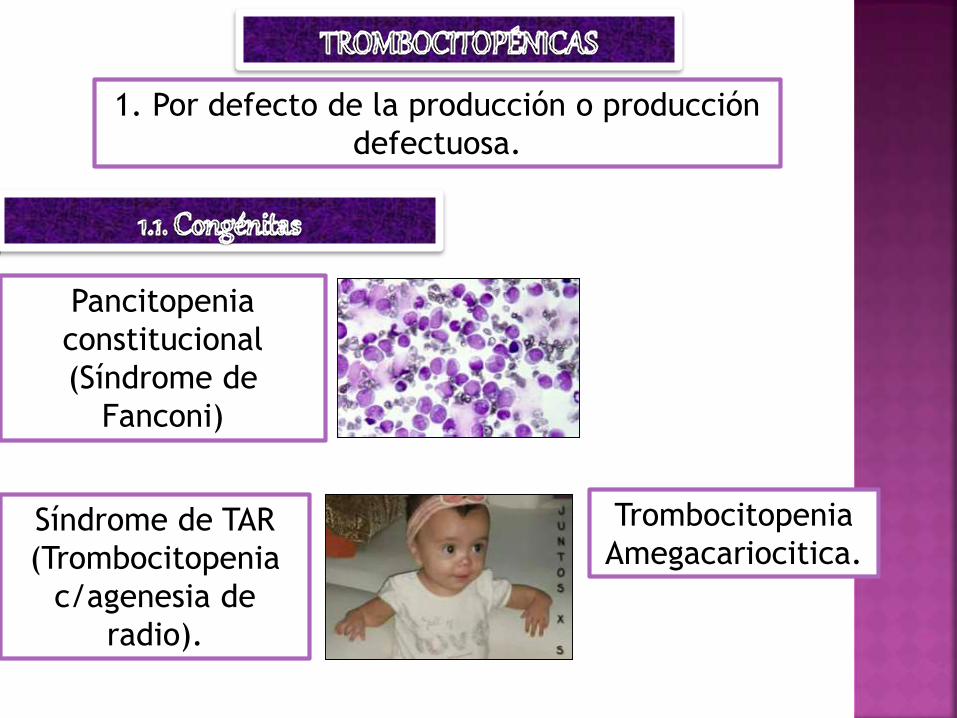
1. Por defecto de la producción o producción
defectuosa.
Pancitopenia
constitucional
(Síndrome de
Fanconi)
Trombocitopenia
Amegacariocitica.Síndrome de TAR
(Trombocitopenia
c/agenesia de
radio).
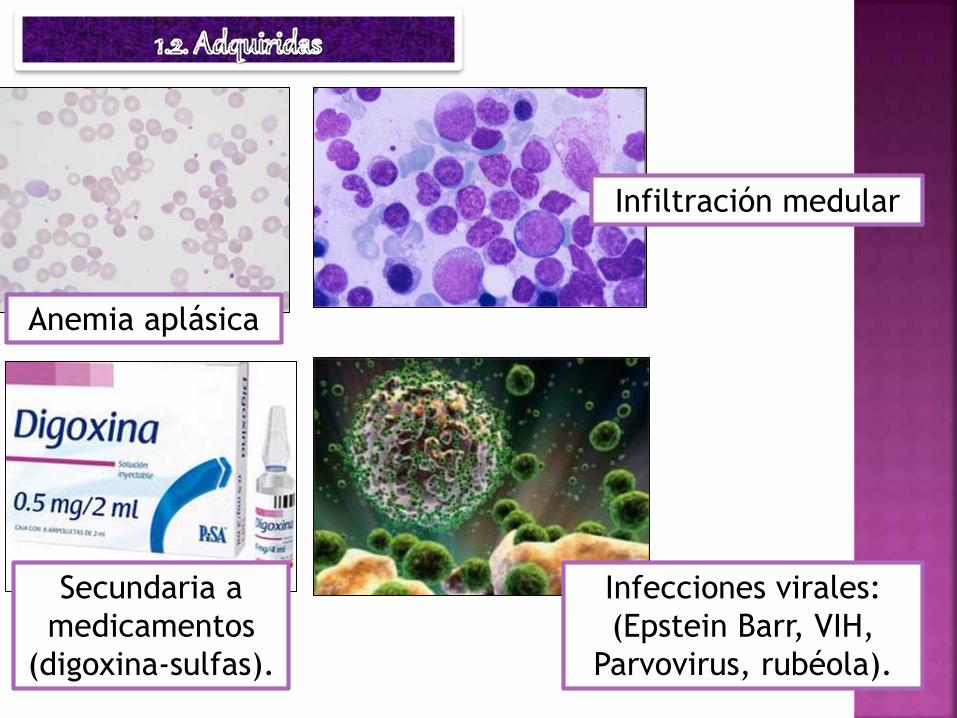
Anemia aplásica
Secundaria a
medicamentos
(digoxina-sulfas).
Infiltración medular
Infecciones virales:
(Epstein Barr, VIH,
Parvovirus, rubéola).
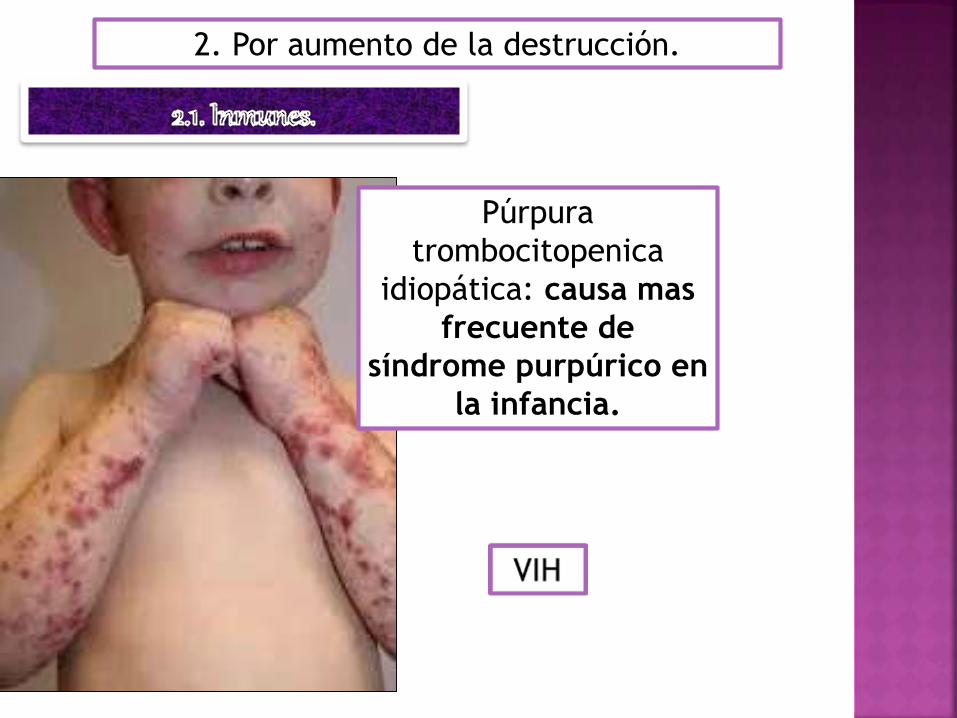
2. Por aumento de la destrucción.
Púrpura
trombocitopenica
idiopática: causa mas
frecuente de
síndrome purpúrico en
la infancia.
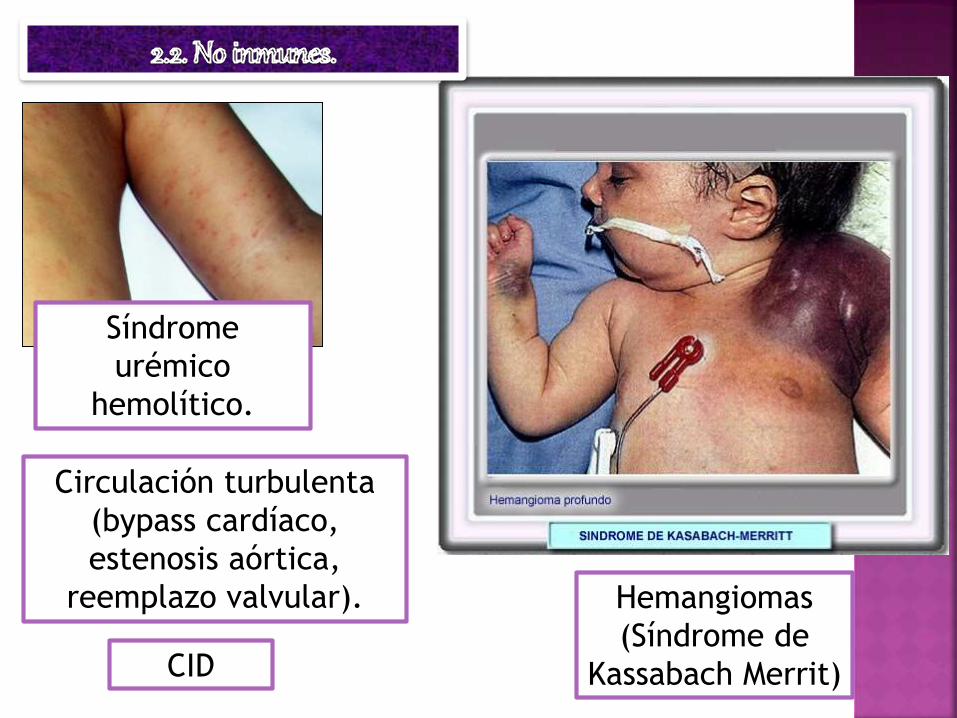
Hemangiomas
(Síndrome de
Kassabach Merrit)
Circulación turbulenta
(bypass cardíaco,
estenosis aórtica,
reemplazo valvular).
CID
Síndrome
urémico
hemolítico.
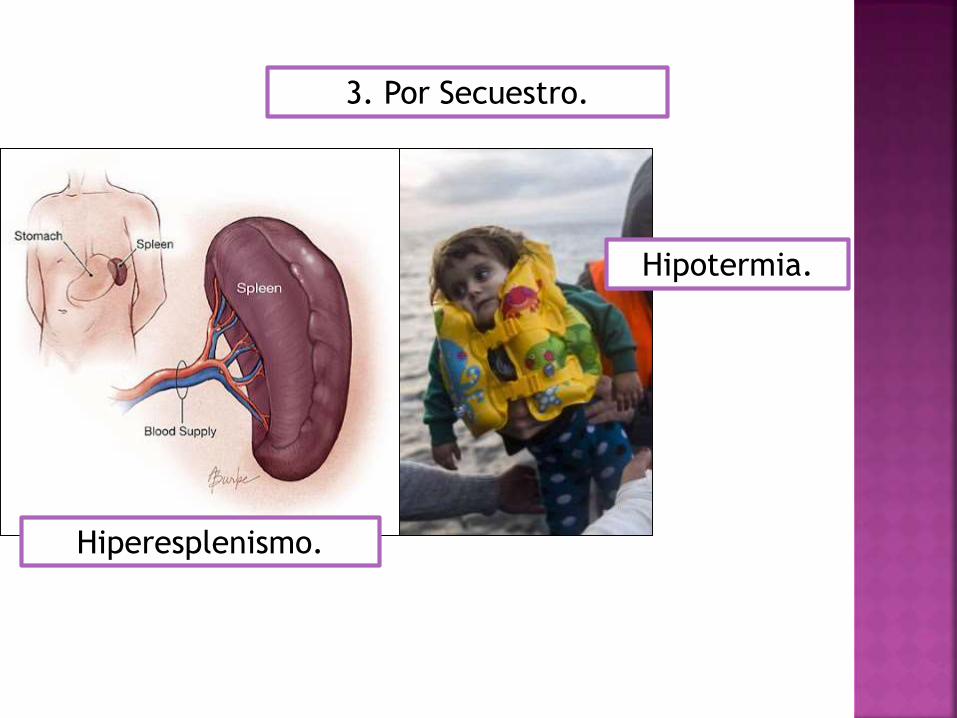
3. Por Secuestro.
Hiperesplenismo.
Hipotermia.

Recuento plaquetario menor de
100,000/mm3.
Ausencia de enfermedad
infecciosa aguda intercurrente.*
Ausencia de enfermedad de
base.*
Megacariocitos normales o
aumentados en la PAMO.*

Hipótesis:
Infección viral 1-3 semanas antes.
Verdadera enfermedad autoinmune con producción deautoanticuerpos (IgG).
Se han descrito casos de PTI tras infección por virusEB, VZ, CMV, rubeola, hepatitis, parvovirus e inclusotras administración de vacunas con virus vivosatenuados.
Desconocida.

Los factores que inician la producción de
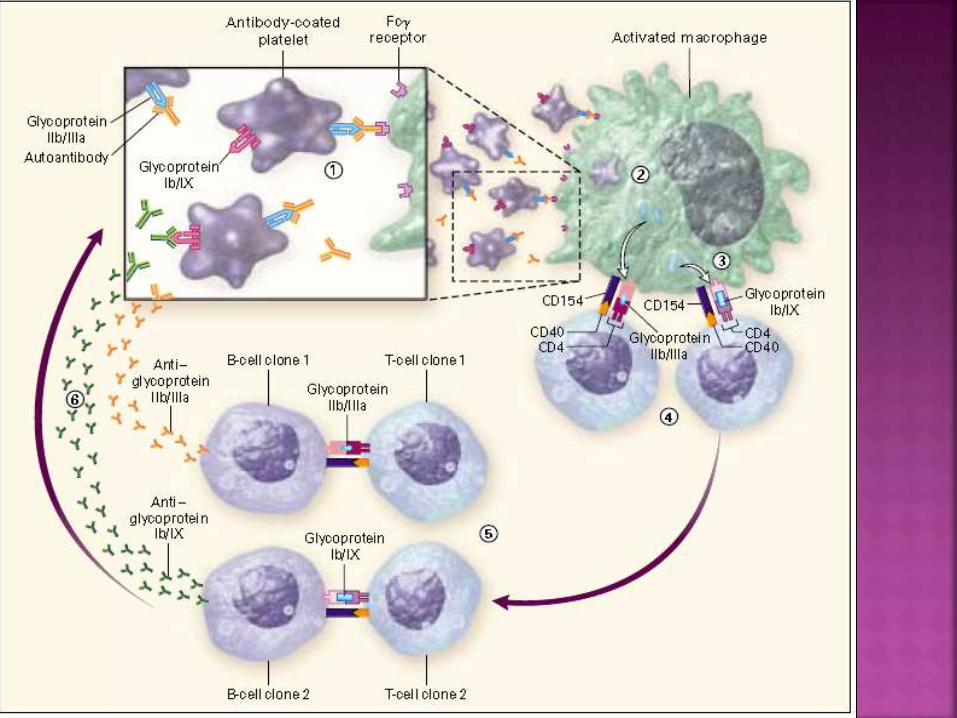
autoanticuerpos es desconocida.
Las plaquetas recubiertas de Ac (tipo IgG) se
unen a los macrófagos a través de sus
receptores Fcy, siendo internalizadas y
degradadas.
La presentación de epítopes derivados de GP
IIB/IIIa amplifican la respuesta inmune.

PTI de reciente
diagnostico.
PTI prolongada.
PTI crónica.PTI
recidivante
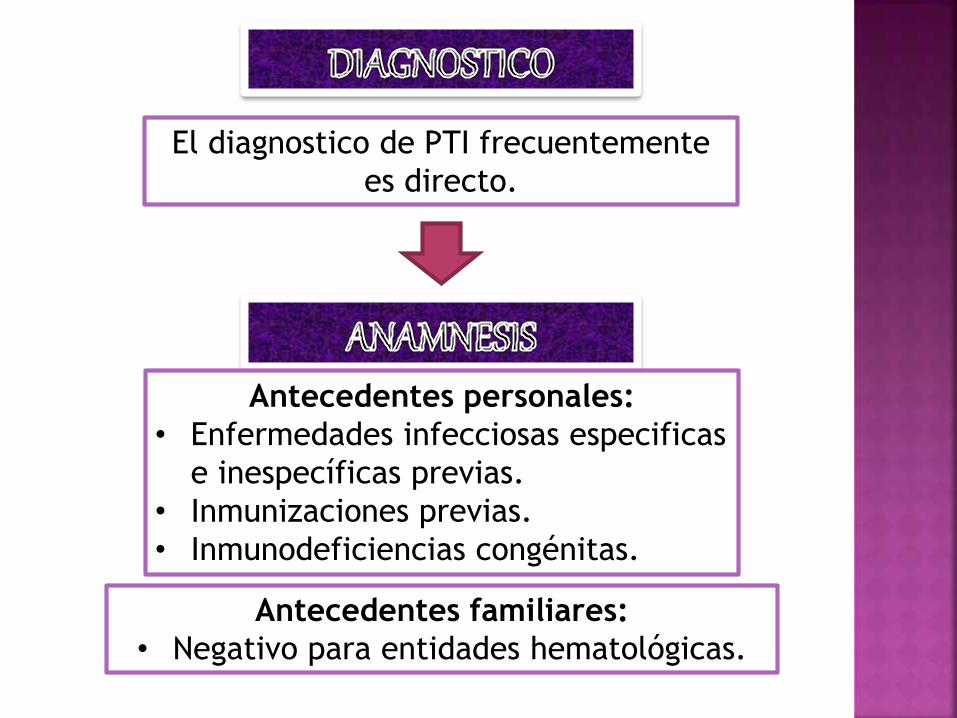
El diagnostico de PTI frecuentemente
es directo.
Antecedentes personales:
• Enfermedades infecciosas especificas
e inespecíficas previas.
• Inmunizaciones previas.
• Inmunodeficiencias congénitas.
Antecedentes familiares:
• Negativo para entidades hematológicas.
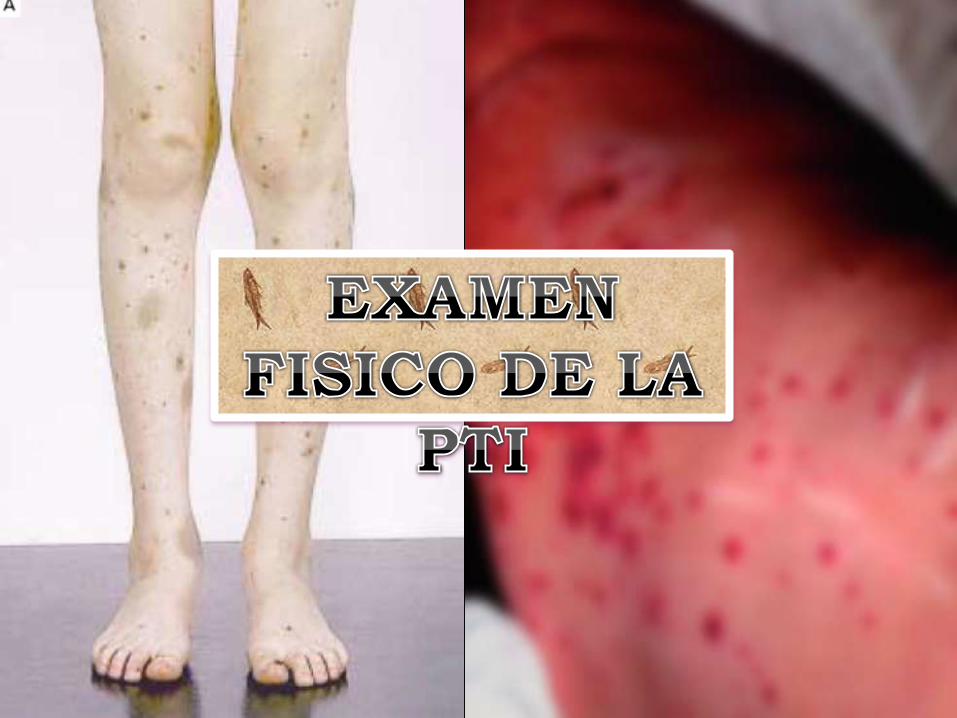
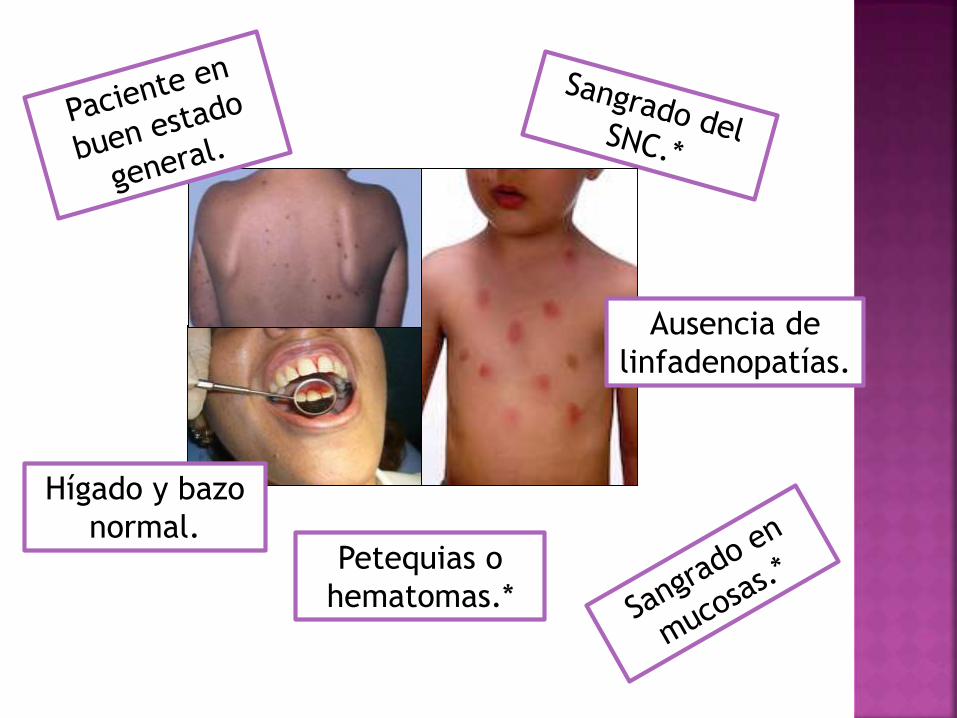
Petequias o
hematomas.*
Hígado y bazo
normal.
Ausencia de
linfadenopatías.
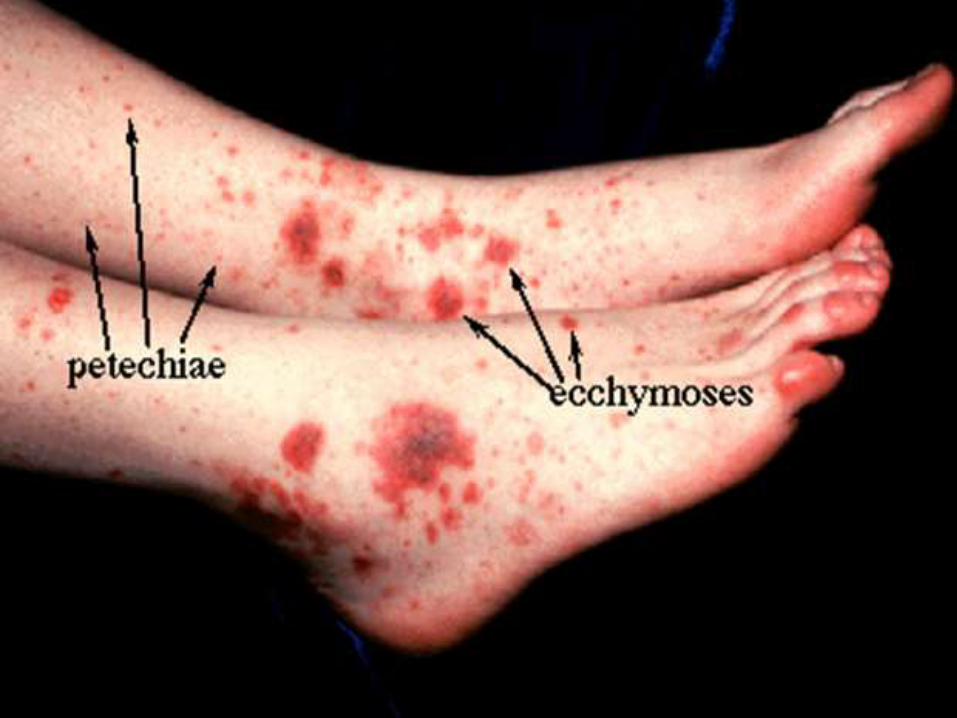

REQUISITOS para el diagnostico…
Sx purpúrico con trombocitopenia
menor a 100,000/ mm3.
Ausencia de enfermedad
infecciosa aguda concomitante.
Ausencia de patología de
base.
Megacariocitos normales o
aumentados en medula ósea.
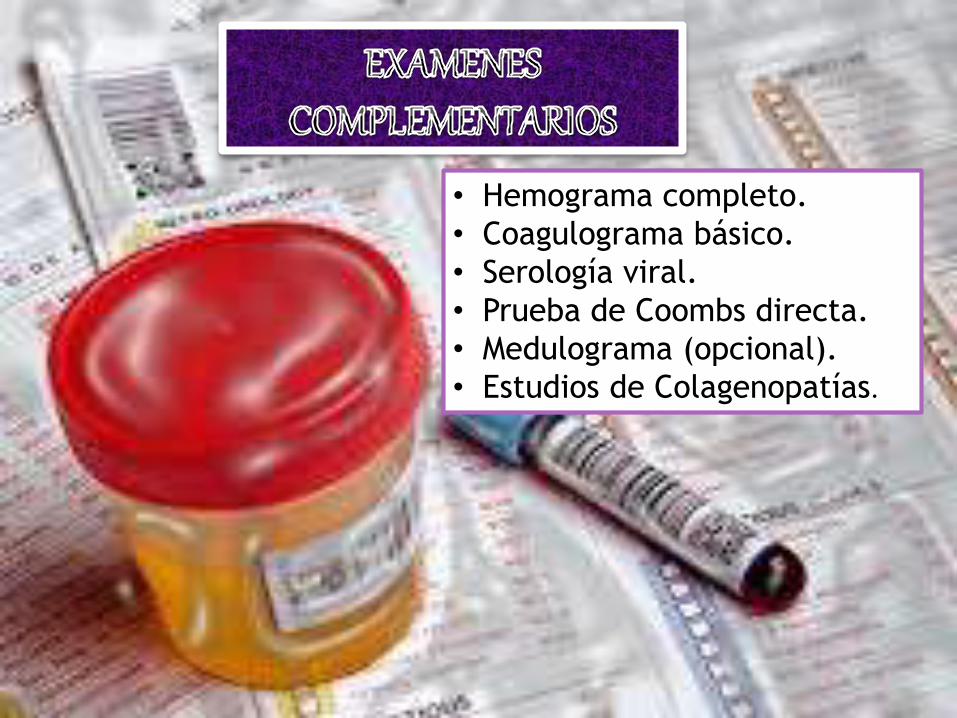
• Hemograma completo.
• Coagulograma básico.
• Serología viral.
• Prueba de Coombs directa.
• Medulograma (opcional).
• Estudios de Colagenopatías.
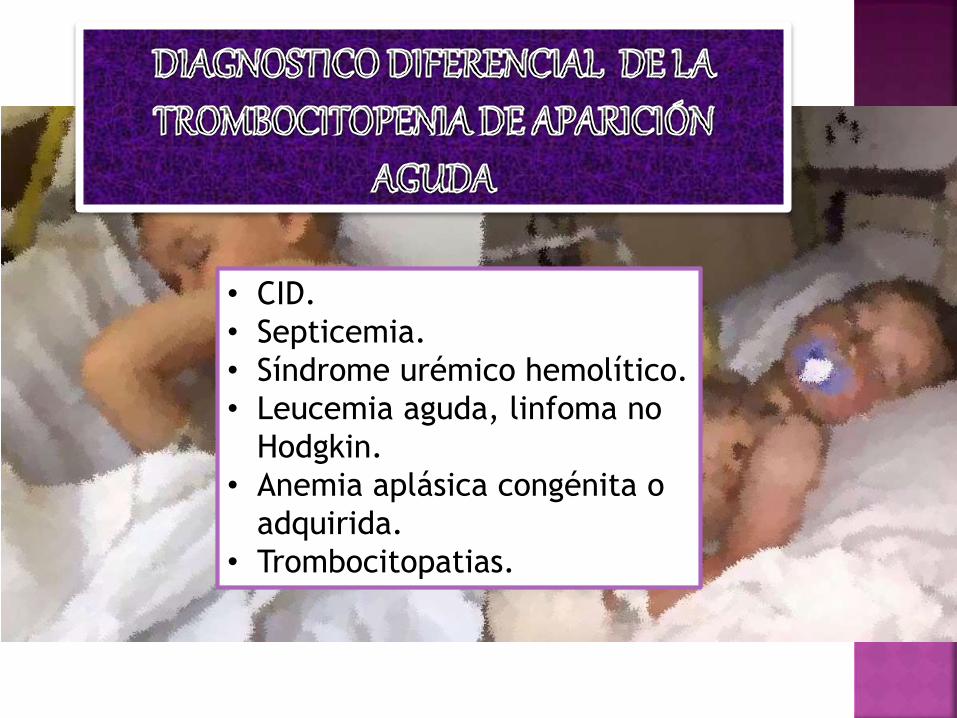
• CID.
• Septicemia.
• Síndrome urémico hemolítico.
• Leucemia aguda, linfoma no
Hodgkin.
• Anemia aplásica congénita o
adquirida.
• Trombocitopatias.

• Niños con manifestaciones de
sangrado húmedo.*
• Recuento de plaquetas menor
de 20,000/ mm3.*
• Niños menores de un año.
• Medio sociofamiliar bajo.
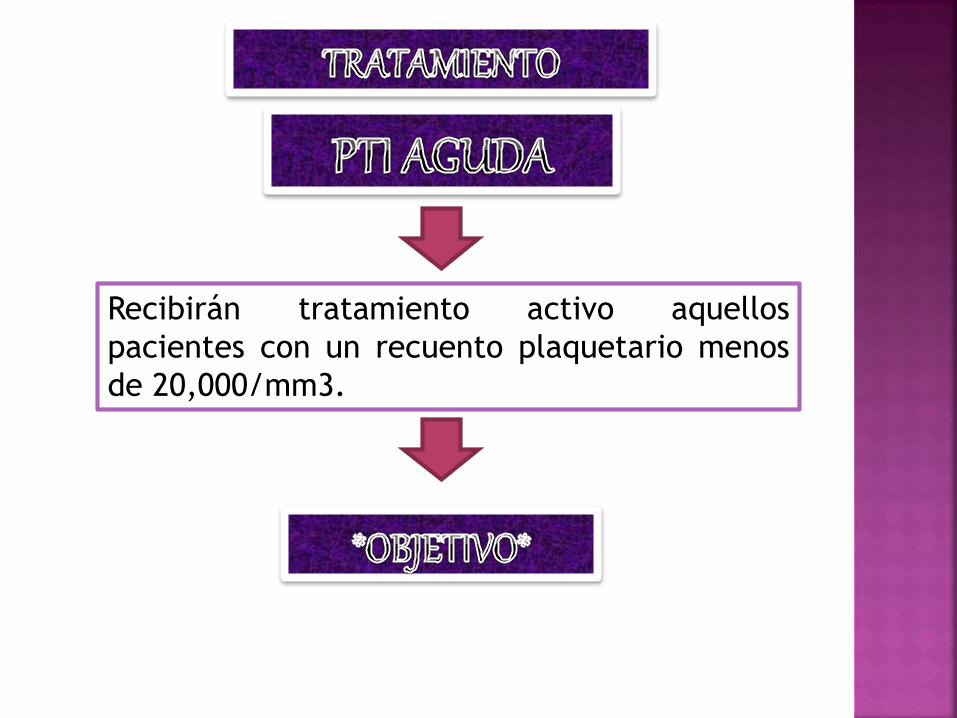
Recibirán tratamiento activo aquellos
pacientes con un recuento plaquetario menos
de 20,000/mm3.

La terapéutica será determinada…
IgG IV: 1g/kg/día por dos dias
consecutivos.*
IgG IV: 0.8g/kg, dosis única.*
Prednisona oral: 4 mg/kg/día por
cuatro días consecutivos.*
Prednisona oral: 1-2 mg/kg/día por un periodo de dos a tres semanas.*

Metilprednisolona IV: 30 mg/kg/día por un lapso
de dos a tres días consecutivos.*
Inmunoglobulina anti-D IV: dosis única de 50-75 ug/kg (solo en pacientes
Rh positivos). * Puede reducir la 0.5 a 2 g/dL
la HGB
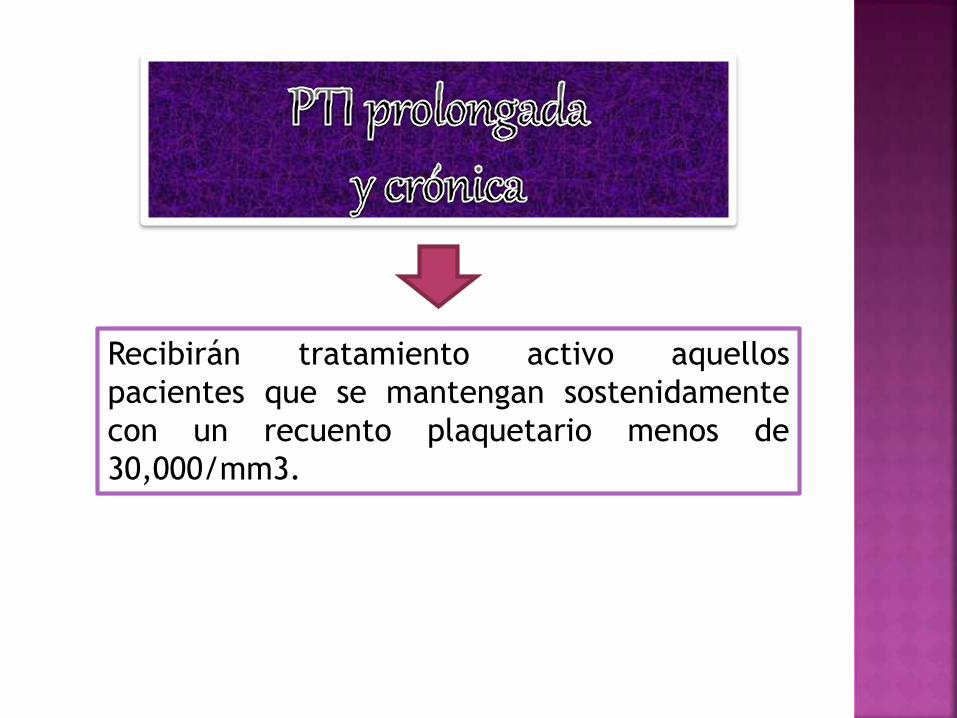
Recibirán tratamiento activo aquellos
pacientes que se mantengan sostenidamente
con un recuento plaquetario menos de
30,000/mm3.
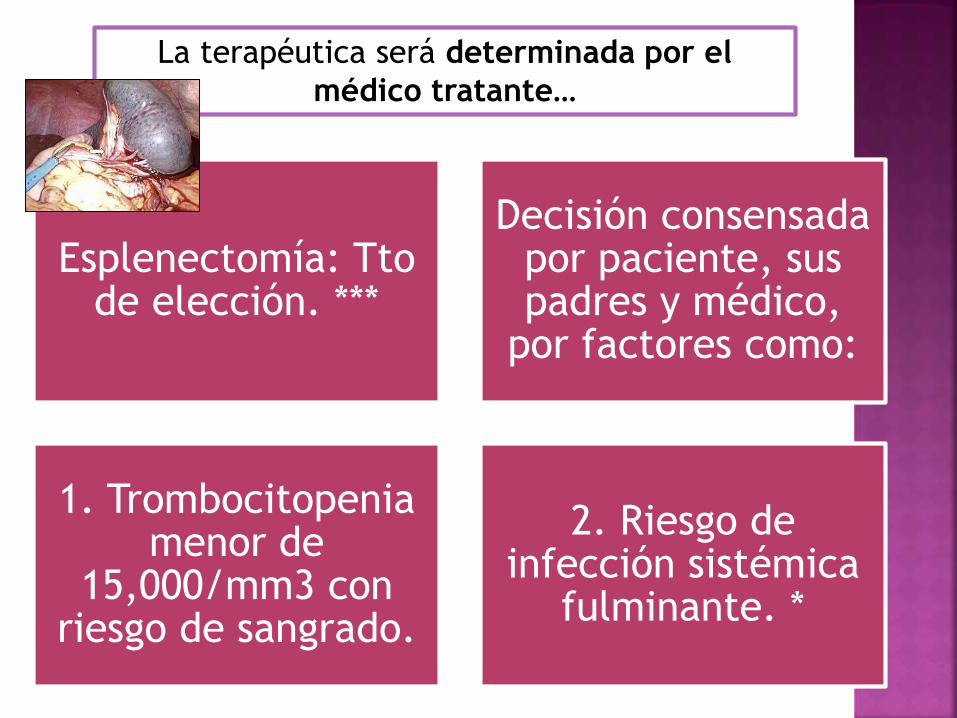
La terapéutica será determinada por el
médico tratante…
Esplenectomía: Tto de elección. ***
Decisión consensada por paciente, sus padres y médico,
por factores como:
1. Trombocitopenia menor de
15,000/mm3 con riesgo de sangrado.
2. Riesgo de infección sistémica
fulminante. *
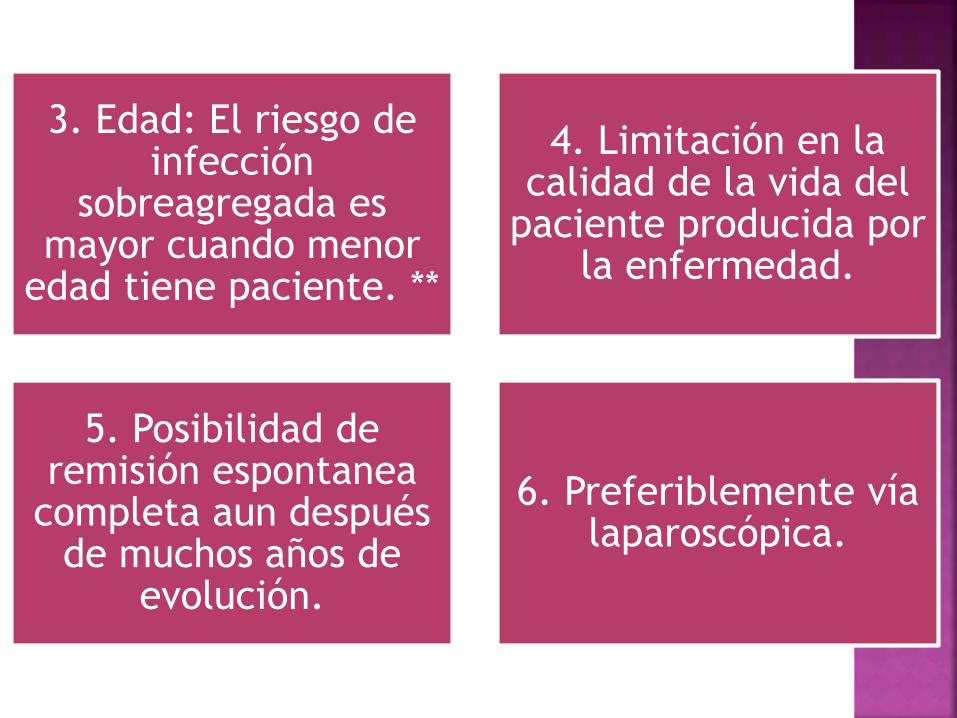
3. Edad: El riesgo de infección
sobreagregada es mayor cuando menor
edad tiene paciente. **
4. Limitación en la calidad de la vida del
paciente producida por la enfermedad.
5. Posibilidad de remisión espontanea
completa aun después de muchos años de
evolución.
6. Preferiblemente vía laparoscópica.

7. En pacientes con riesgo de sepsis menores de 6 años
postesplenectomía o la esplenectomía haya
fracasado rituximab: 375 mg/m2/dosis una vez por semana por 4 semanas.
Si la esplenectomía y el Rituximab han fracasado y
el paciente sigue con trombocitopenia menor de
15,000/mm3 se podrá realizar algunas opciones
terapéuticas.
1. Inmunoglobulina anti-D IV: 50-75 ug/kg, dosis única
(Solo Rh +).
2. IgG IV: 1 g/kg/día por dos días consecutivos o dosis
única de 0.8g/kg.

Pulsos periódicos de Corticosteroides: 30mg/kg/día de Metilprednisolona IV dos o
tres días consecutivos o 4mg/kg/día de prednisona oral por 4 dias consecutivas, 20-40 mg/m2/día de Dexametasona oral por 4 días consecutivos.
Si todas las medidas anteriores no dieron resultado y el
paciente tiene sangrado y plaquetas menores de
15,000/mm3. **
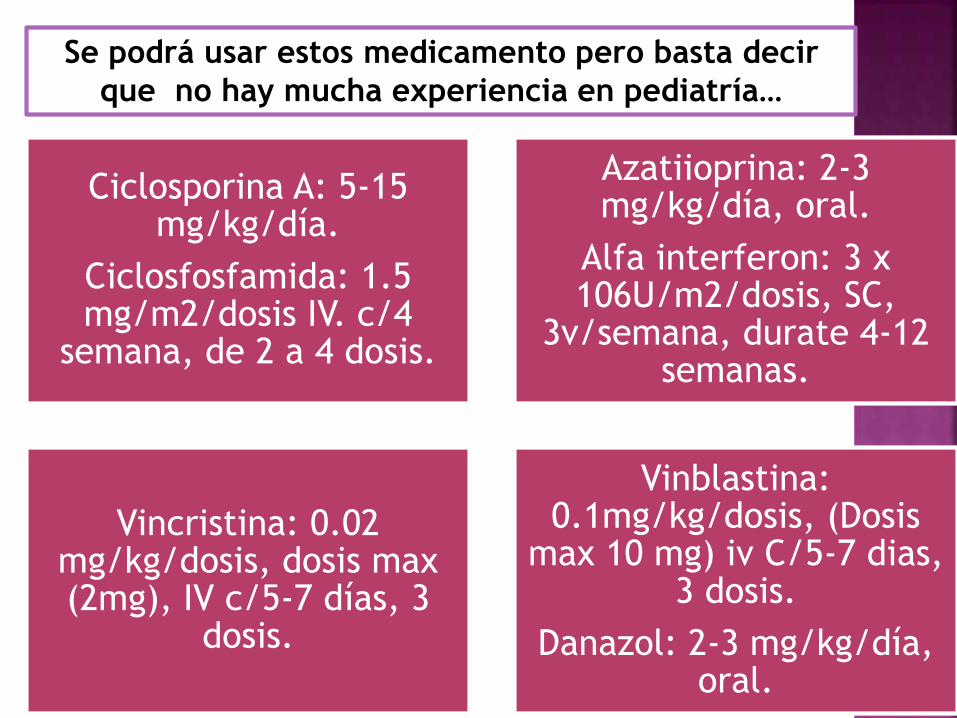
Se podrá usar estos medicamento pero basta decir
que no hay mucha experiencia en pediatría…
Ciclosporina A: 5-15 mg/kg/día.
Ciclosfosfamida: 1.5 mg/m2/dosis IV. c/4
semana, de 2 a 4 dosis.
Azatiioprina: 2-3 mg/kg/día, oral.
Alfa interferon: 3 x 106U/m2/dosis, SC,
3v/semana, durate 4-12 semanas.
Vincristina: 0.02 mg/kg/dosis, dosis max (2mg), IV c/5-7 días, 3
dosis.
Vinblastina: 0.1mg/kg/dosis, (Dosis
max 10 mg) iv C/5-7 dias, 3 dosis.
Danazol: 2-3 mg/kg/día, oral.
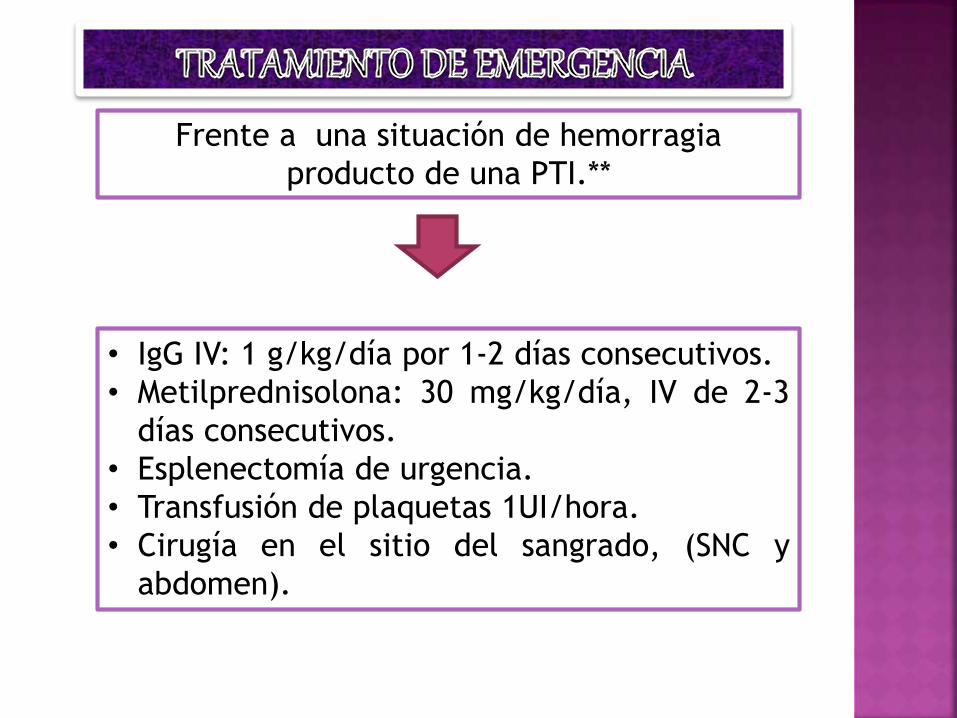
Frente a una situación de hemorragia
producto de una PTI.**
• IgG IV: 1 g/kg/día por 1-2 días consecutivos.
• Metilprednisolona: 30 mg/kg/día, IV de 2-3
días consecutivos.
• Esplenectomía de urgencia.
• Transfusión de plaquetas 1UI/hora.
• Cirugía en el sitio del sangrado, (SNC y
abdomen).
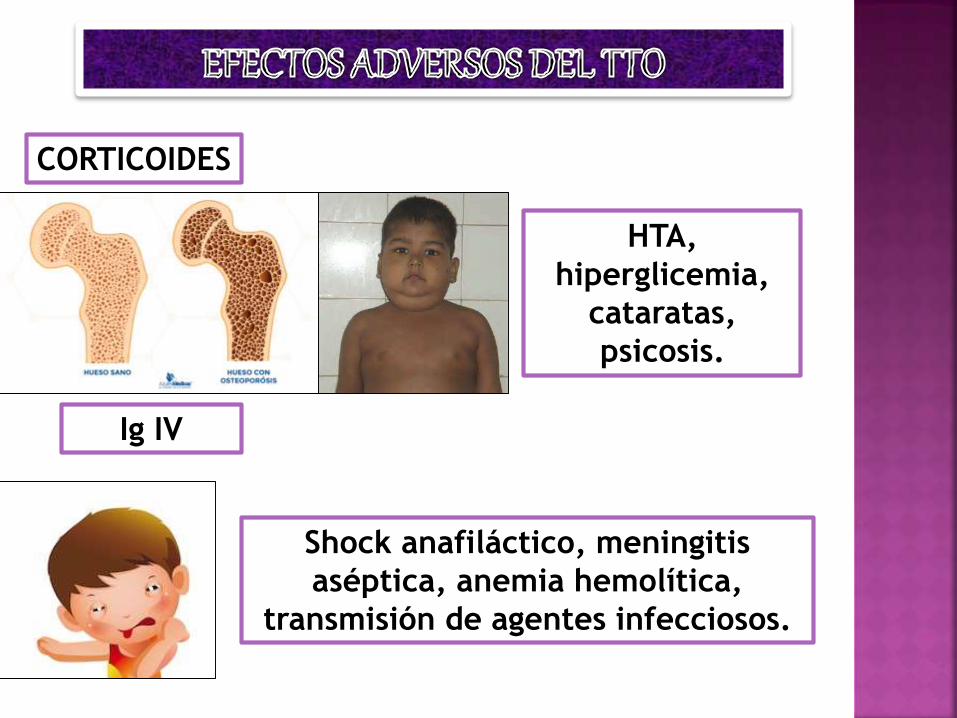
CORTICOIDES
HTA,
hiperglicemia,
cataratas,
psicosis.
Ig IV
Shock anafiláctico, meningitis
aséptica, anemia hemolítica,
transmisión de agentes infecciosos.
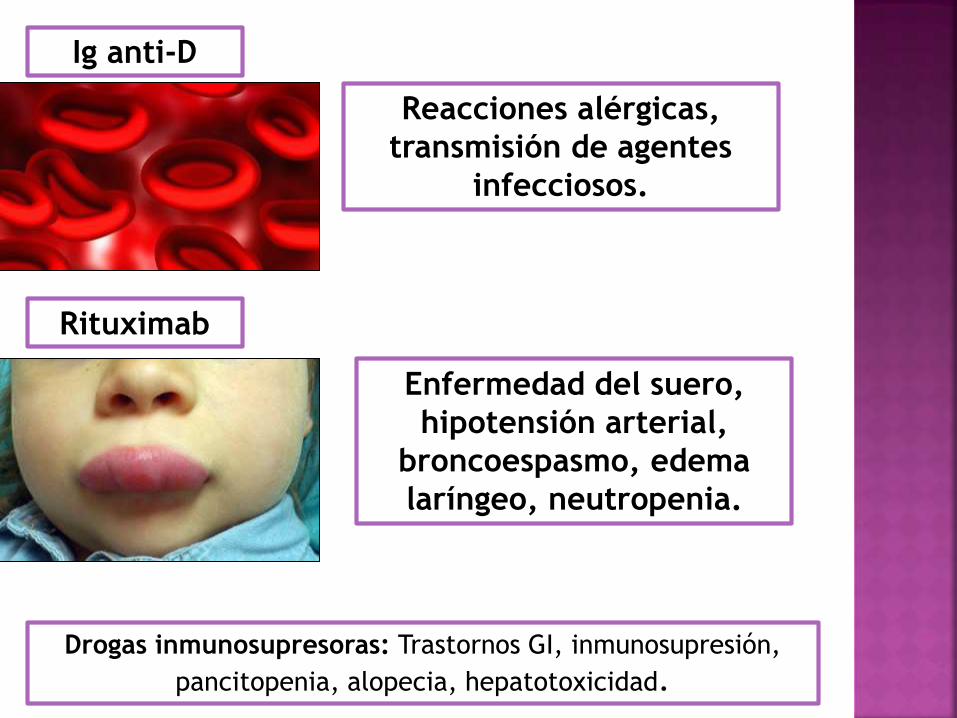
Ig anti-D
Reacciones alérgicas,
transmisión de agentes
infecciosos.
Rituximab
Enfermedad del suero,
hipotensión arterial,
broncoespasmo, edema
laríngeo, neutropenia.
Drogas inmunosupresoras: Trastornos GI, inmunosupresión,
pancitopenia, alopecia, hepatotoxicidad.
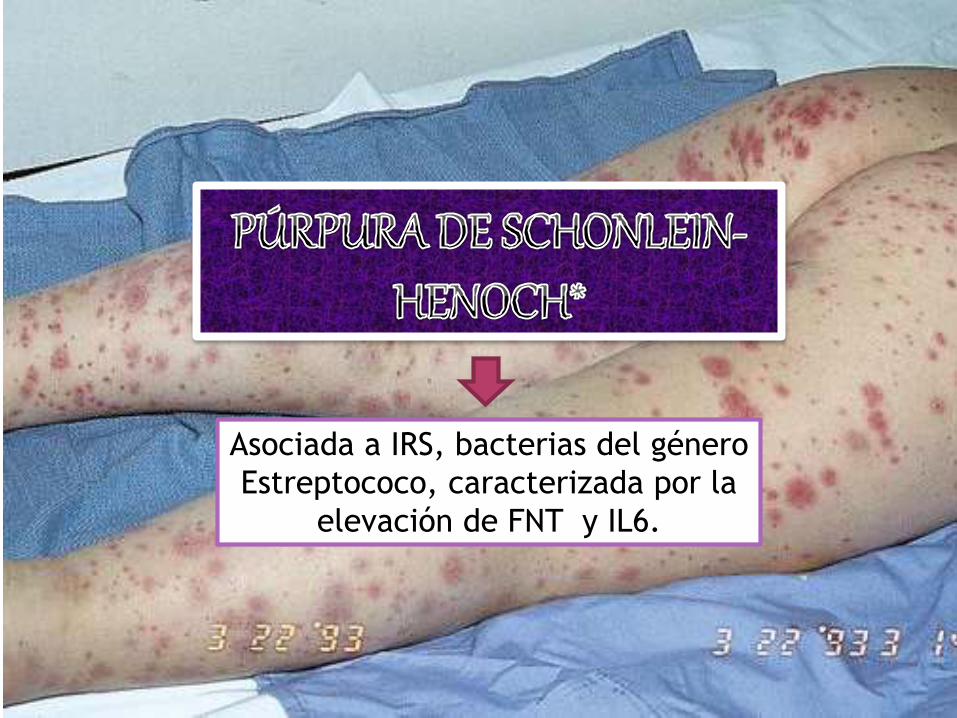
Asociada a IRS, bacterias del género
Estreptococo, caracterizada por la
elevación de FNT y IL6.
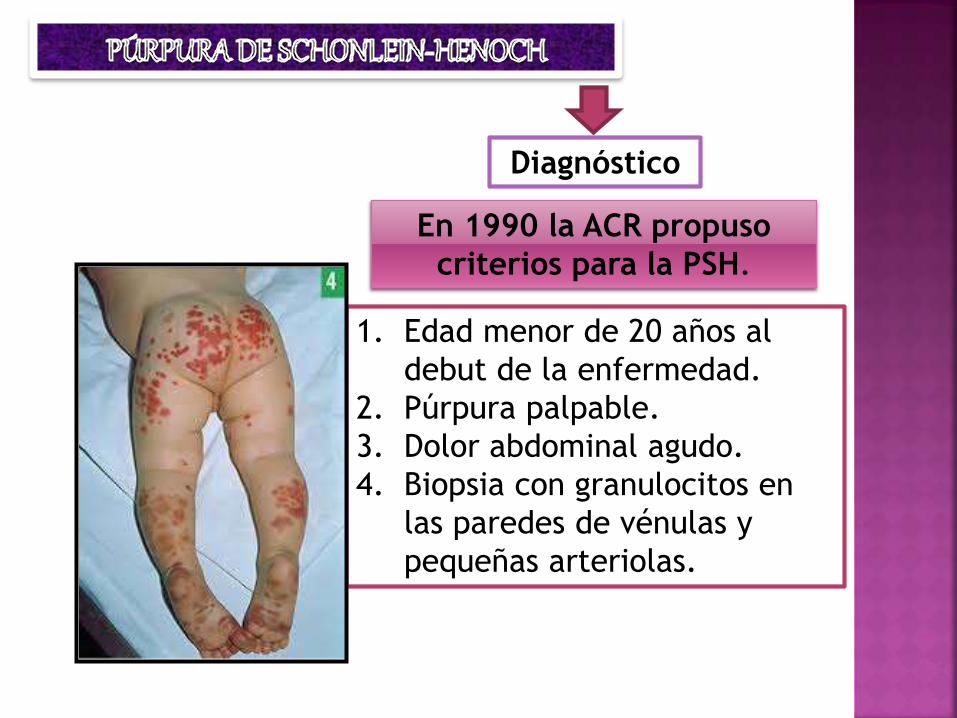
Diagnóstico
En 1990 la ACR propuso
criterios para la PSH.
1. Edad menor de 20 años al
debut de la enfermedad.
2. Púrpura palpable.
3. Dolor abdominal agudo.
4. Biopsia con granulocitos en
las paredes de vénulas y
pequeñas arteriolas.
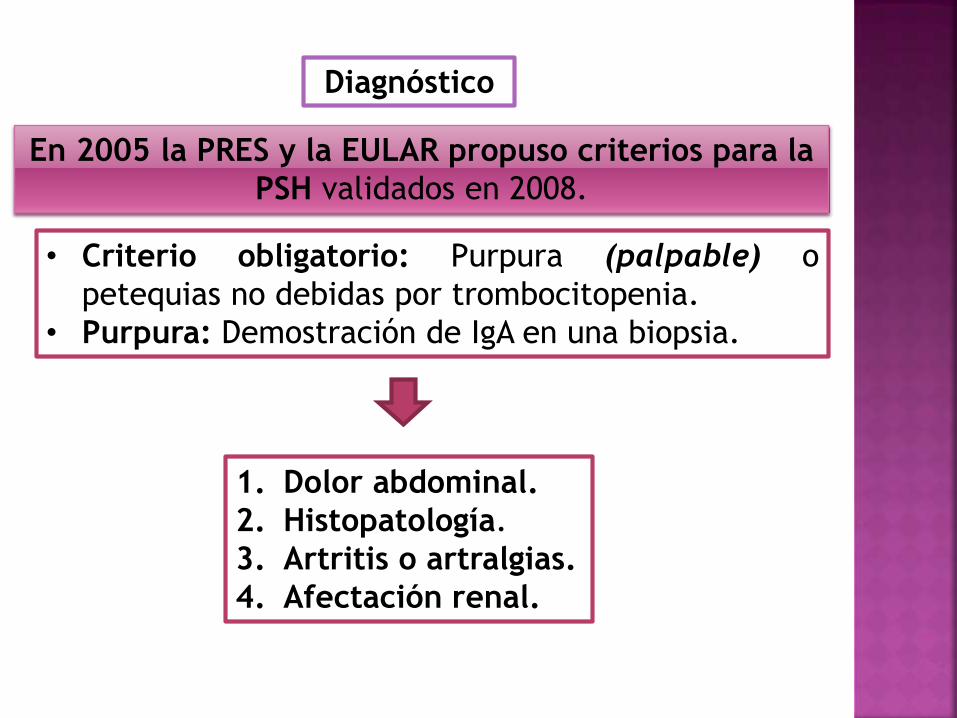
Diagnóstico
En 2005 la PRES y la EULAR propuso criterios para la
PSH validados en 2008.
• Criterio obligatorio: Purpura (palpable) o
petequias no debidas por trombocitopenia.
• Purpura: Demostración de IgA en una biopsia.
1. Dolor abdominal.
2. Histopatología.
3. Artritis o artralgias.
4. Afectación renal.
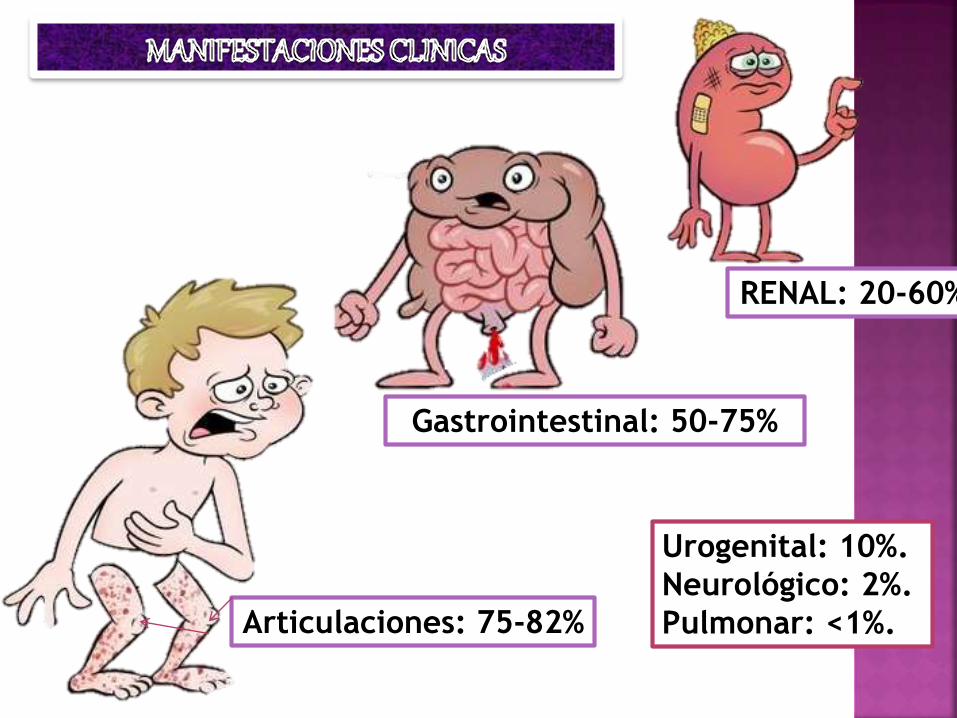
RENAL: 20-60%
Articulaciones: 75-82%
Gastrointestinal: 50-75%
Urogenital: 10%.
Neurológico: 2%.
Pulmonar: <1%.
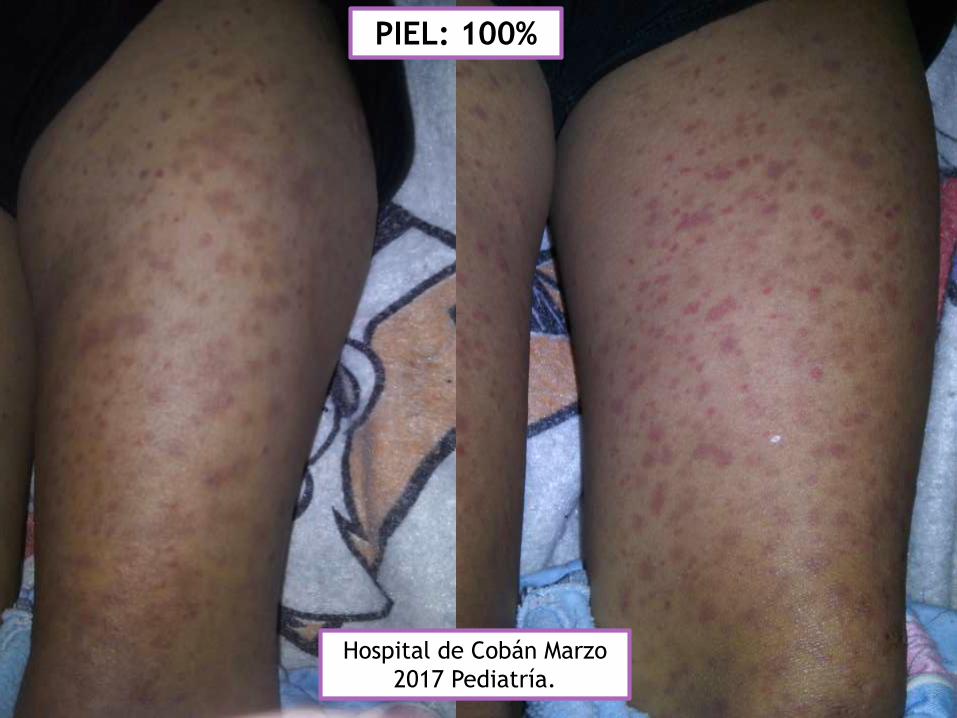
Hospital de Cobán Marzo
2017 Pediatría.
PIEL: 100%
Hospital de Cobán Marzo
2017 Pediatría.
Hospital de Cobán Marzo
2017 Pediatría.
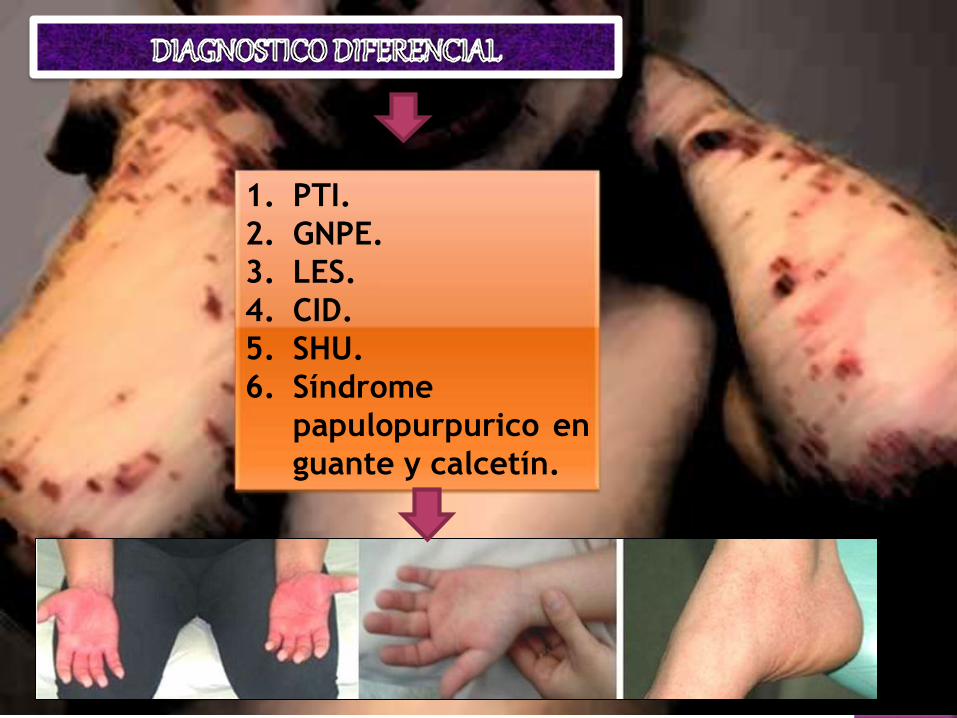
1. PTI.
2. GNPE.
3. LES.
4. CID.
5. SHU.
6. Síndrome
papulopurpurico en
guante y calcetín.
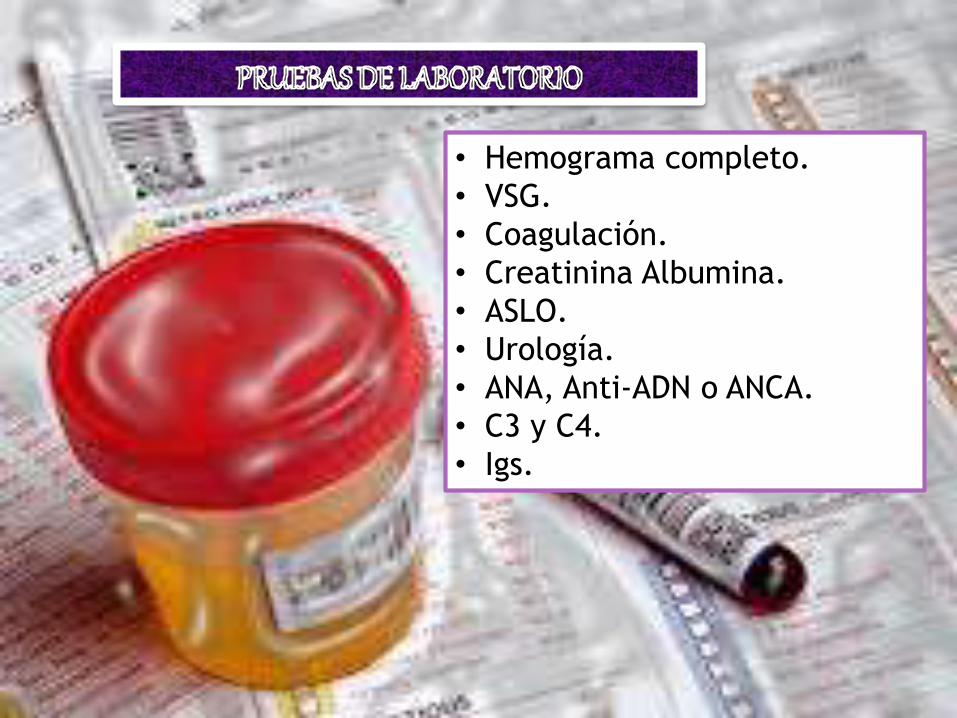
• Hemograma completo.
• VSG.
• Coagulación.
• Creatinina Albumina.
• ASLO.
• Urología.
• ANA, Anti-ADN o ANCA.
• C3 y C4.
• Igs.

• Ecografía renal.*
• Radiografía abdominal.*
• Ecografía abdominal.*
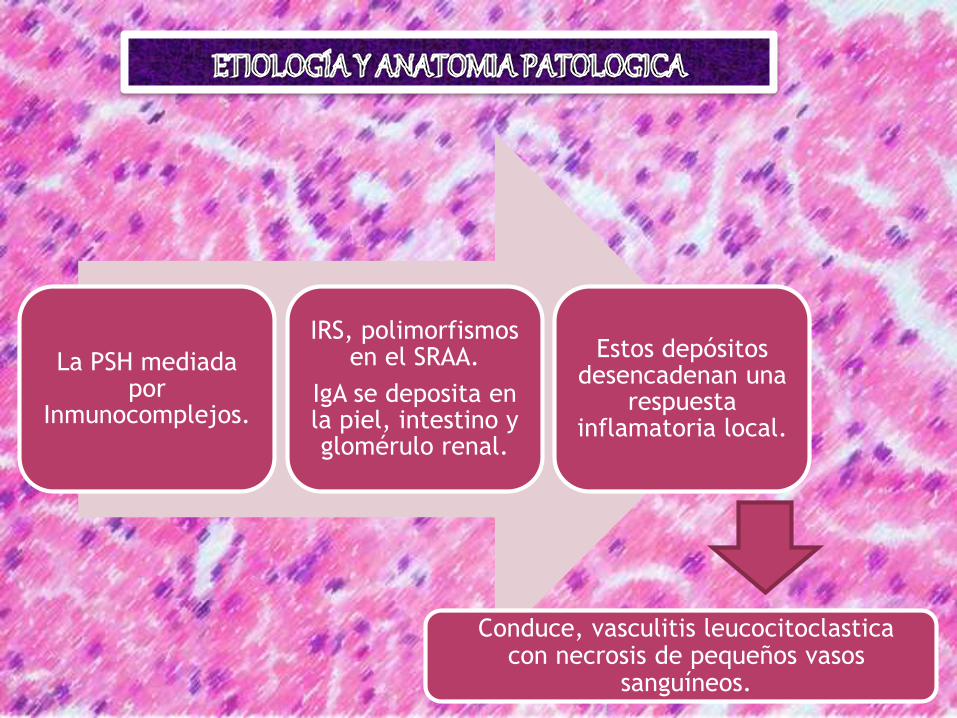
La PSH mediada por
Inmunocomplejos.
IRS, polimorfismos en el SRAA.
IgA se deposita en la piel, intestino y glomérulo renal.
Estos depósitos desencadenan una
respuesta inflamatoria local.
Conduce, vasculitis leucocitoclastica con necrosis de pequeños vasos
sanguíneos.
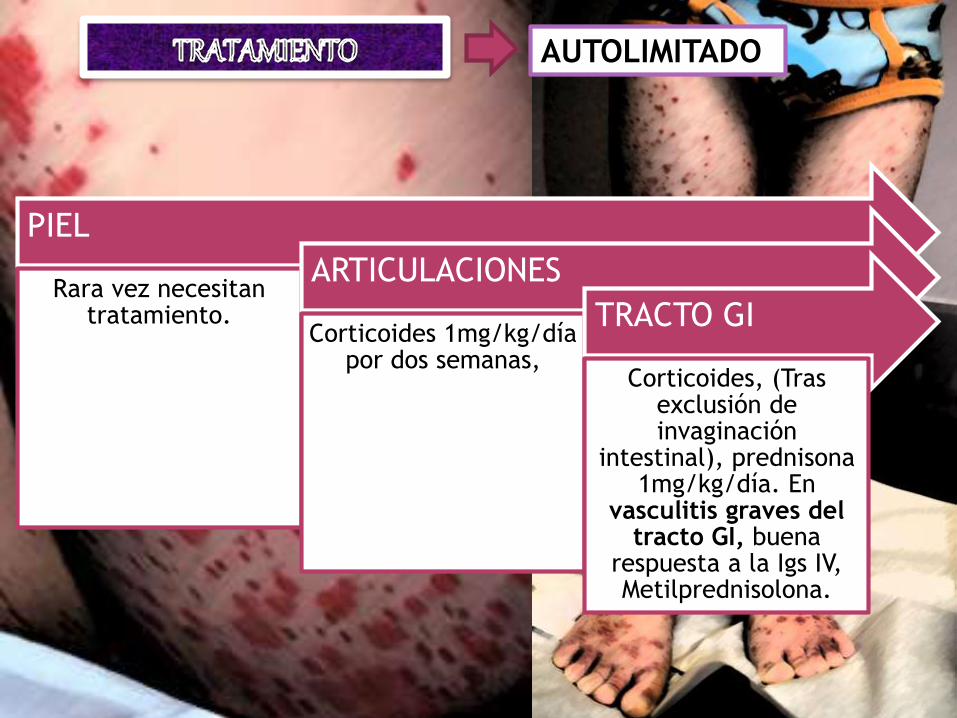
AUTOLIMITADO
PIEL
Rara vez necesitan tratamiento.
ARTICULACIONES
Corticoides 1mg/kg/día por dos semanas,
TRACTO GI
Corticoides, (Tras exclusión de invaginación
intestinal), prednisona 1mg/kg/día. En
vasculitis graves del tracto GI, buena
respuesta a la Igs IV, Metilprednisolona.
CORTICOIDES
No ha demostrado una reducción en la prevalencia de
nefropatía.
Biopsia renal
1. Síndrome nefrítico.
2. Síndrome nefrótico.
3. Proteinuria en rango nefrótico.
4. Proteinuria persistente.
Nefritis grave
1. Metilprednisolona IV bolus 30mg/kg/día por 3 días, seguido de
corticoterapia.
2. IECAS disiminuye la proteinuria.
La PSH sin nefritis es autolimitada.
Se desarrolla a los 6 meses daño renal, por lo que se evalúa de 6 a
12 meses, si hubo alteración renal en el
debut.
Seguimientos periódicos con urología, PA,
durante los primeros 6 meses. **
Recuento plaquetario < 150,000/mm3,
grave < 50,000/mm3.
SE CLASIFICA EN FETAL
• Precoz: primeras 72 hrs.
• Tardía: pasadas las 72 hrs.
Principal causa de trombocitopenia
precoz, a hipoxia fetal secundario a
preclamsia o RCIU.
Leve-moderada 50-100,000/mm3
Autolimitada y se recupera en los
primeros 7-10 días.
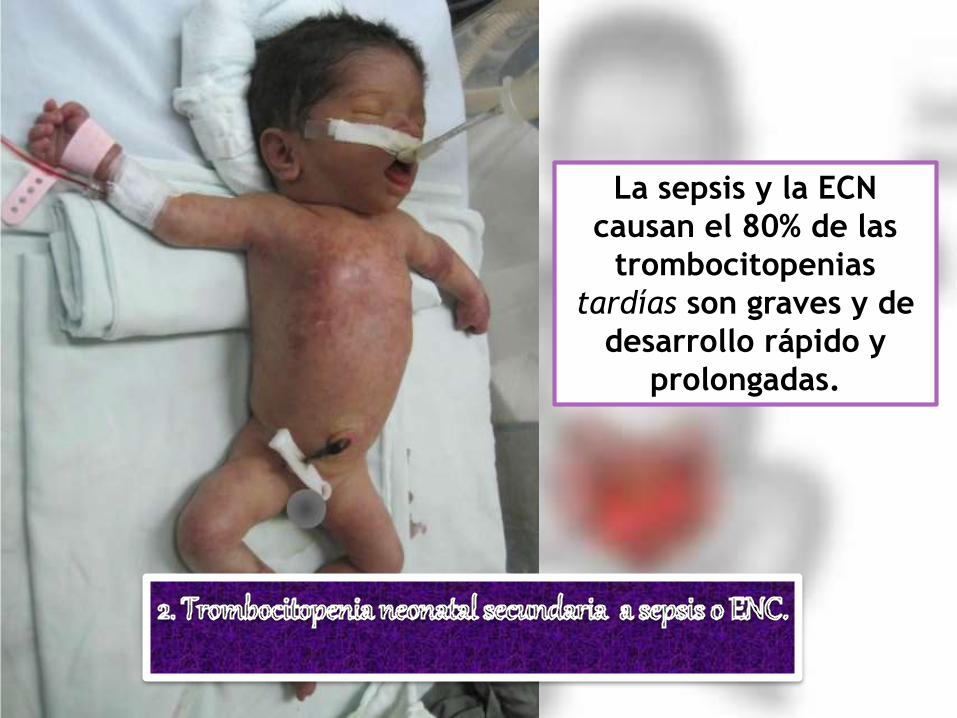
La sepsis y la ECN
causan el 80% de las
trombocitopenias
tardías son graves y de
desarrollo rápido y
prolongadas.

La TNA es la causa mas frecuente
de trombocitopenia grave
precoz.

Similar a la EHRN, solo que esta ocurre en el 50%
del primer embarazo.
Acción de un aloanticuerpo plaquetario especifico
materno que reacciona contra el antígeno
plaquetario paterno HPA* heredado del padre.

Se ha observado, trombocitopenia a
21 SG, sospecharse en aquellos fetos
con HIC, ventriculomegalia,
hidrocefalia.
Clínica de HIC (intraparenquimatosa
que intraventricular), purpuras y
hematomas en las primeras horas de
vida sin otra sintomatología asociada.
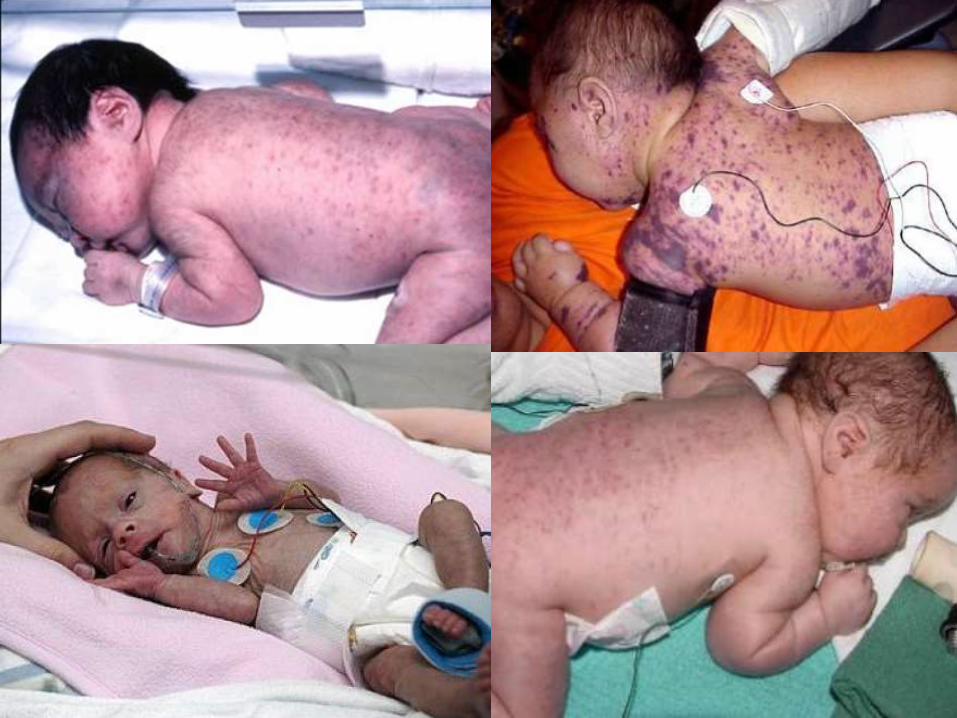

Se basa en la identificación de los
aloanticuerpos maternos dirigidos contra
los HPA fetales y la determinación del
genotipo HPA materno y paterno
mediante técnicas moleculares.
CLINICA
1. Trombocitopenia <20,000/mm3.**
2. HIC: APGAR en el minuto 1 mayor de 5,
w de 2.2kg, sangrado prenatal,
petequias.
3. Sin otros problemas médicos
adicionales.
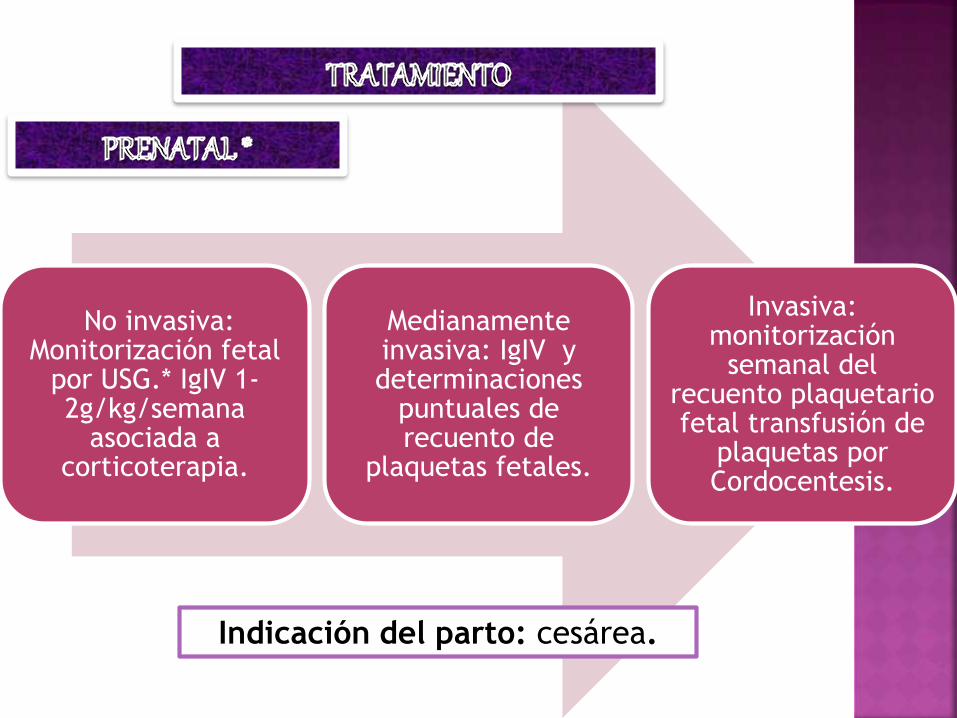
No invasiva: Monitorización fetal
por USG.* IgIV 1-2g/kg/semana
asociada a corticoterapia.
Medianamente invasiva: IgIV y determinaciones
puntuales de recuento de
plaquetas fetales.
Invasiva: monitorización
semanal del recuento plaquetario fetal transfusión de
plaquetas por Cordocentesis.
Indicación del parto: cesárea.
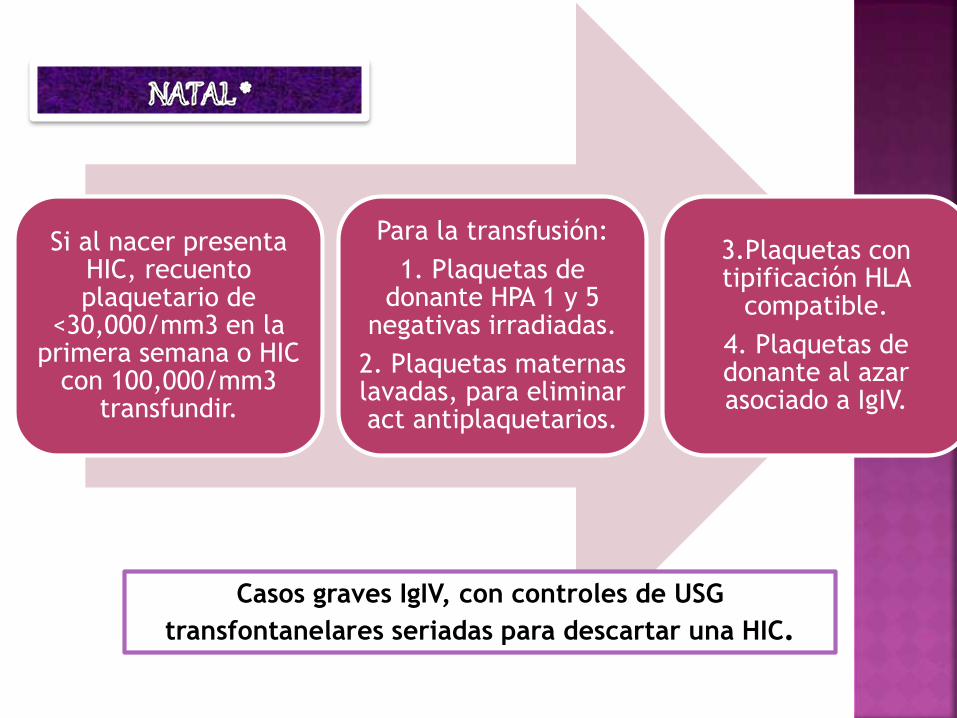
Si al nacer presenta HIC, recuento plaquetario de
<30,000/mm3 en la primera semana o HIC
con 100,000/mm3 transfundir.
Para la transfusión:
1. Plaquetas de donante HPA 1 y 5
negativas irradiadas.
2. Plaquetas maternas lavadas, para eliminar act antiplaquetarios.
3.Plaquetas con tipificación HLA
compatible.
4. Plaquetas de donante al azar asociado a IgIV.
Casos graves IgIV, con controles de USG
transfontanelares seriadas para descartar una HIC.

Es secundaria al paso de autoanticuerpos
maternos ocurre principalmente en gestantes
afectadas de LES o purpura trombocitopenica.
Independientemente del recuento
plaquetario no existe indicación de
transfusión, solo TBP grave <30,000 con IgIV
2mg/kg/2-5 días.

Las principales causas infección por
Citomegalovirus (10%), y la toxoplasmosis
(40%). Se presenta en el nacimiento o en los
primeros días de vida.

Por el desarrollo de los progenitores hematopoyeticas que
da lugar a una disminución en la producción plaquetaria
fetal y neonatal.
Estrategia neuroprotectora en APN, se
encuentra trombocitopenia no amerita tto.
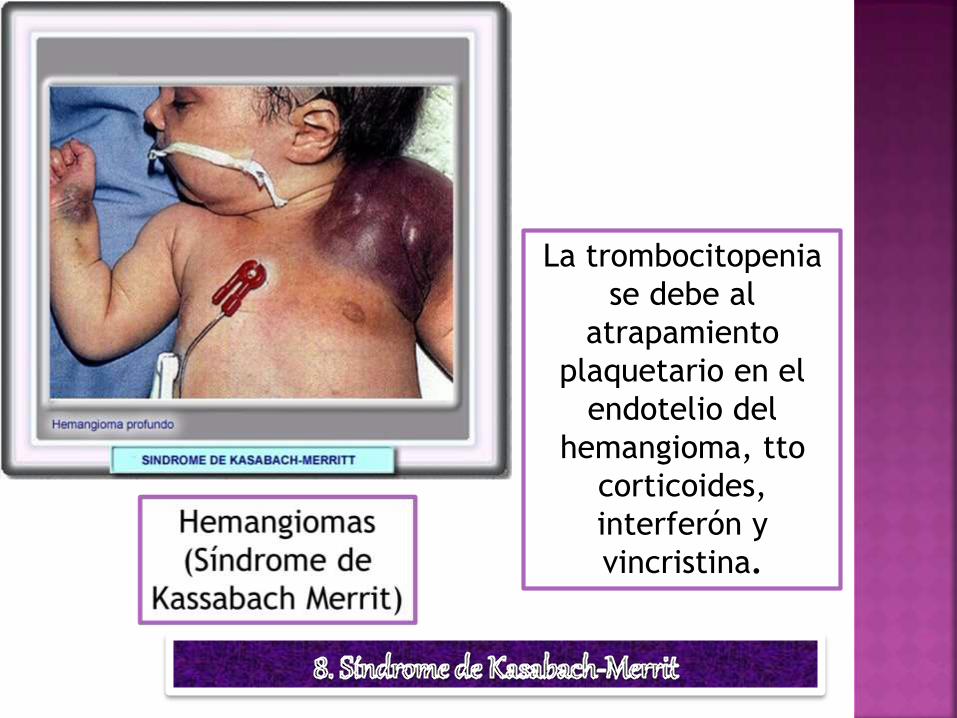
La trombocitopenia
se debe al
atrapamiento
plaquetario en el
endotelio del
hemangioma, tto
corticoides,
interferón y
vincristina.
Prevalencia desconocida. Posiblemente
alteraciones en la producción.
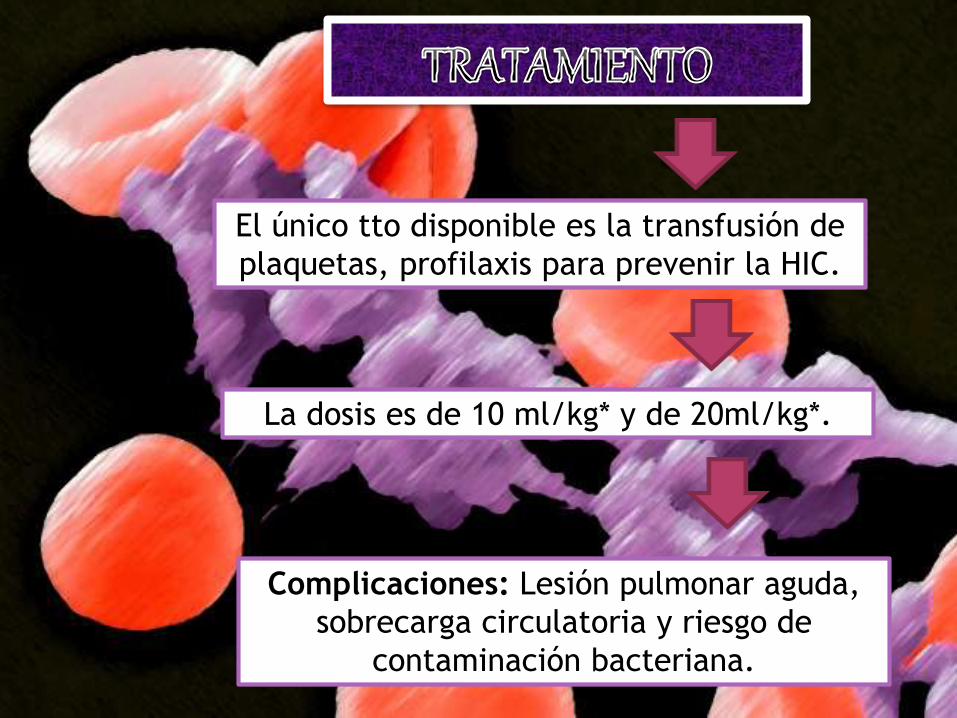
El único tto disponible es la transfusión de
plaquetas, profilaxis para prevenir la HIC.
La dosis es de 10 ml/kg* y de 20ml/kg*.
Complicaciones: Lesión pulmonar aguda,
sobrecarga circulatoria y riesgo de
contaminación bacteriana.



















