
UNIVERSIDAD DE LAS AMÉRICAS PUEBLA catarina.udlap.mx/u_dl_a/tales/documentos/lbi/vargas... ·...
68
1 UNIVERSIDAD DE LAS AMÉRICAS PUEBLA <<Escuela de Ciencias>> <<Departamento de Ciencias Químico Biológicas>> “Inhibición de las enzimas alfa – glucosidasas mediante compuestos orgánicos extraídos de la herbolaría mexicana” Tesis que, para completar los requisitos del Programa de Honores presenta la estudiante <<Luis Adrian Vargas Tapia>> <<151898>> <<Licenciatura en Biología>> <<Dr. Julio Lenin Domínguez Ramírez >> San Andrés Cholula, Puebla. <<Primavera 2019>>
Transcript of UNIVERSIDAD DE LAS AMÉRICAS PUEBLA catarina.udlap.mx/u_dl_a/tales/documentos/lbi/vargas... ·...

1
UNIVERSIDAD DE LAS AMÉRICAS PUEBLA
<<Escuela de Ciencias>> <<Departamento de Ciencias Químico Biológicas>>
“Inhibición de las enzimas alfa – glucosidasas mediante compuestos orgánicos extraídos de la herbolaría mexicana”
Tesis que, para completar los requisitos del Programa de Honores presenta la estudiante
<<Luis Adrian Vargas Tapia>>
<<151898>>
<<Licenciatura en Biología>>
<<Dr. Julio Lenin Domínguez Ramírez >>
San Andrés Cholula, Puebla. <<Primavera 2019>>

2
Tesis que, para completar los requisitos del Programa de Honores presenta el
estudiante <<Luis Adrian Vargas Tapia 151898>>
Director de Tesis
<<Dr. Julio Lenin Domínguez Ramírez>>
Presidente de Tesis
<<Dr. Eugenio Sánchez Arreola>>
Secretario de Tesis
<<Irene Vergara Bahena>>

3
Introducción
La diabetes es un grupo de desórdenes de metabólicos caracterizados por la hiperglucemia
como resultado de una secreción defectuosa de insulina, insensibilidad a la acción de la
insulina, o ambas (American Diabetes Association, 2014). La hiperglucemia crónica está
asociada con el daño a largo plazo, disfunción y fallo de diferentes órganos como: los ojos,
nervios, riñones, y vasos sanguíneos (American Diabetes Association, 2014).
La mayoría de los casos de diabetes se encuentran dentro de dos categorías
etiopatogenéticas: diabetes tipo I y diabetes tipo II (American Diabetes Association, 2014).
La diabetes tipo II es causada por una combinación de resistencia a la acción de la insulina
y de una respuesta secretora inadecuada y poco compensatoria de la insulina (American
Diabetes Association, 2014). Se estima que esta forma de diabetes involucra a un 90-95%
de todos los pacientes con diabetes (American Diabetes Association, 2014). El tratamiento
de este tipo de diabetes involucra el uso de hipoglucémicos que se complementa con un
estilo de vida saludable. Los hipoglucémicos orales son el tratamiento más común y existe
una gran diversidad de ellos. Las sulfonilureas, por ejemplo, fueron ampliamente usadas
durante los años cincuenta, y su mecanismo de acción consistía en estimular la secreción de
insulina por la célula beta pancreática mediante su unión a un canal potasio-dependiente de
ATP (Alfaro et al, 2000). Las biguanidas son otra clase de hipoglucémicos que actúan
principalmente a dos niveles: en el músculo, aumentando la entrada de glucosa a las
células, y en el hígado reduciendo los niveles de glucosa a través de la anulación de la
glucogenolisis (Alfaro et al, 2000). Sin embargo, ambas clases de fármacos causan efectos
secundarios a nivel gastrointestinal, así como estados de hipoglucemia graves que
conllevan tratamiento hospitalario (Alfaro et al, 2000). Por ello, la búsqueda de nuevos

4
fármacos sin estos efectos secundarios que puedan ser usados para controlar los niveles de
glucosa en sangre sigue siendo una prioridad en el tratamiento de la diabetes (Hans
Reinauer & Philip D. Home, 2003). En este contexto, a búsqueda de este tipo de
compuestos basados en la farmacopea autóctona o de los pueblos originarios ha llevado a la
selección de las α-glucosidasas como una diana para el tratamiento de la diabetes.
Las enzimas α-glucosidasas son enzimas localizadas en el borde en el cepillo del instestino
delgado en donde catalizan el paso final en el proceso digestivo de los sacáridos (Mata et
al, 2013) (Fig.1). La maltasa glucoamilasa y la sucrosa isomaltasa son enzimas trans-
membranales, las cuales poseen dos subunidades catalíticas en el mismo polipéptido: una
subunidad N-terminal próximo a la membrana, y una subunidad C-terminal (Sim, Quezada-
Calvillo, Sterchi, Nichols, & Rose, 2008) (Fig.2). De acuerdo al sistema de clasificación de
enzimas hidrolizadoras de sacáridos, por sus siglas en inglés (CAZY), todas estas
subunidades pertenecen a la familia de hidrolasas glucosídicas 31 (GH31) subgrupo 1 (Ren
et al., 2011). Este grupo de enzimas opera a través de un mecanismo que resulta en la
retención de la configuración en el centro anomérico (Sim et al., 2010). Las subunidades de
maltasa glucoamilasa y la sucrosa isomaltasa, son idénticas aproximadamente en uno 40-
60% en su secuencia aminoacil (Sim et al., 2008) (Fig.3).
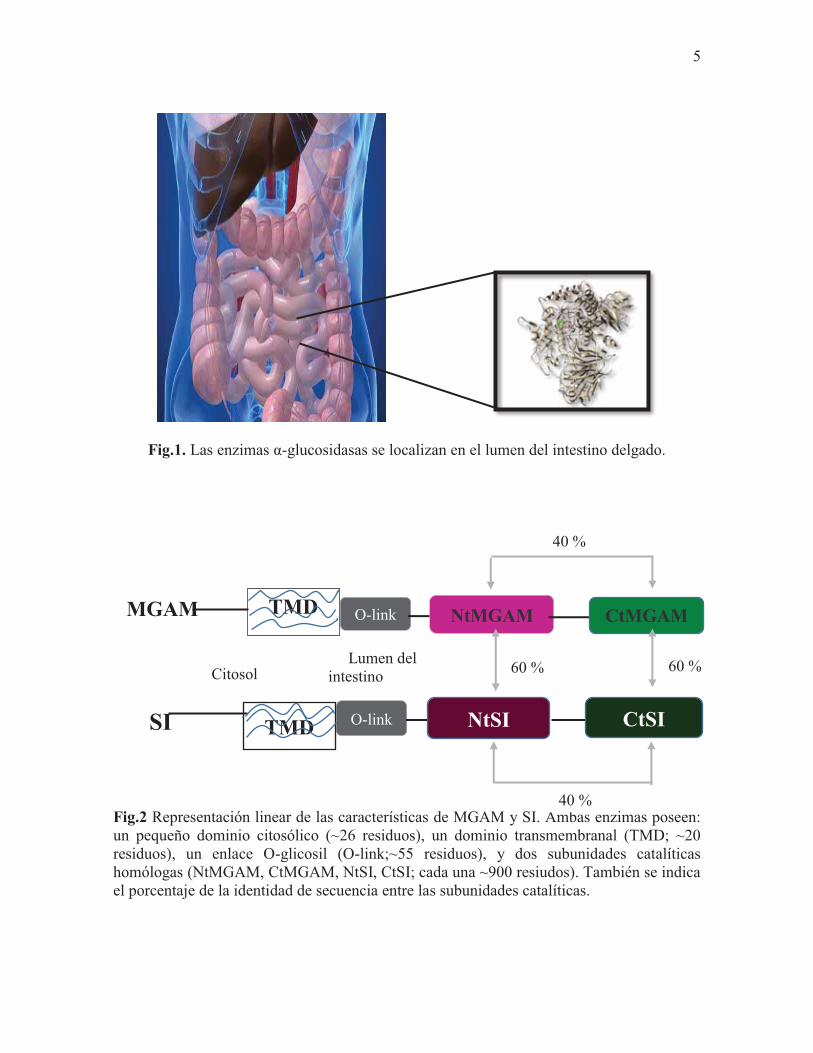
5
Fig.1. Las enzimas α-glucosidasas se localizan en el lumen del intestino delgado.
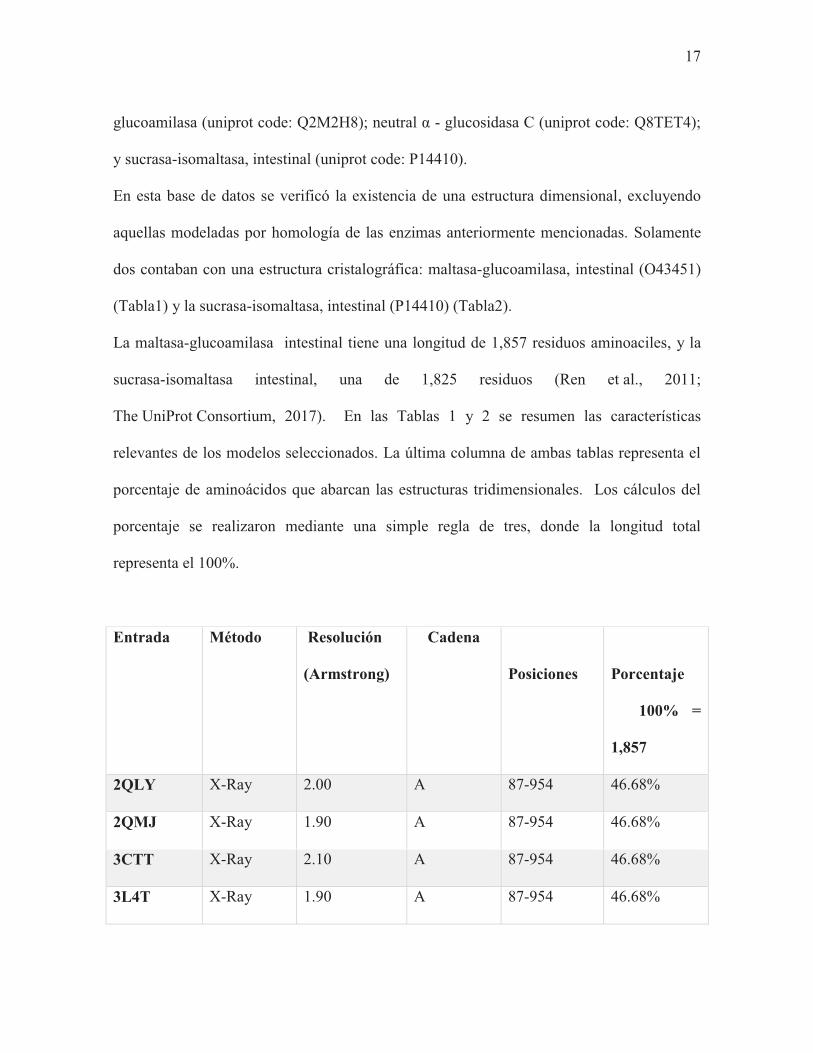
Fig.2 Representación linear de las características de MGAM y SI. Ambas enzimas poseen: un pequeño dominio citosólico (~26 residuos), un dominio transmembranal (TMD; ~20 residuos), un enlace O-glicosil (O-link;~55 residuos), y dos subunidades catalíticas homólogas (NtMGAM, CtMGAM, NtSI, CtSI; cada una ~900 resiudos). También se indica el porcentaje de la identidad de secuencia entre las subunidades catalíticas.
40 %
40 %
MGAM TMD O-link NtMGAM CtMGAM
SI O-link NtSI CtSI TMD
60 % 60 % Citosol Lumen del intestino

6

7
Figura3. Secuencias aminoaciles alineadas de ambos dominios catalíticos (34LW N-terminal) y (3TOP C-terminal) de la enzima maltasa glucoamilasa.
El objetivo de crear fármacos inhibidores de su actividad catalítica ha reavivado
recientemente el interés respecto al estudio de las enzimas α-glucosidasas y sus posibles
formas de inhibición; así, se han determinado la cristalización de las enzimas MGMA y SI,
y de sus dos respectivos dominios en presencia de dos inhibidores, acarbosa (Drug Bank
code: DB00284) y miglitol (Drug Bank code: DB00491) (Wishart et al., 2018) (Fig.4 y
Fig.5).

8
Fig.4 Estructura química del inhibidor Acarbosa.
Fig.5 Estructura química del inhibidor Miglitol.
Así mismo, gracias a los avances en catálisis enzimática computacional, es posible conocer
las interacciones de los fármacos miglitol y acarbosa con los aminoácidos del sitio
catalítico de la maltasa glucoamilasa en sus dos respectivos dominios catalíticos; N-

9
terminal y C-terminal. Para el miglitol, compuesto que generalmente se encuentra
cocristalizado con el dominio C-terminal en las bases datos de proteínas (PDB Bank, por
ejemplo), conocemos que interactúa principalmente con dominio N-terminal a través de
puentes de hidrógeno con D327, R526, D542, y H600, formando en total ocho enlaces de
hidrógeno con los aminoácidos del sitio catalítico del dominio N-terminal (PDB:3L4W)
(Laskowski, 2004). Otra interacción significativa es la del anillo nitrogenado, el cual se
encuentra dentro de una distancia de enlaces de hidrógeno de 2.8 Armstrongs del D443
nucleófilo catalítico (Sim et al., 2010) (Fig.6)
Fig.6 Interacción del miglitol con el sitio catalítico del dominio N-terminal.
En cuanto a la acarbosa, así como el miglitol, interacciona principalmente a través de los
grupos hidroxilo que forman puentes de hidrógeno con las cadenas laterales de H1589,
D1526, R1516, D1157 y D1279, estableciendo un total de once puentes de hidrógeno con

10
el sitio catalítico del dominio C terminal de la maltasa-glucoamilasa (PDB:3TOP)
(Laskowski, 2004) (Fig.7)
Fig.7 Interacción de la acarbosa con el sitio catalítico del dominio C-terminal.
Los inhibidores de α-glucosidasas también son medicamentos hipoglucémicos; estos
inhiben las enzimas localizadas en el borde del enterocito del intestino que hidrolizan los
oligosacáridos a disacáridos y monosacáridos que posteriormente son absorbidos. (Alfaro et
al, 2000). Un incremento en la absorción de los oligosacáridos se traduce en una elevación
dramática de los niveles de glucosa en la sangre (van de Laar, 2008). La hiperinsulinemia o
hiperglucemia posprandial, por sus siglas en inglés (PPHG), son factores propios de riesgo

11
para el desarrollo de complicaciones macrovasculares en la diabetes mellitus II (van de
Laar, 2008). Por lo tanto, los inhibidores de α-glucosidasas, como la acarbosa, se han
utilizado para reducir principalmente los niveles de PPHG, a través de la interferencia en el
proceso digestivo de las enzimas glucosidasas, además de modificar la secreción endógena
de GLP1 “glucagon-like-peptide 1”, el cual es un análogo de la insulina (Mata, Cristians,
Escandón-Rivera, Juárez-Reyes, & Rivero-Cruz, 2013), lo que disminuye la incorporación
de glucosa en la sangre (Kim, Kwon, & Son, 2000). Sin embargo, los efectos secundarios
de los inhibidores más comunes, como el miglitol y la acarbosa, son significativos, por
ejemplo: dolores estomacales, flatulencias, diarrea, y aumento de peso (Kim et al., 2000)
(van de Laar, 2008). Tales efectos secundarios, sin embargo, podrían evitarse con el uso de
flavonoides y otros compuestos extraídos de la herbolaria tradicional, cuyas plantas que la
conforman han sido reportadas con escasos efectos secundarios por sus usuarios asiduos de
diferentes culturas y épocas. (Kim et al., 2000) (Mata et al., 2013). Uno de los
inconvenientes que existen respecto al desarrollo de estos fármacos es la necesidad de tener
una amplia gama de estos inhibidores con el fin de evitar que el paciente genere una rápida
resistencia a los más recurrentes (Kim et al., 2000). Y no sólo eso, en el caso particular de
las alfa-glucosidasas, también es necesario que dichos inhibidores logren bloquear ambos
dominios (N-terminal y C-terminal) de las enzimas glucosidasas.
El diseño de fármacos capaces de atacar varios objetivos de manera simultánea, que sean
eficaces y sin efectos secundarios, constituye una de las tendencias en investigación
farmacológica, y es así mismo un punto de inflexión en dicha tendencia de la investigación
debido a que propone que la solución a una patología sea sistemática en lugar de específica
(Montero, 2016). El modelaje in silico en años recientes, ha adquirido un considerable

12
interés por parte de la comunidad científica enfocada en el desarrollo de fármacos (Fox &
Kriegl, 2006). El término in silico hace alusión a la estructura interna del computador que
está compuesto aproximadamente en un 90% por materiales de sílice (Scior, Martínez
Morales, & Salinas Stefanón, 2007), por lo que al hacer mención de esta palabra se debe
entender que todo el proceso de simulación experimental ha ocurrido dentro del ordenador,
y que no comparte característica física alguna con el proceso original a partir del cual fue
creado sílice (Scior et al., 2007). Las ventajas que ofrecen los métodos de modelado in
silico, son el costo económico para su realización, así como el tiempo que requieren para
obtener resultados, en comparación, por ejemplo, con los métodos in vivo e in vitro
(Mensch et al, 2009), ya que los métodos in silico requerirán principalmente un ordenador,
y el tiempo que requieran para obtener resultados puede estimarse en horas dependiendo
del logaritmo empleado por el software de nuestra elección. Por lo tanto, los modelos in
silico serán capaces de asesorarnos sobre las propiedades que posean determinadas
colecciones constituidas por un gran número de compuestos para los cuales el análisis
experimental in vivo o in vitro de sus propiedades no podría ser realizado con facilidad,
debido a razones de tiempo, dinero, o falta de personal en el laboratorio; o bien, podrían
ofrecernos un panorama comprensivo de las propiedades de dichos compuestos previo a su
síntesis (Fox & Kriegl, 2006) . Los métodos de modelado in silico pueden clasificarse en
función del conocimiento que se tenga del sitio de unión o del receptor. Podría decirse que,
las dos estrategias seguidas de manera habitual en modelaje son las denominadas directas e
indirectas (Montero, 2016). En la primera es necesario conocer las características
tridimensionales de un receptor generalmente a partir de datos cristalográficos y la
actividad o inactividad de las moléculas se interpreta en términos de complementariedad

13
con el receptor. A esto se le conoce por el término de “docking” (Montero, 2016)
(Bursulaya, Totrov, Abagyan, & Brooks, 2003).
El objetivo principal de los programas de docking es la obtención de la estructura del
complejo receptor-ligando con la energía más baja (se debe cumplir que ΔΕ <0), y el
análisis de las principales interacciones en la unión ligando-receptor (Montero, 2016). Las
primeras aproximaciones hacia la problemática del docking consideraban un algoritmo en
el que tanto el ligando como el receptor son rígidos, sin embargo, tal aproximación no es
realista debido a que la mayoría de los ligandos pequeños son flexibles con varios enlaces
rotativos. (Bursulaya et al., 2003). Para evitar esta problemática, se han diseñado varios
algoritmos que complementan la naturaleza del docking rígido con otras características que
beneficien a la flexibilidad de los ligandos, a la vez que se ha mejorado en el poder de
cómputo y en las técnicas de computación. Esto se demuestra, específicamente, con la
introducción de una rejilla basada en las dimensiones del receptor, así como el uso de
coordenadas internas que hacen de la simulación de ligandos flexibles algo
computacionalmente más factible. (Bursulaya et al., 2003; Park, Lee, & Lee, 2006).
Los métodos de modelado in silico como el docking involucran principalmente dos pasos:
el primero, que predice la posición del ligando (donde una unión correcta del ligando es
considerada como aquella lo más similar posible a la del ligando cocristalizado con la
enzima o proteína). El segundo paso tiene como objetivo predecir las afinidades de unión lo
más cercanas posibles a las observaciones experimentales (Houston & Walkinshaw, 2013).
No obstante una de las limitaciones de la técnica de docking es que algunas poses de unión
de los ligandos que son significativamente diferentes, pueden tener valores de afinidad
similares, denominados en inglés “docking scores” lo que conlleva a que no haya una

14
manera precisa de distinguir cuál unión es la correcta (Houston & Walkinshaw, 2013) (Park
et al., 2006). Comprar o probar dichos resultados falsos positivos puede ser perjudicial para
un laboratorio de baja producción en el que los recursos económicos son limitados
(Houston & Walkinshaw, 2013).
Una manera de reducir el grado de error en los análisis docking por las razones antes
mencionadas es combinando los resultados de distintos algoritmos encargados de calcular
el “score” o valor de afinidad de los compuestos con respecto al receptor dentro de un
esquema consensado de los “scores” o valores de afinidad. Este esquema consensado es un
método a partir del cual las afinidades de ciertos compuestos por una diana en particular se
predicen usando más de un algoritmo. Varios estudios han demostrado que usando este
método consensado de prueba se pueden obtener resultados más precisos que usando un
solo algoritmo (Houston & Walkinshaw, 2013). Por ejemplo, uno de los estudios más
ambiciosos y completos en cuanto a comparación de distintos algoritmos de docking
realizado por Warren et al en 2006, encontró que ninguno de los softwares de docking
usados por sí solos realizaron una predicción útil y precisa de la afinidad del ligando sobre
ocho proteínas distintas (Warren et al., 2006).
Otro estudio de comparación entre distintos algoritmos de docking fue el realizado por
Houston et al en 2013 en el cual compararos dos softwares distintos: Autodock y Vina
(Houston & Walkinshaw, 2013). Ambos softwares emplean diferentes algoritmos de
docking. Podría decirse que Autodock pertenece al primer grupo de programas basados en
algoritmos que tratan de encontrar una conformación óptima del ligando resolviendo el
problema de la optimización de la energía global (Bursulaya et al., 2003). Por el contrario,
Vina corresponde al segundo grupo de programas que se basan en algoritmos que tratan de

15
embonar el ligando en el sitio de unión de la proteína mediante un emparejamiento que
puede ser geométrico, químico, energético, etc. (Bursulaya et al., 2003). Para tal estudio
usaron 231 receptores y un total de 228 ligandos. Parte de los resultados demostraron que el
uso de Autodock y Vina en conjunto puede incrementar significativamente la precisión
sobre la pose de unión del ligando con el receptor, cuando ambos programas coinciden
(Houston & Walkinshaw, 2013). Es decir, cuando se consideraban aquellas de poses de
unión en las que ambos programas coincidían a la par que se rechazaban aquellas poses que
no eran lo suficientemente similares entre sí, la proporción de ligandos correctamente
unidos al receptor fue del 82% comparado con un 64% obtenido al usarse un solo programa
(Houston & Walkinshaw, 2013).
Por todo lo anteriormente expuesto, el objetivo del presente trabajo fue realizar
experimentos de docking con dos programas basados en algoritmos diferentes para reducir
el grado de error en los valores de afinidad de los ligandos por el receptor (Bursulaya et al.,
2003; Houston & Walkinshaw, 2013; Warren et al., 2006). Los programas empleados
fueron DOCK6 y Vina. DOCK6 en su modalidad “Rigid” (ligando y receptor rígido) y
“Anchor and Grow” (ligando flexible y receptor rígido) (Brozell et al.,2012) que
corresponde al primer grupo de programas anteriormente mencionado, en tanto que Vina
posee características correspondientes al segundo grupo de programas (Bursulaya et al.,
2003). La diana o receptor del experimento de docking fue la enzima maltasa-glucoamilasa
de la familia de enzimas α-glucosidasas con sus dos respectivas subunidades catalíticas (C-
terminal y N-terminal). Así mismo se usaron cincuenta compuestos con aparente actividad
inhibitoria de las α-glucosidasas extraídos de cuatro especies de plantas empleadas en la
medicina tradicional mexicana, los cuales fueron reportados por Mata et al en 2013:

16
Hintonia latiflora y Hintonia standleyana (Rubiaceae), Lingusticum porteri (Apiaceae), y
Brickellia cavanillesii (Asteraceae) (Mata et al., 2013). La elección de compuestos
extraídos de plantas se basó en el hecho de que estas plantas usadas como remedios en la
medicina tradicional mexicana han sido reportadas con escasos o nulos afectos adversos en
la salud (Mata et al, 2013), efectos que se buscan solucionar en la creación de nuevos
fármacos con actividad inhibidora de las α-glucosidasas con el objetivo de reemplazar a los
actuales fármacos más empleadas como el miglitol y la acarbosa, los cuales tienen serios
efectos adversos (Kim et al., 2000; van de Laar, 2008). De resultar que uno o varios de los
compuestos extraídos de alguna de las especies de plantas mencionadas posea una adecuada
actividad inhibitoria de las enzimas α-glucosidasas con base en los experimentos de
docking realizados con dos algoritmos distintos cuyo objetivo es reducir el grado de error
en los valores de afinidad por el receptor (Houston & Walkinshaw, 2013; Warren et al.,
2006), tendríamos potenciales fármacos inhibidores de α-glucosidasas de origen natural con
nulos o escasos efectos adversos en la salud, lo que haría de este tipo de fármacos la mejor
opción como tratamiento de la Diabetes tipo 2.
Métodos
1.1 Obtención informática de estructuras cristalográficas.
En el sitio web expasy.org se inició la búsqueda de las enzimas α-glucosidasas en humano,
encontrando seis resultados posibles con sus respectivos códigos de Uniprot
knowledgebase (The UniProt Consortium, 2017). Las enzimas encontradas fueron:
Lisosomal α -glucosidasa (uniprot code: P10253); neutral α -glucosidasa AB (uniprot code:
Q14697); maltasa-glucoamilasa, intestinal (uniprot code: O43451); probable maltasa-
17
glucoamilasa (uniprot code: Q2M2H8); neutral α - glucosidasa C (uniprot code: Q8TET4);
y sucrasa-isomaltasa, intestinal (uniprot code: P14410).
En esta base de datos se verificó la existencia de una estructura dimensional, excluyendo
aquellas modeladas por homología de las enzimas anteriormente mencionadas. Solamente
dos contaban con una estructura cristalográfica: maltasa-glucoamilasa, intestinal (O43451)
(Tabla1) y la sucrasa-isomaltasa, intestinal (P14410) (Tabla2).
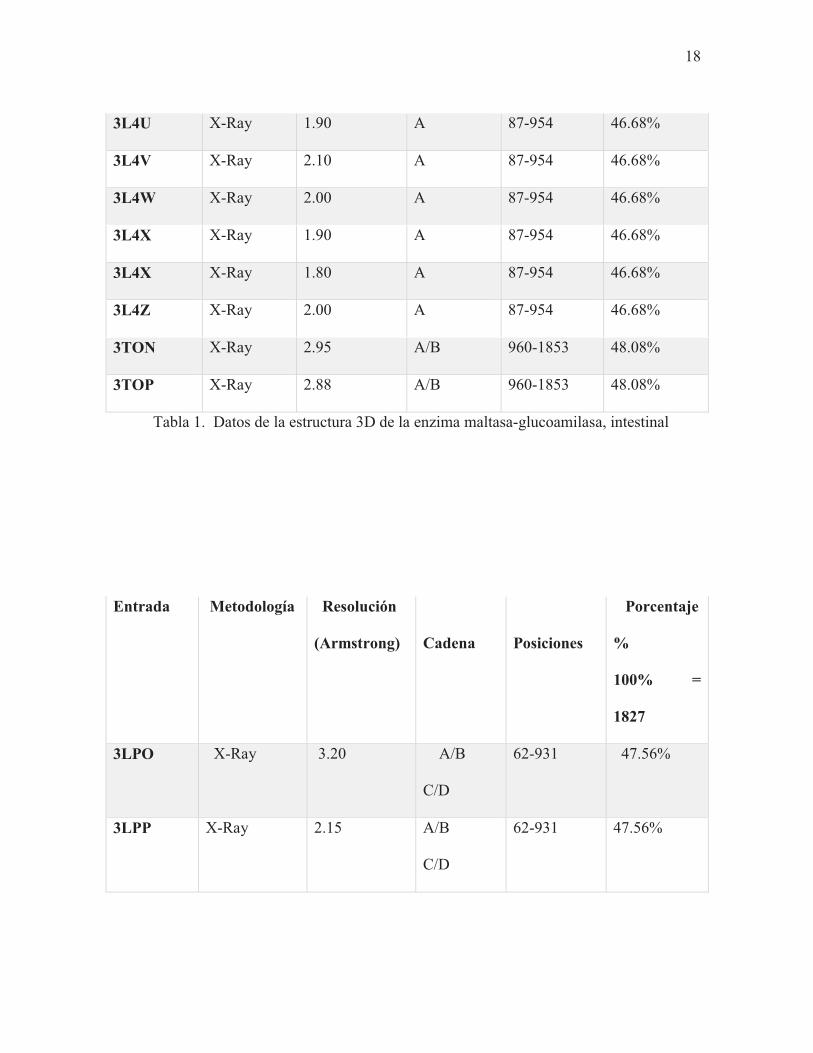
La maltasa-glucoamilasa intestinal tiene una longitud de 1,857 residuos aminoaciles, y la
sucrasa-isomaltasa intestinal, una de 1,825 residuos (Ren et al., 2011;
The UniProt Consortium, 2017). En las Tablas 1 y 2 se resumen las características
relevantes de los modelos seleccionados. La última columna de ambas tablas representa el
porcentaje de aminoácidos que abarcan las estructuras tridimensionales. Los cálculos del
porcentaje se realizaron mediante una simple regla de tres, donde la longitud total
representa el 100%.
Entrada Método Resolución
(Armstrong)
Cadena
Posiciones
Porcentaje
100% =
1,857
2QLY X-Ray 2.00 A 87-954 46.68%
2QMJ X-Ray 1.90 A 87-954 46.68%
3CTT X-Ray 2.10 A 87-954 46.68%
3L4T X-Ray 1.90 A 87-954 46.68%
18
3L4U X-Ray 1.90 A 87-954 46.68%
3L4V X-Ray 2.10 A 87-954 46.68%
3L4W X-Ray 2.00 A 87-954 46.68%
3L4X X-Ray 1.90 A 87-954 46.68%
3L4X X-Ray 1.80 A 87-954 46.68%
3L4Z X-Ray 2.00 A 87-954 46.68%
3TON X-Ray 2.95 A/B 960-1853 48.08%
3TOP X-Ray 2.88 A/B 960-1853 48.08%
Tabla 1. Datos de la estructura 3D de la enzima maltasa-glucoamilasa, intestinal
Entrada Metodología Resolución
(Armstrong)
Cadena
Posiciones
Porcentaje
%
100% =
1827
3LPO X-Ray 3.20 A/B
C/D
62-931 47.56%
3LPP X-Ray 2.15 A/B
C/D
62-931 47.56%

19
Tabla 2. Datos de la estructura 3D de la enzima sucrasa-isomaltasa, intestinal
De igual manera, en la base de datos UCSF ZINC (Irwin, Sterling, Mysinger, Bolstad, &
Coleman, 2012) se buscaron los estereoisómeros de los dos fármacos inhibidores de α-
glucosidasas más usados actualmente: miglitol y acarbosa (Sim et al., 2010; van de Laar,
2008). Para el miglitol se encontraron veinte estereoisómeros naturales de los cuales ocho
han sido empleados en el ser humano, principalmente asociados a la maltasa-glucoamilasa
(Irwin et al., 2012). En cuanto a la acarbosa se encontraron siete estereoisómeros que en su
totalidad han sido empleados en el ser humano y asociados de igual forma principalmente a
la maltasa-glucoamilasa (Irwin et al., 2012).
1.2 Análisis de las estructuras cristalográficas reportadas en las bases de datos
públicas.
Para el análisis estereoquímico de las estructuras cristalográficas reportadas en las tablas 1
y 2, se accedió al sitio web MolProbity (Chen et al., 2010; Davis et al., 2007) que tiene
como objetivo la validación de estructuras tridimensionales de proteínas, ácidos nucleicos y
sus complejos. Este sitio provee análisis detallados de todos los contactos entre los átomos
identificando cualquier problema estérico dentro de las moléculas. También calcula y
exhibe los puentes de hidrógeno y las fuerzas de van der Walls presentes en las interfaces
entre los componentes (Chen et al., 2010; Davis et al., 2007), además de permitir la adición
y optimización de todos los átomos de hidrógenos tanto polares como no polares. Una vez
finalizado este análisis, los resultados son reportados en múltiples formatos; como
puntuaciones numéricas generales (listas y tablas), y como archivos PDB descargables y
gráficas manipulables (Chen et al., 2010; Davis et al., 2007).

20
Del total de catorce estructuras cristalográficas (Tabla 1 y Tabla 2) analizadas con
MolProbity, se eligieron dos estructuras cristalográficas de la enzima maltasa-glucoamilasa
con los códigos 3L4W y 3TOP. Esto debido a que sólo estas dos estructuras fueron
cocristalizadas con los inhibidores miglitol y acarbosa, además de haber contado con
adecuados parámetros estructurales según los análisis realizados en MolProbity. La
estructura 3L4W cocristalizada con el miglitol cuenta con una longitud de 863 aminoácidos
y corresponde a la subunidad N-terminal con actividad maltasa (posiciones 87-954)
(Laskowski, 2004; The UniProt Consortium, 2017) (Fig.8). La estructura 3TOP
cocristalizada con la acarbosa cuenta con una longitud de 890 aminoácidos y corresponde a
la subunidad C-terminal con actividad glucoamilasa (posiciones 960-1853) (Fig.9).
Fig.8 Estructura Cristalográfica 3L4W correspondiente a la subunidad N-terminal de la Maltasa-glucoamilasa. Como puede observarse se encuentra cocristalizada con miglitol el cual se representa con esferas.

21
Fig.9 Estructura Cristalográfica 3TOP correspondiente a la subunidad C-terminal de la Maltasa-glucoamilasa. Como puede observarse se encuentra cocristalizada con acarbosa la cual se representa con esferas.
Ambas estructuras cristalográficas (3L4W) y (3TOP) se sometieron a una minimización
energética con el software UCSF Chimera, el cual es un software de modelaje de
biomoléculas en tercera dimensión (Pettersen et al., 2004) . Dicha minimización se realizó
para posibilitar que la energía de interacción entre ligando y receptor en los experimentos
de docking fuera la menor posible.
El primer paso para la minimización de la energía fue eliminar todos los átomos que no
eran estándar (excepto el inhibidor), y después se estableció el número de pasos
descendentes en 1000. Una vez finalizada la minimización se guardaba la estructura y se

22
evaluaban nuevamente en MolProbity las condiciones de la estructura ahora con una
minimización de su energía.
1.2 Diseño computacional de los compuestos inhibidores y preparación previa al
docking
Se realizó el diseño computacional de cincuenta compuestos con aparente actividad
inhibitoria de las enzimas α-glucosidasas extraídos de cuatro especies plantas usadas en la
herbolaria mexicana: Hintonia latiflora y Hintonia standleyana (Rubiaceae), Lingusticum
porteri (Apiaceae), y Brickellia cavanillesii (Asteraceae), tomando como base los datos
publicados en. (Mata et al., 2013)
El diseño se llevó a cabo con el software MarvinSketch, software especializado en el diseño
de moléculas orgánicas en dos dimensiones (Mihala, 2017). Además se empleó el software
Avogadro, programa que ofrece un diseñador químico semántico con fines de edición
molecular avanzada y una plataforma para visualización y modelaje (Hanwell, 2012) para
asignar una estructura tridimensional a todos los compuestos diseñados así como para
implementar un análisis de mecánica molecular que optimizara la geometría de las
moléculas, y nos permitiera a su vez mediante la preparación de un campo de un fuerza
virtual calcular la energía por cantidad de materia (kJ/mol). El campo de fuerza empleado
fue GAFF y el algoritmo empleado fue “Conjugate gradients”. Con los valores de energía
resultantes se creó una base de datos, y a la finalización de los análisis los archivos se
guardaron en formato mol2.
Se diseñó una organización esquemática para clasificar a los cincuenta compuestos
diseñados; esquema 1, esquema 2, esquema 3, esquema 4, y controles. Es necesario
enfatizar que la información estructural contenidad en (Mata, 2013: 473) se limita a

23
esquemas de las estructuras bidimensionales generales, parte importante del presente
trabajo es la preparación de estas moléculas para estudios in silico. Por ello, cabe aclarar
que en aquellos compuestos que poseían varias isoformas con ligeras modificaciones, sólo
se consideró para su diseño la primera isoforma, a excepción de las moléculas 23,24-
dihydrocucurbitacina, 25-acetil-23,24-dihidrocucurbitacina, 3-O-Beta-D-glocupiranosil-
23,24-dihidrocucurbitacina, y 25-O-acetil-3º-Beta-D-glucopiranosil-23,24-
dihidrocurbitacina (identificadas en el artículo como los compuestos 4,5,6 y 7 del esquema
1 respectivamente) (Mata, 2013: 473) , al igual que los compuesto denominado 7-metoxi-
5,3’,4’-trihidroxi-4-fenilcoumarina (10 a en el esquema 1) y el compuesto identificado
como 10 de igual forma en el esquema 1 (Mata, 2013: 473), debido a que nuestro parecer
presentaban modificaciones considerables con galactopiranosil y glucopiranosil, glucósidos
que se asemejan en estructura al inhibidor acarbosa. Por otra parte, la molécula caleina B,
una de las dos sesquitepernlactonas (identificada en el artículo como 46 en el esquema 3)
fue omitida de los experimentos de docking debido a problemas con el diseño de su
estructura, en específico con el componente angeloyloxy que la conforma. En cuanto a la
molécula identificada en el artículo como 57 en el esquema 4, se reemplazó la extensión del
B-D-alopiranosa por alfa-D-alopiranosa debido a que era el único isómero disponible en el
sitio web Chemspider (Royal Society of Chemistry, 2015).
Respecto a la preparación de los compuestos previo a los experimentos de docking, estos se
convirtieron a archivos con extensión pdbqt, formato que lee Vina para identificar a los
ligandos en los experimentos de docking. Para ello se usó el software Racoon de Autodock
Tools (Forli, 2016) el cual convierte simultáneamente varios archivos con extensión mol2
a archivos con extensión pdbqt.

24
1.3 Preparación de los receptores para Docking en Vina
Debido a que UCSF Chimera requiere especificar las medidas del tamaño y las
coordenadas en un plano cartesiano de 3 ejes (X, Y, Z) se creó una rejilla o caja con el
software AutoDock Tools 4.2 (Morris et al., 2009), con el objetivo de encerrar en su
totalidad a la proteína y a su ligando correspondiente para realizar posteriormente en los
experimentos de docking la unión entre ambos.. AutoDock Tools es un software nos
permite editar las cargas Gasteiger y aquellas de los residuos totales, así como remover los
hidrógenos no polares, con el fin de tener nuestra estructura proteica en óptimas
condiciones, y poder definir de manera adecuada las medidas y coordenadas de nuestra
gradilla respecto a la proteína a un espacio de 1 angstrom, que es la longitud máxima que
reconoce Vina. Una vez definidas las medidas y coordenadas, se prosiguió a realizar el
Docking en Vina.
1.3.1. Docking en Vina
Vina es un software que nos permite realizar docking en dos modalidades, rigido y flexible.
Para llevar a cabo la primera modalidad es necesario facilitar un receptor y un ligando en
formato pdbqt, proporcionar las medidas de la rejilla creada en AutoDock Tools, cuyas
dimensiones sean adecuadas para la interacción del ligando con el receptor y especificar un
nivel de exhaustividad. En nuestro caso los experimentos se realizaron con un nivel de
exhaustividad de 3000. La ejecución del programa se llevó a cabo mediante la terminal de
UNIX en Ubuntu.

25
1.4. Preparación de los receptores y ligandos para docking en DOCK6
1.4.1. Preparación de los receptores 3L4W Y 3TOP
Para la realización del docking, Dock6 (Brozell et al.,2012) requiere de dos archivos del
receptor con características distintas. El primero consiste en un archivo en fomato pdb sin
hidrógenos, y un segundo en formato mol2 con hidrógenos. Para la preparación del primero
archivo se abrieron en UCSF Chimera ambos receptores y a través de la acción
(Select>>Residue) se seleccionaron todos los residuos de aminoácidos, las especies
químicas que no pertenecieran al receptor, así como aquellos ligandos que no fueran de
nuestro interés. Para la selección de los hidrógenos se llevó a cabo el siguiente paso
(Select>>Chemistry>>element>>H). Posteriormente se eliminaron todos los objetos
seleccionados a través de la acción (Actions>>Atoms/bonds>>Delete) y se guardaron
ambos receptores en formado pdb.
En cuanto a la preparación del segundo archivo, se abrió el archivo anterior en formato pdb,
y mediante la acción (Tools >> Structure editing>>Dock Prep) se añadieron los Hidrógenos
y las cargas AMBER principalmente. Luego se guardó el archivo en formato mol2.
1.4.2 Preparación de los ligandos
Para la preparación de los ligandos se abrieron todos los archivos de cada esquema en una
ventana de UCSF Chimera con el objetivo de crear cuatro archivos en formato mol2 (uno
por cada esquema). Posteriormente a través de la acción (Tools >> Structure
editing>>Dock Prep) se añadieron los Hidrógenos y las cargas AMBER. Para guardar el
archivo se eligió la opción “single file”, y se especificó que fuera con extensión mol2.

26
1.4.3 Creación de la rejilla en el sitio activo y ligando.
La creación de la rejilla requirió que se abriera en UCSF Chimera ambos receptores
desprotonados (3L4W Y 3TOP), y que se escribiera un archivo DMS a través de la acción
(Actions >> Surface >>) y (Structure >> Editing >>Write DMS). Una vez creado el DMS
ejecutó en la terminal una instrucción llamada INSPH.in mediante el programa “sphgen”,
que creó un archivo con extensión sph. Posteriormente se usó una instrucción de
“sphgen.in” en el programa “showspheres” para generar un archivo pdb que al ejecutarlo
muestras las posibles esferas de interacción en la superficie del receptor. En 3L4W el
cluster o conjunto de esferas seleccionado fue el cinco, en tanto que en 3TOP fue el ocho.
Como último paso para la creación de la rejilla se crearon dos instrucciones “box” y “grid”
que se ejecutaron en la terminal con los programas “showbox” y “grid”.
1.4.4 Docking rígido con DOCK 6
Al igual que VINA, DOCK 6 tiene la capacidad de realizar docking rígido. Este docking
consiste en la unión del ligando con el receptor teniendo sólo el primero dicha movilidad
durante el experimento. Para su realización se creó una instrucción que indicara el archivo
que contenía los ligandos de cada esquema en formato mol2, el receptor sin hidrógenos, y
el archivo de la rejilla previamente creado. Dicha instrucción se ejecutó en la terminal con
el programa “DOCK 6”.
1.5 Docking “anchor and grow” con DOCK 6
El docking “anchor and grow” es una variación del docking rígido que de igual forma se
lleva a cabo con DOCK 6. La realización de este tipo de docking requirió el uso de los

27
receptores y ligandos con el mismo formato empleado en el docking rígido con DOCK 6,
los archivos de retícula, así como las instrucciones (archivos con formato in) y sus
respectivos programas necesarios para su ejecución. La instrucción final de igual forma se
ejecutó desde la terminal con el software DOCK 6.
2. Análisis Estadístico
Se realizó una regresión lineal entre los diferentes tipos de tratamientos de Vina y DOCK 6,
así como la estadística descriptiva de todos los resultados por cada tratamiento con el
software GraphPad Prism versión 6.00 para Windows.
RESULTADOS.
1. Experimentos de docking en Vina
1.1 Experimentos con la estructura cristalográfica 3L4W (N-terminal)
Para la realización de los experimentos de docking en Vina, se emplearon los cincuenta
ligandos diseñados computacionalmente extraídos en un inicio de herbolaria mexicana
(Mata,2013). Los compuestos que mostraron tener mayor afinidad por el receptor fueron las
4 fenil-coumarinas, identificadas como: RM 17 (-9.0 kcal/mol), RM19 (-9.5 kcal/mol),
RM20 (-9.3 kcal/mol) (Fig.10). Así mismo las afinidades de estos compuestos superaron la
afinidad promedio calculada para todos (-7.39 kcal/mol) (Fig.11). Esto coincide con
resultados obtenidos de forma experimental en que los compuestos RM19 y RM20
extraídos de H. standleyana demostraron tener mayores efectos hipoglucémicos que los
controles en ratas diabéticas (Mata, 2013). Respecto a los compuestos control, en nuestro

28
experimento, el miglitol mostró tener afinidad por el receptor (-6.1 kcal/mol) (Fig.10),
mientras que la acarbosa tuvo una pobre afinidad por receptor.
RM1
RM2
RM3
RM4
RM5
RM6
RM7
RM8
RM9
RM10
RM10a
RM11
RM12
RM15
RM17
RM18
RM19
RM20
RM22
RM23
RM24
RM25
RM26
RM27
RM28
RM29
RM30
RM31
RM32
RM33
RM34
RM35
RM36
RM37
RM38
RM39
RM42
RM43
RM47
RM53
RM54
RM55
RM56
RM59
RM57
RM58
RM60
RM65
RM66
RM67
Miglitol
Acarbosa
-1 0
-9
-8
-7
-6
-5
-4
-3
-2
-1
0
1
Afin
idad
(kcal/m
ol)
Figura 10. Afinidad del panel de compuestos contra el receptor subunidad N-terminal (3L4W) por Vina. Las afinidades se comportan de manera extrema: algunos compuestos no se unen al sitio catalítico. Aquellos que sí lo hacen despliegan afinidades iguales o mayores que el compuesto control (Miglitol). En el eje de las X se representan los ligandos usando los los colores y códigos alfabéticos propuestos en los anexos 1 al 4. En el eje de las Y se localizan las afinidades de los compuestos por el receptor expresadas en kcal/mol. La afinidad calculada por vina para el miglitol, cocristalizado con la estructura 3L4W, se señala con un círculo.

29
Afin
idad
(kcal/m
ol)
RM1
RM2
RM3
RM4
RM5
RM6
RM7
RM10a
RM11
RM12
RM17
RM18
RM19
RM20
RM22
RM25
RM26
RM29
RM30
RM31
RM32
RM33
RM36
RM37
RM43
RM47
RM53
RM55
RM60
Miglitol
Acarbosa
- 1 0
-9
-8
-7
-6
-5
-4
-3
-2
-1
0
1
Figura 11. Estadística descriptiva de la afinidad de los resultados computacionales con 3L4W por Vina. La energía de afinidad promedio es representada por la línea gruesa punteada (-7.390 kcal/mol). La desviación estándar (1.645) el promedio se representa por las líneas punteadas delgadas (-9.035 kcal/mol y -5.74 kcal/mol). Todos aquellos ligandos con energía de afinidad igual a 0 fueron omitidos del análisis. Todos aquellos puntos que rebasan la desviación estándar menos el promedio (-9.035 kcal/mol) son considerados buenos inhibidores potenciales.
1.2 Experimentos con la estructura cristalográfica 3TOP (C-terminal)
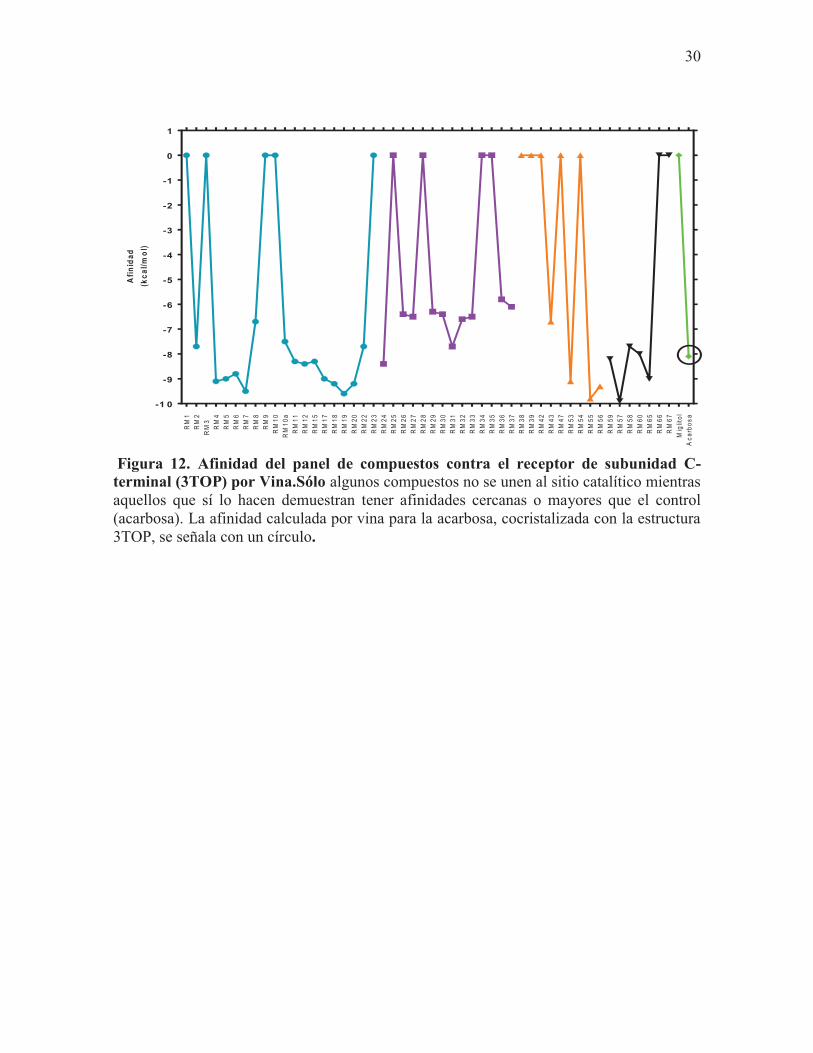
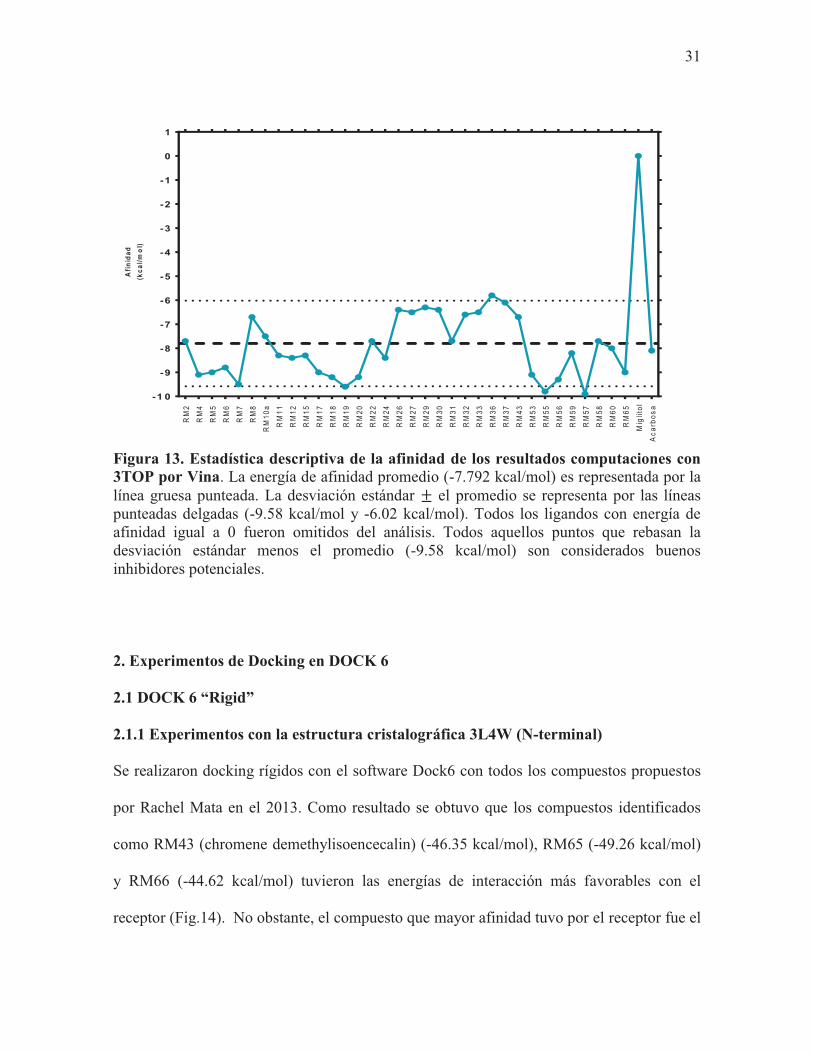
Los compuestos que tuvieron mayor afinidad por el receptor fueron aquellos identificados
como RM19 (4-fenil coumarina) (-9.6 kcal/mol), RM55 (-9.8 kcal/mol) y RM57 (9.9
kcal/mol) (Fig.12). De igual forma, tuvieron una afinidad mayor al promedio de todos los
compuestos (-7.8 kcal/mol) (Fig.13) Respecto a los controles, sólo la acarbosa mostró tener
afinidad por el receptor (-8.1 kcal/mol) (Fig.12).
30
RM1
RM2
RM3
RM4
RM5
RM6
RM7
RM8
RM9
RM10
RM10a
RM11
RM12
RM15
RM17
RM18
RM19
RM20
RM22
RM23
RM24
RM25
RM26
RM27
RM28
RM29
RM30
RM31
RM32
RM33
RM34
RM35
RM36
RM37
RM38
RM39
RM42
RM43
RM47
RM53
RM54
RM55
RM56
RM59
RM57
RM58
RM60
RM65
RM66
RM67
Miglitol
Acarbosa
-1 0
-9
-8
-7
-6
-5
-4
-3
-2
-1
0
1Afin
idad
(kcal/m
ol)
Figura 12. Afinidad del panel de compuestos contra el receptor de subunidad C-terminal (3TOP) por Vina.Sólo algunos compuestos no se unen al sitio catalítico mientras aquellos que sí lo hacen demuestran tener afinidades cercanas o mayores que el control (acarbosa). La afinidad calculada por vina para la acarbosa, cocristalizada con la estructura 3TOP, se señala con un círculo.
31
Afin
idad
(kcal/m
ol)
RM2
RM4
RM5
RM6
RM7
RM8
RM10a
RM11
RM12
RM15
RM17
RM18
RM19
RM20
RM22
RM24
RM26
RM27
RM29
RM30
RM31
RM32
RM33
RM36
RM37
RM43
RM53
RM55
RM56
RM59
RM57
RM58
RM60
RM65
Miglitol
Acarbosa
-1 0
-9
-8
-7
-6
-5
-4
-3
-2
-1
0
1
Figura 13. Estadística descriptiva de la afinidad de los resultados computaciones con 3TOP por Vina. La energía de afinidad promedio (-7.792 kcal/mol) es representada por la línea gruesa punteada. La desviación estándar el promedio se representa por las líneas punteadas delgadas (-9.58 kcal/mol y -6.02 kcal/mol). Todos los ligandos con energía de afinidad igual a 0 fueron omitidos del análisis. Todos aquellos puntos que rebasan la desviación estándar menos el promedio (-9.58 kcal/mol) son considerados buenos inhibidores potenciales.
2. Experimentos de Docking en DOCK 6
2.1 DOCK 6 “Rigid”
2.1.1 Experimentos con la estructura cristalográfica 3L4W (N-terminal)
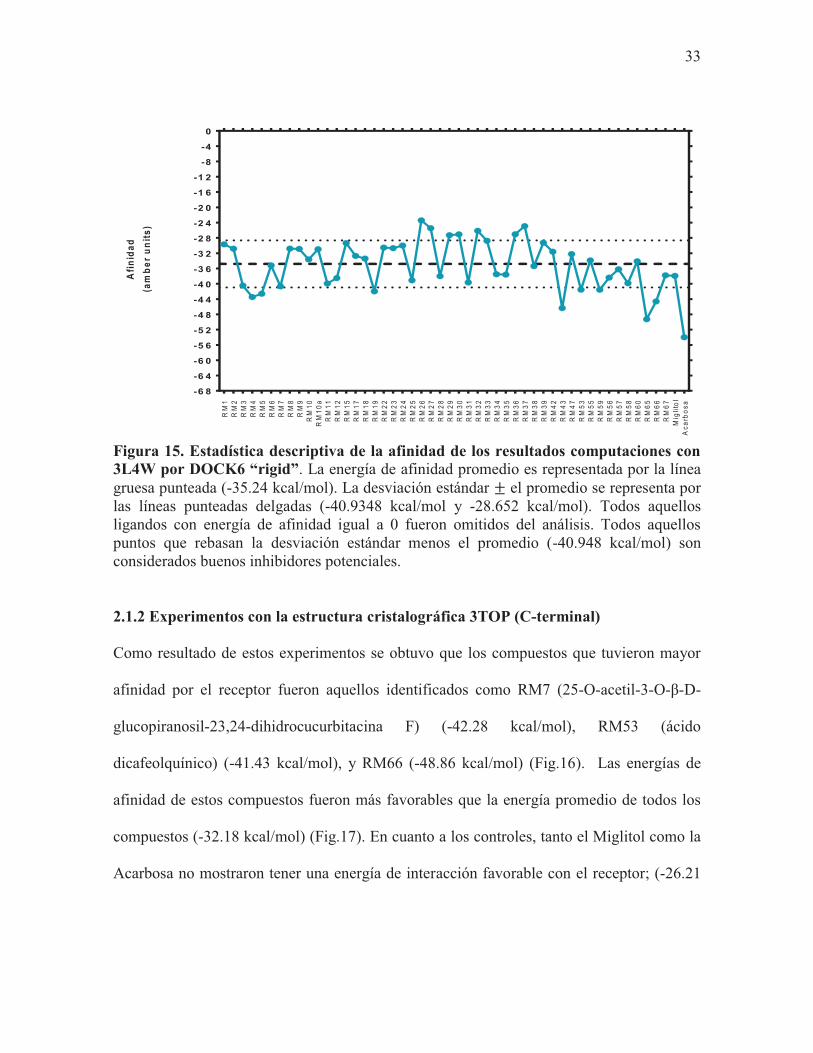
Se realizaron docking rígidos con el software Dock6 con todos los compuestos propuestos
por Rachel Mata en el 2013. Como resultado se obtuvo que los compuestos identificados
como RM43 (chromene demethylisoencecalin) (-46.35 kcal/mol), RM65 (-49.26 kcal/mol)
y RM66 (-44.62 kcal/mol) tuvieron las energías de interacción más favorables con el
receptor (Fig.14). No obstante, el compuesto que mayor afinidad tuvo por el receptor fue el

32
control Acarbosa (-53.98 kcal/mol), en tanto que el Miglitol no mostró tener afinidad por el
receptor (Fig.14). Estos compuestos incluyendo la acarbosa tuvieron una afinidad mayor
que el promedio de los compuestos (-35.24 kcal/mol) (Fig.15).
RM1
RM2
RM3
RM4
RM5
RM6
RM7
RM8
RM9
RM10
RM10a
RM11
RM12
RM15
RM17
RM18
RM19
RM20
RM22
RM23
RM24
RM25
RM26
RM27
RM28
RM29
RM30
RM31
RM32
RM33
RM34
RM35
RM36
RM37
RM38
RM39
RM42
RM43
RM47
RM53
RM54
RM55
RM59
RM56
RM57
RM58
RM60
RM65
RM66
RM67
Miglitol
Acarbosa
-6 8
-6 4
-6 0
-5 6
-5 2
-4 8
-4 4
-4 0
-3 6
-3 2
-2 8
-2 4
-2 0
-1 6
-1 2
-8
-4
0
Afinidad
(Amber
units)
Figura 14. Afinidad del panel de compuestos contra el receptor de subunidad N-terminal (3L4W) por DOCK 6 “rigid”. La mayoría de las afinidades se comportan de manera discreta: sólo un compuesto (RM 20) no se une al sitio catalítico mientras que aquellos que sí lo hacen muestran afinidades similares o sutilmente mayores que el control (miglitol). La afinidad calculada por DOCK 6 “rigid” para el miglitol cocristalizado con la estructura 3L4W, se señala con un círculo.
33
Afin
idad
(amberun
its)
RM1
RM2
RM3
RM4
RM5
RM6
RM7
RM8
RM9
RM10
RM10
aRM11
RM12
RM15
RM17
RM18
RM19
RM22
RM23
RM24
RM25
RM26
RM27
RM28
RM29
RM30
RM31
RM32
RM33
RM34
RM35
RM36
RM37
RM38
RM39
RM42
RM43
RM47
RM53
RM55
RM59
RM56
RM57
RM58
RM60
RM65
RM66
RM67
Miglitol
Acarbosa
-6 8
-6 4
-6 0
-5 6
-5 2
-4 8
-4 4
-4 0
-3 6
-3 2
-2 8
-2 4
-2 0
-1 6
-1 2
-8
-4
0
Figura 15. Estadística descriptiva de la afinidad de los resultados computaciones con 3L4W por DOCK6 “rigid”. La energía de afinidad promedio es representada por la línea gruesa punteada (-35.24 kcal/mol). La desviación estándar el promedio se representa por las líneas punteadas delgadas (-40.9348 kcal/mol y -28.652 kcal/mol). Todos aquellos ligandos con energía de afinidad igual a 0 fueron omitidos del análisis. Todos aquellos puntos que rebasan la desviación estándar menos el promedio (-40.948 kcal/mol) son considerados buenos inhibidores potenciales. 2.1.2 Experimentos con la estructura cristalográfica 3TOP (C-terminal)
Como resultado de estos experimentos se obtuvo que los compuestos que tuvieron mayor
afinidad por el receptor fueron aquellos identificados como RM7 (25-O-acetil-3-O-β-D-
glucopiranosil-23,24-dihidrocucurbitacina F) (-42.28 kcal/mol), RM53 (ácido
dicafeolquínico) (-41.43 kcal/mol), y RM66 (-48.86 kcal/mol) (Fig.16). Las energías de
afinidad de estos compuestos fueron más favorables que la energía promedio de todos los
compuestos (-32.18 kcal/mol) (Fig.17). En cuanto a los controles, tanto el Miglitol como la
Acarbosa no mostraron tener una energía de interacción favorable con el receptor; (-26.21
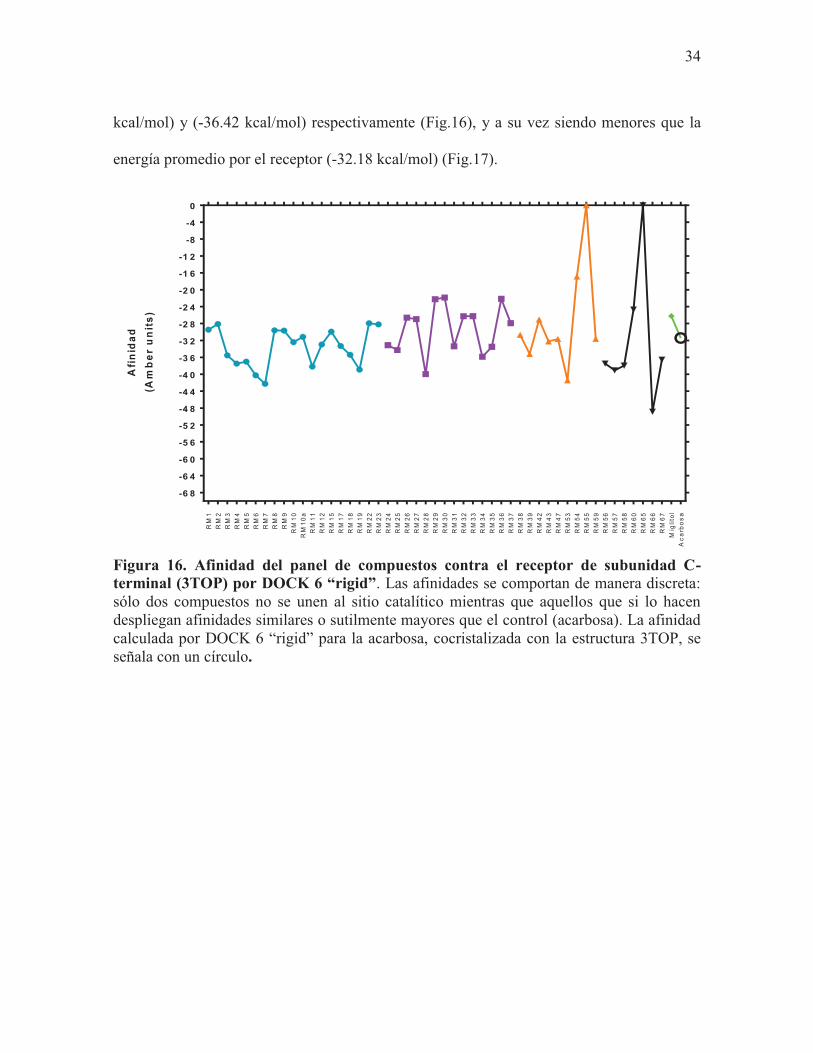
34
kcal/mol) y (-36.42 kcal/mol) respectivamente (Fig.16), y a su vez siendo menores que la
energía promedio por el receptor (-32.18 kcal/mol) (Fig.17).
RM1
RM2
RM3
RM4
RM5
RM6
RM7
RM8
RM9
RM10
RM10a
RM11
RM12
RM15
RM17
RM18
RM19
RM22
RM23
RM24
RM25
RM26
RM27
RM28
RM29
RM30
RM31
RM32
RM33
RM34
RM35
RM36
RM37
RM38
RM39
RM42
RM43
RM47
RM53
RM54
RM55
RM59
RM56
RM57
RM58
RM60
RM65
RM66
RM67
Miglitol
Acarbosa
-6 8
-6 4
-6 0
-5 6
-5 2
-4 8
-4 4
-4 0
-3 6
-3 2
-2 8
-2 4
-2 0
-1 6
-1 2
-8
-4
0
Afinidad
(Amber
units)
Figura 16. Afinidad del panel de compuestos contra el receptor de subunidad C-terminal (3TOP) por DOCK 6 “rigid”. Las afinidades se comportan de manera discreta: sólo dos compuestos no se unen al sitio catalítico mientras que aquellos que si lo hacen despliegan afinidades similares o sutilmente mayores que el control (acarbosa). La afinidad calculada por DOCK 6 “rigid” para la acarbosa, cocristalizada con la estructura 3TOP, se señala con un círculo.

35
Afin
idad
(amberun
its)
RM1
RM2
RM3
RM4
RM5
RM6
RM7
RM8
RM9
RM10
RM10a
RM11
RM12
RM15
RM17
RM18
RM19
RM22
RM23
RM24
RM25
RM26
RM27
RM28
RM29
RM30
RM31
RM32
RM33
RM34
RM35
RM36
RM37
RM38
RM39
RM42
RM43
RM47
RM53
RM54
RM59
RM56
RM57
RM58
RM60
RM66
RM67
Miglitol
Acarbosa
-6 8
-6 4
-6 0
-5 6
-5 2
-4 8
-4 4
-4 0
-3 6
-3 2
-2 8
-2 4
-2 0
-1 6
-1 2
-8
-4
0
Figura 17. Estadística descriptiva de la afinidad de los resultados computacionales con 3TOP por DOCK 6 “rigid”. La energía de afinidad promedio se representa por la línea gruesa interrumpida (-32.18 kcal/mol). La desviación estándar el promedio es representada por las líneas punteadas delgadas (-38.39 kcal/mol y 26.11 kcal/mol). Todos aquellos ligandos con energía de afinidad igual a 0 fueron omitidos del análisis. Todos aquellos puntos que rebasan la desviación estándar menos el promedio (-38.18 kcal/mol) son considerados buenos inhibidores potenciales.
2.2 DOCK 6 “anchor and grow”
2.2.1 Experimentos con la estructura cristalográfica 3L4W (N-terminal)
Se realizó docking flexible con el software DOCK 6. Los compuestos que tuvieron una
mayor afinidad por el receptor fueron la 4-fenilcoumarina identificada como RM19 (-54.72
kcal/mol), el ácido dicafeoilquínico (RM53) (-54.82 kcal/mol) y el compuesto RM65 (-
66.78 kcal/mol) (Fig.18). De igual forma que con los experimentos realizados en Vina, el
compuesto RM19 mostró tener una energía de interacción favorable con el receptor,
resultado que coincide nuevamente con los resultados experimentales en los que este
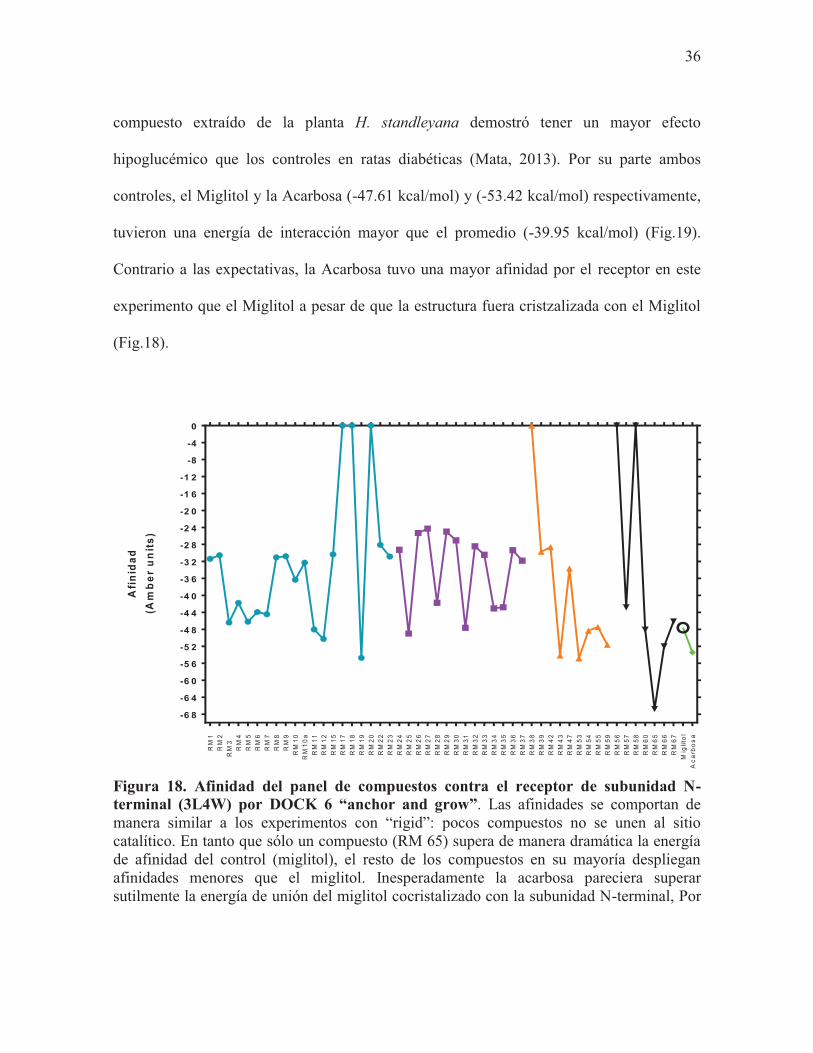
36
compuesto extraído de la planta H. standleyana demostró tener un mayor efecto
hipoglucémico que los controles en ratas diabéticas (Mata, 2013). Por su parte ambos
controles, el Miglitol y la Acarbosa (-47.61 kcal/mol) y (-53.42 kcal/mol) respectivamente,
tuvieron una energía de interacción mayor que el promedio (-39.95 kcal/mol) (Fig.19).
Contrario a las expectativas, la Acarbosa tuvo una mayor afinidad por el receptor en este
experimento que el Miglitol a pesar de que la estructura fuera cristzalizada con el Miglitol
(Fig.18).
RM1
RM2
RM3
RM4
RM5
RM6
RM7
RM8
RM9
RM10
RM10a
RM11
RM12
RM15
RM17
RM18
RM19
RM20
RM22
RM23
RM24
RM25
RM26
RM27
RM28
RM29
RM30
RM31
RM32
RM33
RM34
RM35
RM36
RM37
RM38
RM39
RM42
RM43
RM47
RM53
RM54
RM55
RM59
RM56
RM57
RM58
RM60
RM65
RM66
RM67
Miglitol
Acarbosa
-6 8
-6 4
-6 0
-5 6
-5 2
-4 8
-4 4
-4 0
-3 6
-3 2
-2 8
-2 4
-2 0
-1 6
-1 2
-8
-4
0
Afin
idad
(Amber
units)
Figura 18. Afinidad del panel de compuestos contra el receptor de subunidad N-terminal (3L4W) por DOCK 6 “anchor and grow”. Las afinidades se comportan de manera similar a los experimentos con “rigid”: pocos compuestos no se unen al sitio catalítico. En tanto que sólo un compuesto (RM 65) supera de manera dramática la energía de afinidad del control (miglitol), el resto de los compuestos en su mayoría despliegan afinidades menores que el miglitol. Inesperadamente la acarbosa pareciera superar sutilmente la energía de unión del miglitol cocristalizado con la subunidad N-terminal, Por

37
último, la afinidad calculada por DOCK 6 “anchor and grow” para el miglitol se señala con un círculo.
Afin
idad
(amberun
its)
RM1
RM2
RM3
RM4
RM5
RM6
RM7
RM8
RM9
RM10
RM10
aRM11
RM12
RM15
RM19
RM22
RM23
RM24
RM25
RM26
RM27
RM28
RM29
RM30
RM31
RM32
RM33
RM34
RM35
RM36
RM37
RM39
RM42
RM43
RM47
RM53
RM54
RM55
RM59
RM57
RM60
RM65
RM66
RM67
Miglitol
Acarbosa
-6 8-6 4-6 0-5 6-5 2-4 8-4 4-4 0-3 6-3 2-2 8-2 4-2 0-1 6-1 2-8-40
Figura 19. Estadística descriptiva de la afinidad de los resultados computacionales con 3L4W por DOCK 6 “anchor and grow”. La energía de afinidad promedio es representada por la línea gruesa punteada (-39.95 kcal/mol). La desviación estándar el promedio se representa por las líneas punteadas delgadas (-49.7 kcal/mol y -29.0 kcal/mol). Todos aquellos ligandos con energía de afinidad igual a 0 fueron omitidos del análisis. Todos aquellos puntos que rebasan la desviación estándar menos el promedio (-49.7 kcal/mol) son considerados buenos inhibidores potenciales.
2.2.2 Experimentos con la estructura cristalográfica 3TOP (C-terminal)
Los compuestos identificados como RM19 (4-fenilcoumarina), RM 53 (ácido
dicafeoilquínico) y RM 59, tuvieron la misma energía de afinidad para el receptor (-53.31
kcal/mol) (Fig.20). El ligando identificado como RM43 (cromeno, -54.3 kcal/mol) junto
con los otros tres compuestos también tuvo una energía de unión favorable (Fig.20).
Respecto a los controles sólo el miglitol (-59.93 kcal/mol) tuvo una energía de unión mayor

38
que superaba a la energía promedio (-37.89 kcal/mol) (Fig.21), contrario a la acarbosa (-
35.39 kcal/mol). Este resultado fue inesperado debido a que el isómero 3TOP de la α-
glucosidasa fue cristalizado con la acarbosa como principal ligando inhibidor. RM1
RM2
RM3
RM4
RM5
RM6
RM7
RM8
RM9
RM10
RM10a
RM11
RM12
RM15
RM17
RM18
RM19
RM20
RM22
RM23
RM24
RM25
RM26
RM27
RM28
RM29
RM30
RM31
RM32
RM33
RM34
RM35
RM36
RM37
RM38
RM39
RM42
RM43
RM47
RM53
RM54
RM55
RM59
RM56
RM57
RM58
RM60
RM65
RM66
RM67
Miglitol
Acarbosa
-6 8
-6 4
-6 0
-5 6
-5 2
-4 8
-4 4
-4 0
-3 6
-3 2
-2 8
-2 4
-2 0
-1 6
-1 2
-8
-4
0
Afin
idad
(Amberun
its)
Figura 20. Afinidad del panel de compuestos contra el receptor de subunidad C-terminal (3TOP) por DOCK 6 “anchor and grow”. Las afinidades se comportan de manera similar al experimento con 3L4W. Sólo algunos compuestos no se unen al sitio catalítico.La cantidad de compuestos que despliegan una energía de afinidad mayor que el control, es similar a la cantidad de compuestos que muestra una afinidad menor que el control (acarbosa). Inesperadamente en este experimento el miglitol exhibe una energía de unión mayor que la acarbosa cocristalizada con la subunidad C-terminal. Por último, la afinidad calculada por DOCK 6 “anchor and grow” para la acarbosa se señala con un círculo.

39
Afin
idad
(amberun
its)
RM1
RM2
RM3
RM4
RM5
RM6
RM7
RM8
RM9
RM10
RM10
aRM11
RM12
RM15
RM19
RM22
RM23
RM24
RM25
RM26
RM27
RM28
RM29
RM30
RM31
RM32
RM33
RM34
RM35
RM36
RM37
RM39
RM42
RM43
RM47
RM53
RM54
RM59
RM57
RM60
RM67
Miglitol
Acarbosa
-6 8-6 4-6 0-5 6-5 2-4 8-4 4-4 0-3 6-3 2-2 8-2 4-2 0-1 6-1 2-8-40
Figura 21. Estadística descriptiva de la afinidad de los resultados computacionales con 3TOP por DOCK 6 “anchor and grow”. La línea punteada gruesa representa la energía promedio de unión (-37.89 kcal/mol). La desviación estándar el promedio representada por las líneas punteadas delgadas (-46.34 kcal/mol y -28.29 kcal/mol). Todos aquellos ligandos con energía de afinidad igual a 0 fueron omitidos del análisis. Todos aquellos puntos que rebasan la desviación estándar menos el promedio (-46.34 kcal/mol) son considerados buenos inhibidores potenciales.
3. Resultados de Vina vs DOCK 6 “Rigid”
3.1 Estructura 3L4W (N-terminal)
Para investigar la posible relación existente entre los resultados de ambos métodos, se
realizó un análisis de regresión lineal. Los resultados del análisis sugieren que las energías
de afinidad propuestas por ambos métodos están pobremente relacionadas entre sí (F26,
=8.984, r2=0.2568, p= 0.005; (Fig.22). No obstante, la distribución mayoritaria de los
datos, dentro de los límites de la banda de confianza de 99% alude a que, a pesar del bajo

40
coeficiente de correlación, los datos poseen una distribución uniforme y coherente con el
modelo de regresión lineal. Por ende, esta distribución sugiere que hay algunos resultados
que coinciden con alta afinidad en ambos métodos a pesar de ser métodos ortogonales,
siendo el compuesto RM 19 (4-fenil cumarina) aquél con mayor afinidad por el receptor en
ambos métodos, y diametralmente opuesto al compuesto control miglitol que, también en
ambos métodos, posee una pobre afinidad por el receptor. Por último, aunque ambos
métodos despliegan resultados de alta afinidad que coinciden, podemos deducir que el
método de Vina en esta comparación de métodos fue mejor, lo cual puede entenderse de
mejor manera con el valor de la pendiente de la recta obtenida (m= 3.818), que representa
una pendiente positiva inclinada sobre el eje X equivalente al eje de los datos de Vina,
sobre el que se distribuye el modelo de regresión lineal (Fig.22)

41
V in a
Rigid
- 1 0 -9 -8 -7 -6 -5
-6 0
-4 0
-2 0
0
Figura 22. Regresión lineal de los resultados de unión de Vina vs DOCK 6 “rigid” con 3L4W. Las energías de unión calculadas en kcal/mol por Vina se localizan en el eje de las X, mientras que las energías de unión calculadas en kcal/mol por DOCK 6 “rigid” se localizan en el eje de las Y. Los resultados calculados por ambos métodos están pobremente relacionadas como se muestra en la recta de regresión lineal. La mayoría de los ligandos se concentran en la zona de alta afinidad calculada por Rigid pero en la zona de mediana afinidad calculada por Vina. El control miglitol se representa con un triángulo blanco con una alta afinidad por el receptor calculada por “rigid pero una baja afinidad calculada por Vina .
3.2 Estructura 3TOP (C-terminal)
De igual forma se realizó un análisis de regresión lineal con los resultados de energía de
unión para la estructura cristalográfica 3TOP. Los resultados de la regresión sugieren que
existe una relación medianamente fuerte entre los resultados de ambos métodos (F29,
=69.67, r2=0.7061, p< 0.001); (Fig.23). No obstante, a pesar de que el coeficiente de
correlación no es demasiado alto, las bandas de confianza en 99%, representadas con las
líneas punteadas, nos sugieren que nuestra modelo es altamente confiable, y que la mayoría

42
de los datos se distribuyen a lo largo de la pendiente (m= 4.152), la cual también sugiere
que usando cualquiera de ambos métodos, se obtendrían afinidades muy similares por el
receptor, y que si se usan en conjunto, la calidad en cuanto a precisión de los resultados
mejora. En cuanto al compuesto con mayor afinidad por el receptor, el compuesto
identificado como RM57 lo fue para ambos métodos, seguido del compuesto RM19 (4-fenil
cumarina). De igual forma, considerando cualitativamente los resultados obtenidos con la
estructura 3L4W, esto refuerza los resultados que correlacionan en ambos métodos. Por
último, también es posible apreciar diferencias significativas, pero no evidentes, entre los
sitios de unión de 3L4W y 3TOP (Fig.23).
V in a
Rigid
-1 1 -1 0 -9 -8 -7 -6 -5-8 0
-6 0
-4 0
-2 0
0
Figura 23. Regresión lineal de los resultados de unión de Vina vs DOCK 6 “rigid” con 3TOP Los resultados calculados por ambos métodos despliegan una relación medianamente fuerte como se muestra en la recta de regresión lineal. La distribución de los ligandos en la recta de correlación es homogénea. El control acarbosa se representa con un triángulo blanco invertido con una alta afinidad por el receptor calculada por “rigid pero una baja afinidad calculada por Vina.

43
4. Resultados de Vina vs DOCK6 “anchor and grow”
4.1 Estructura 3L4W (N-terminal)
De igual forma para explorar la posible relación existente entre los resultados calculados
por Vina y aquellos calculados por DOCK 6 “anchor and grow”, se realizó un análisis de
regresión lineal. El análisis sugiere que los resultados obtenidos de ambos métodos están
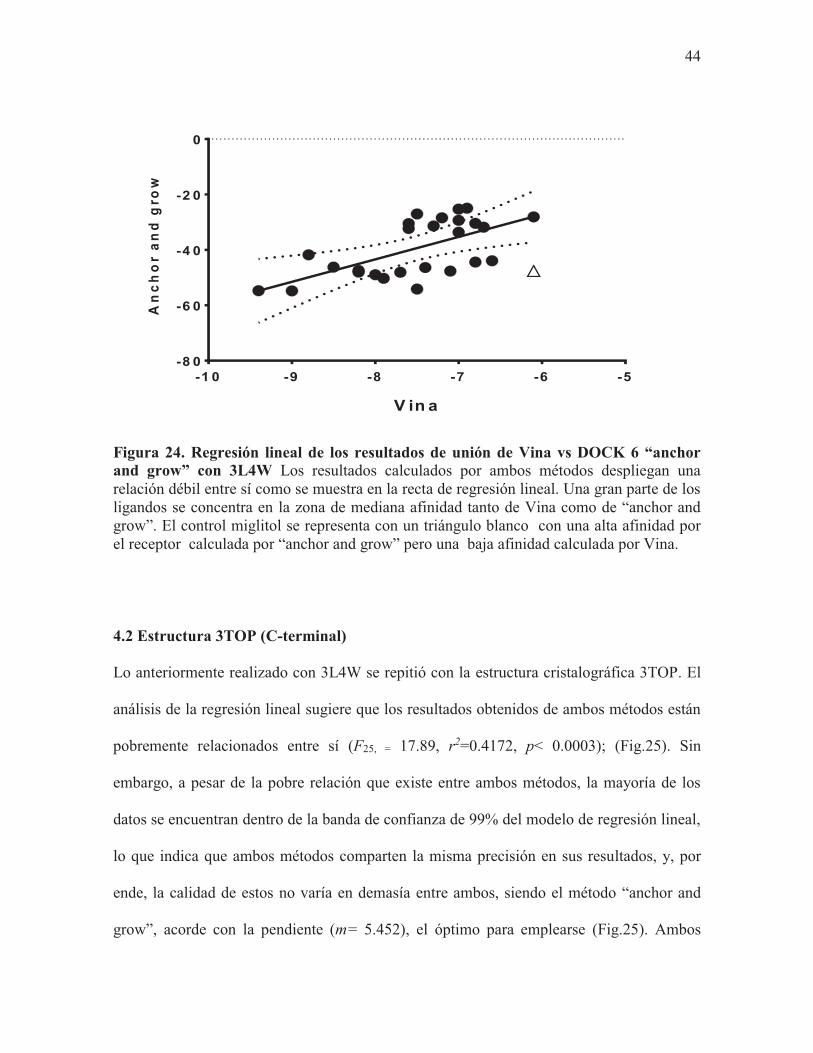
pobremente relacionados entre sí (F24, =15.94, r2=0.3991, p< 0.0005); (Fig.24). La banda de
confianza del 99% nos confirma que, en efecto, los resultados de ambos métodos no están
relacionados entre sí, y que hay una cantidad considerable de resultados fuera del modelo
de regresión planteado. La pendiente (m=8.112), nos sugiere que, de elegirse un método, el
más oportuno sería el de DOCK6 “anchor and grow”, sin embargo, en cuanto a calidad y
precisión de resultados, DOCK6 para este receptor no sería superior a Vina. Nuevamente,
como en el modelo anterior planteado para 3L4W, el compuesto RM19 (4-fenil cumarina)
es el que posee mayor afinidad por el receptor en ambos métodos, seguido del compuesto
RM53 (ácido dicafeolquínico). Cabe resaltar que, para ambos métodos, el compuesto
control miglitol, fue uno de los que se mostró menos afines por el receptor (Fig.24).
44
V in a
Anch
oran
dgrow
- 1 0 -9 -8 -7 -6 -5-8 0
-6 0
-4 0
-2 0
0
Figura 24. Regresión lineal de los resultados de unión de Vina vs DOCK 6 “anchor and grow” con 3L4W Los resultados calculados por ambos métodos despliegan una relación débil entre sí como se muestra en la recta de regresión lineal. Una gran parte de los ligandos se concentra en la zona de mediana afinidad tanto de Vina como de “anchor and grow”. El control miglitol se representa con un triángulo blanco con una alta afinidad por el receptor calculada por “anchor and grow” pero una baja afinidad calculada por Vina.
4.2 Estructura 3TOP (C-terminal)
Lo anteriormente realizado con 3L4W se repitió con la estructura cristalográfica 3TOP. El
análisis de la regresión lineal sugiere que los resultados obtenidos de ambos métodos están
pobremente relacionados entre sí (F25, = 17.89, r2=0.4172, p< 0.0003); (Fig.25). Sin
embargo, a pesar de la pobre relación que existe entre ambos métodos, la mayoría de los
datos se encuentran dentro de la banda de confianza de 99% del modelo de regresión lineal,
lo que indica que ambos métodos comparten la misma precisión en sus resultados, y, por
ende, la calidad de estos no varía en demasía entre ambos, siendo el método “anchor and
grow”, acorde con la pendiente (m= 5.452), el óptimo para emplearse (Fig.25). Ambos
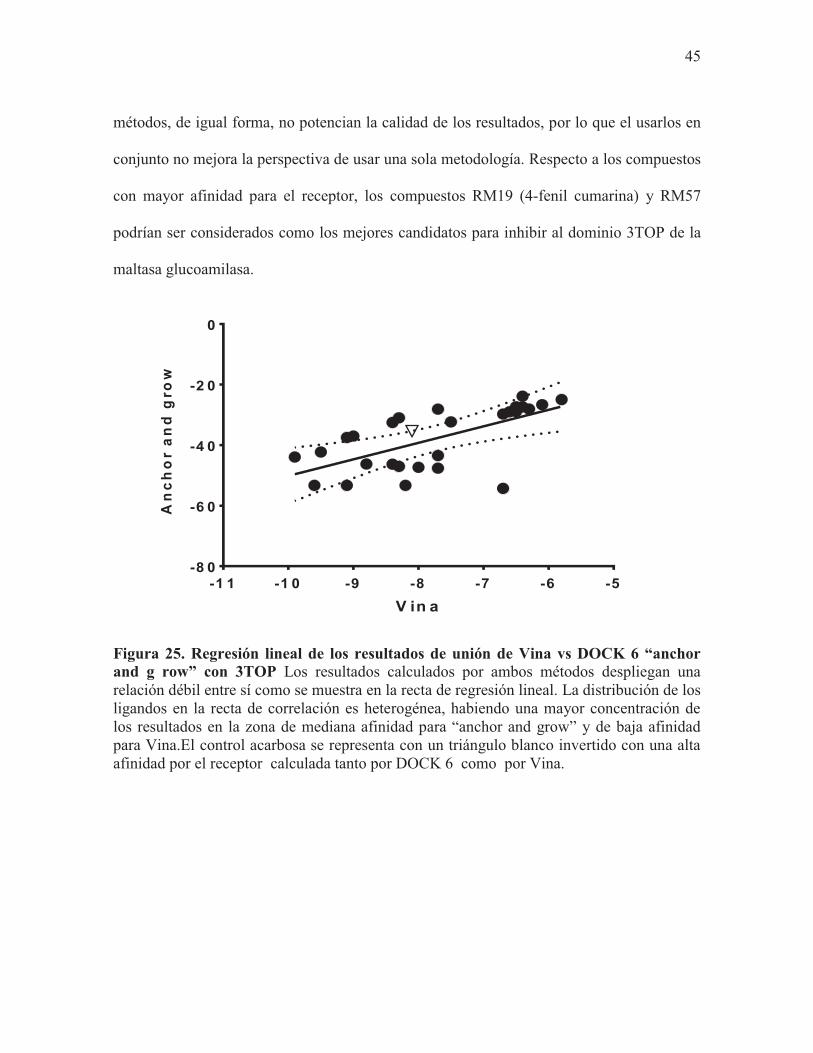
45
métodos, de igual forma, no potencian la calidad de los resultados, por lo que el usarlos en
conjunto no mejora la perspectiva de usar una sola metodología. Respecto a los compuestos
con mayor afinidad para el receptor, los compuestos RM19 (4-fenil cumarina) y RM57
podrían ser considerados como los mejores candidatos para inhibir al dominio 3TOP de la
maltasa glucoamilasa.
V in a
Anch
oran
dgrow
-1 1 -1 0 -9 -8 -7 -6 -5-8 0
-6 0
-4 0
-2 0
0
Figura 25. Regresión lineal de los resultados de unión de Vina vs DOCK 6 “anchor and g row” con 3TOP Los resultados calculados por ambos métodos despliegan una relación débil entre sí como se muestra en la recta de regresión lineal. La distribución de los ligandos en la recta de correlación es heterogénea, habiendo una mayor concentración de los resultados en la zona de mediana afinidad para “anchor and grow” y de baja afinidad para Vina.El control acarbosa se representa con un triángulo blanco invertido con una alta afinidad por el receptor calculada tanto por DOCK 6 como por Vina.

46
5. Resultados de DOCK 6 “rigid” vs DOCK 6 “anchor and grow”
5.1 Estructura 3L4W (N-terminal)
Por último, con el objetivo de encontrar la posible relación existente entre los resultados
calculados por DOCK 6 “rigid” y aquellos calculados también por DOCK 6 pero con el
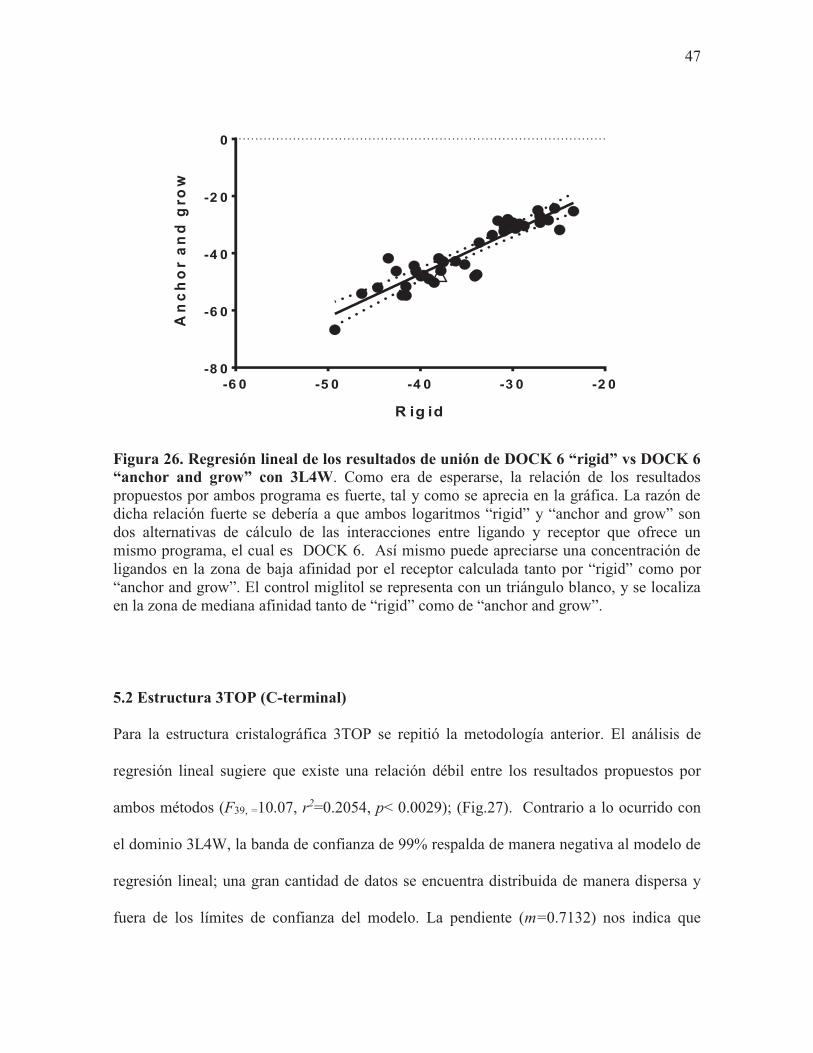
algoritmo “anchor and grow”, se realizó un análisis de regresión lineal. El análisis de los
resultados con la estructura 3L4W sugieren que las energías de unión obtenidas de ambos
métodos están altamente relacionadas entre sí (F41, =233.1, r2=0.8504, p< 0.0001); (Fig.26).
De igual forma, las bandas de confianza de 99%, nos indican que el modelo es altamente
confiable, y que el uso de ambos métodos potencia la posibilidad de obtener mejores
resultados a que si solamente se empleara un método. Aun así, los resultados no son
inesperados; al tratarse de un mismo software DOCK6, pero empleado con distintos
algoritmos (rigid y anchor and grow), es de esperarse que su uso conjunto mejore la
precisión y la calidad de los resultados obtenidos por DOCK6. De igual forma, la pendiente
(m=1.503) nos sugiere que “anchor and grow” y “rigid” funcionan de manera muy similar,
y que los resultados de cada uno poseen la misma calidad, y de usarse ambos mejora aún
más. Respecto a los compuestos con mayor afinidad, contrario a la tendencia de los
anteriores modelos, RM65 junto a RM43 fueron los compuestos más afines por el receptor
en ambos métodos. Por su parte, los ligandos RM19 y RM53 que fueron los más afines por
el receptor en los anteriores modelos, se ubican a la mitad de la pendiente, muy cercanos al
control miglitol (Fig.26).
47
-6 0 -5 0 -4 0 -3 0 -2 0-8 0
-6 0
-4 0
-2 0
0
Anch
oran
dgrow
R ig id
Figura 26. Regresión lineal de los resultados de unión de DOCK 6 “rigid” vs DOCK 6 “anchor and grow” con 3L4W. Como era de esperarse, la relación de los resultados propuestos por ambos programa es fuerte, tal y como se aprecia en la gráfica. La razón de dicha relación fuerte se debería a que ambos logaritmos “rigid” y “anchor and grow” son dos alternativas de cálculo de las interacciones entre ligando y receptor que ofrece un mismo programa, el cual es DOCK 6. Así mismo puede apreciarse una concentración de ligandos en la zona de baja afinidad por el receptor calculada tanto por “rigid” como por “anchor and grow”. El control miglitol se representa con un triángulo blanco, y se localiza en la zona de mediana afinidad tanto de “rigid” como de “anchor and grow”.
5.2 Estructura 3TOP (C-terminal)
Para la estructura cristalográfica 3TOP se repitió la metodología anterior. El análisis de
regresión lineal sugiere que existe una relación débil entre los resultados propuestos por
ambos métodos (F39, =10.07, r2=0.2054, p< 0.0029); (Fig.27). Contrario a lo ocurrido con
el dominio 3L4W, la banda de confianza de 99% respalda de manera negativa al modelo de
regresión lineal; una gran cantidad de datos se encuentra distribuida de manera dispersa y
fuera de los límites de confianza del modelo. La pendiente (m=0.7132) nos indica que

48
“rigid” es ligeramente mejor que “anchor and grow”, sin embargo, el usar de manera
conjunta ambos métodos no mejora la probabilidad de obtener mejores resultados. Al
tratarse de dos algoritmos correspondientes a un mismo programa, lo sugerido por el
modelo de regresión lineal no resulta evidente (Fig,27). Una de las razones podría ser que
DOCK6 resulta ineficiente a la hora de trabajar con receptores de longitud aminioacil
extensa, lo que posiblemente conlleva a que el software experimente dificultades al
momento de encontrar el sitio catalítico, o bien, que la estructura no tenga la misma calidad
cristalográfica que posee 3L4W, y en consecuencia que DOCK6, un software que requiere
más parámetros que Vina, no funcione de manera adecuada. En cuanto a los compuestos
que mostraron tener mayor afinidad por el receptor, se encuentran el compuesto RM53
(ácido dicafeolquínico) a la par que el compuesto RM7 (25-O-acetil-3-O-β-D-
glucopiranosil-23,24-dihidrocucurbitacina F). De igual forma, a pesar de encontrarse fuera
de los límites de confianza del modelo, el compuesto RM19 también suele mostrar una alta
afinidad por el receptor. Por su parte, el compuesto RM6, el cual comparte características
estructurales muy similares al compuesto RM7, también desplegó una alta afinidad en
ambos métodos con una buena correlación entre ellos. Esta clase de ligandos, sin bien no
tuvieron los mejores resultados de unión en los anteriores modelos, siempre desplegaron
una energía de unión relativamente baja, en términos fisicoquímicos, por el receptor.

49
R ig id
Anchorandgrow
- 5 0 -4 0 -3 0 -2 0 -1 0-8 0
-6 0
-4 0
-2 0
0
Figura 27. Regresión lineal de los resultados de unión de DOCK 6 “rigid” vs DOCK 6 “anchor and grow” con 3TOP. Contrario a lo que se esperaría, los resultados propuestos por ambos métodos están pobremente relacionados como se muestra en la gráfica. Esto a pesar de ambos métodos corresponden al programa DOCK 6. La mayoría de los ligadnos como puede apreciarse exhiben una distribución heterogénea a lo largo de la recta de regresión lineal. El compuesto control acarbosa se representa con un triángulo blanco invertido, y se localiza en la zona de mediana afinidad por el receptor calculada tanto por DOCK 6 “rigid” como por DOCK 6 “anchor and grow”.
6.Discusión
Se logró encontrar al menos un inhibidor competitivo de la enzima maltasa-glucoamilasa
para sus dos respectivos dominios catalíticos (N-terminal y C-terminal), el cual fue el
compuesto identificado como RM19 (4-fenil cumarina). Tal compuesto mostró tener una
alta afinidad por el receptor en todos los experimentos de docking con los dos diferentes
softwares empleados; Vina y DOCK6, este último, en su modalidad “rigid” y “anchor and

50
grow”. Por otra parte, si bien RM19, no logró ser el compuesto con la más favorable
energía de unión por el receptor en todos los experimentos, siempre fue uno de los
compuestos con mayor afinidad por el receptor, y en el peor de los casos, su afinidad
mostró ser muy similar al compuesto control en turno (miglitol o acarbosa) cocristalizado
con el dominio catalítico de la enzima maltasa glucoamilasa. No obstante, los demás
compuestos que también desplegaron una alta afinidad por el receptor, no deberían ser
descartados, debido a que los valores de energía de unión de dos de ellos, RM57 Y RM53
(ácido dicafeolquínico), se aproximaron en varios experimentos a la energía de unión
exhibida por RM19. La consideración de estos compuestos como buenos inhibidores
competitivos, radica a su vez, en un factor importante a considerar para futuros estudios;
que es la exploración de posibles efectos adversos sobre la salud de RM19, así como de
RM57 y 5M53. Tal consideración desembocaría, posiblemente, en el diseño de un nuevo
compuesto a partir del conocimiento de estos tres ligandos, con lo cual se buscaría la
eliminación de efectos secundarios que por sí solas estas moléculas pudieran tener. El
rendimiento químico y posterior purificación de estos compuestos versus la cantidad de
planta a usar, sería uno de los aspectos a considerar para el diseño de un fármaco sin efectos
adversos con una actividad inhibitoria superior a los ya empleados actualmente como el
miglitol y la acarbosa. Este aspecto sería evaluado a través de la experimentación in vitro, o
bien, en caso de seguir por la vía de la experimentación in silico, mediante otra técnica
tendría diferente al “docking”, como la técnnica “Quantitative Structure- Activity
Relationship” (QSAR 3D).
En cuanto a la capacidad exploratoria del sitio de unión de los distintos softwares de
docking empleados, Vina y DOCK6, podemos concluir que se aproximaron en gran medida

51
en reproducir la energía de interacción y la afinidad de los compuestos por el receptor, de
tal forma que nuestros resultados coinciden ampliamente con lo ensayado in vitro Mata et
al en el 2013, quienes también reportan al compuesto denominado por nosotros como
RM19, como un inhibidor competitivo con protección gástrica, y de igual forma al
compuesto RM43 (demethylisoencecalin), que en uno de nuestros experimentos con
DOCK6 “rigid” también demostró ser un buen inhibidor de ambos dominios catalíticos de
la enzima glucoamilasa. Sin embargo, si se tuviera que elegir a una metodología en término
de requerimientos de tiempo y poder computacional, Vina proporcionó mejores resultados
para ambos dominios catalíticos de la maltasa glucoamilasa. En cuanto a DOCK6, en su
modalidad “rigid”, generó resultados semejantes a los generados con Vina, para ambos
dominios catalíticos. DOCK 6 “anchor and grow” también tuvo un desempeño muy similar
al de Vina, pero con el dominio catlítico N-terminal (3L4W), y en su uso conjunto con su
completo “rigid”, logró estar altamente correlacionado para este dominio. Por el contrario,
con el dominio C-terminal (3TOP), “anchor and grow”, tuvo resultados disímiles con Vina,
y una pobre correlación con su complemento “rigid”, lo cual nos sugiere que DOCK 6 no
se recomienda para proteínas como 3TOP con una longitud aminoacil extensa debido a que
probablemente, le es difícil encontrar el sitio catalítico, y, por ende, reproducir energías de
interacción.
Por último, podemos concluir que las metodologías in silico, en específico el docking, son
de gran ayuda en modelos de predicción de unión enzima-sustrato, debido a que permiten
ahorrar una cantidad considerable de trabajo, esfuerzo y dinero, previo a la experimentación
in vitro.

52
7.Conclusión
El tratamiento de la diabetes tipo II involucra el uso de hipoglicémicos que se complementa
con un estilo de vida saludable. Los hipoglicémicos orales son el tratamiento más común y
existe una gran diversidad de ellos. Sin embargo, los fármacos con los que se trata causan
efectos secundarios a nivel gastrointestinal, así como estados de hipoglucemia graves que
conllevan tratamiento hospitalario. Por ello, la búsqueda de nuevos fármacos sin estos
efectos secundarios que puedan ser usados para controlar los niveles de glucosa en sangre
sigue siendo una prioridad en el tratamiento de la diabetes. En este trabajo nuestro objetivo
es el de identificar nuevos fármacos inhibidores de su actividad catalítica de las enzimas α-
glucosidasas. Usamos principalmente la herramienta in silico conocida como docking, así
como información de la literatura para muestrear un grupo de compuestos extraídos de la
farmacopea tradicional mexicana. Nuestros resultados sugieren que, al menos, un
compuesto es capaz de inhibir a los dos dominios de la α-glucosidasas que estudiamos.
Además, lo hacen con afinidades comparables a las de dos inhibidores de referencia:
acarbosa y miglitol. Nuestros resultados provienen de dos protocolos ortogonales, es decir,
refuerzan nuestros resultados al ser métodos distintos.
Nuestros resultados son alentadores y conllevan a la planeación de experimentos in vitro
para probar la eficacia de las moléculas identificadas.

53
ANEXOS ESQUEMA 1
RM1 RM2
RM3 R1 = H R2= β-D-glucopiranosil
RM4 R1= H; R2= H RM5 R1= H; R2= Ac RM6 R1= β-D-glucopiranosil; R2= H RM7 R1=β-D-glucopiranosil; R2= Ac
54
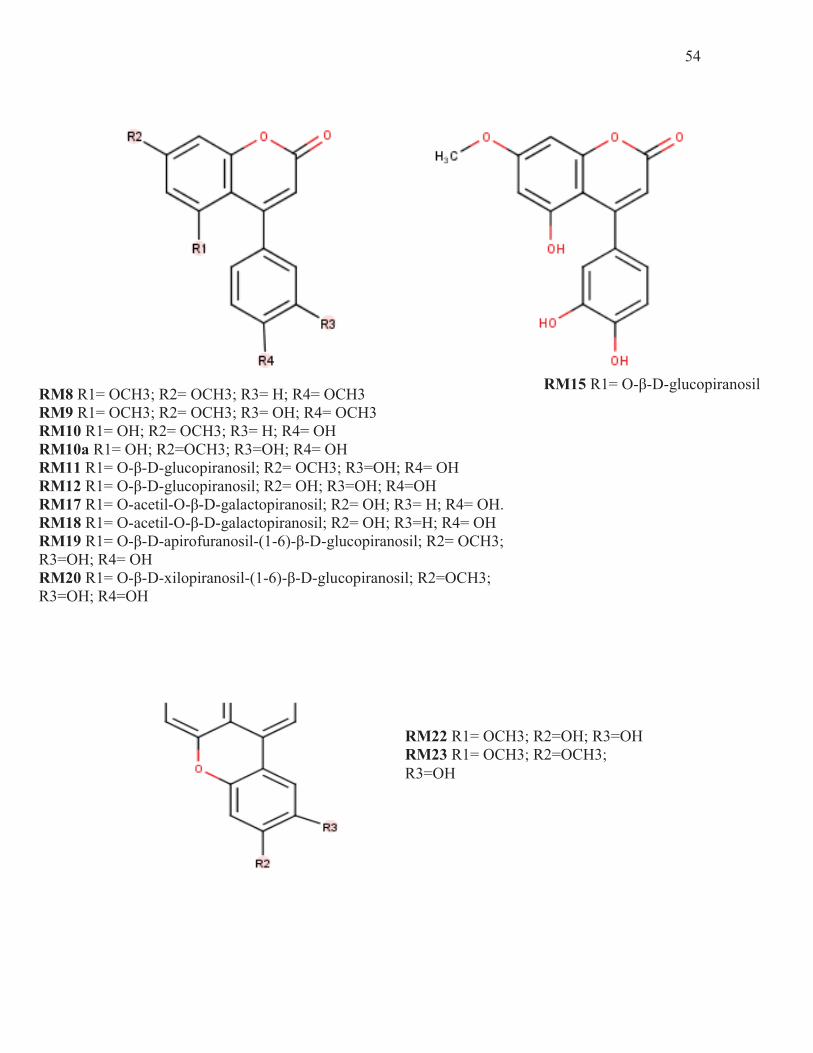
RM8 R1= OCH3; R2= OCH3; R3= H; R4= OCH3 RM9 R1= OCH3; R2= OCH3; R3= OH; R4= OCH3 RM10 R1= OH; R2= OCH3; R3= H; R4= OH RM10a R1= OH; R2=OCH3; R3=OH; R4= OH RM11 R1= O-β-D-glucopiranosil; R2= OCH3; R3=OH; R4= OH RM12 R1= O-β-D-glucopiranosil; R2= OH; R3=OH; R4=OH RM17 R1= O-acetil-O-β-D-galactopiranosil; R2= OH; R3= H; R4= OH. RM18 R1= O-acetil-O-β-D-galactopiranosil; R2= OH; R3=H; R4= OH RM19 R1= O-β-D-apirofuranosil-(1-6)-β-D-glucopiranosil; R2= OCH3; R3=OH; R4= OH RM20 R1= O-β-D-xilopiranosil-(1-6)-β-D-glucopiranosil; R2=OCH3; R3=OH; R4=OH
RM15 R1= O-β-D-glucopiranosil
RM22 R1= OCH3; R2=OH; R3=OH RM23 R1= OCH3; R2=OCH3; R3=OH

55
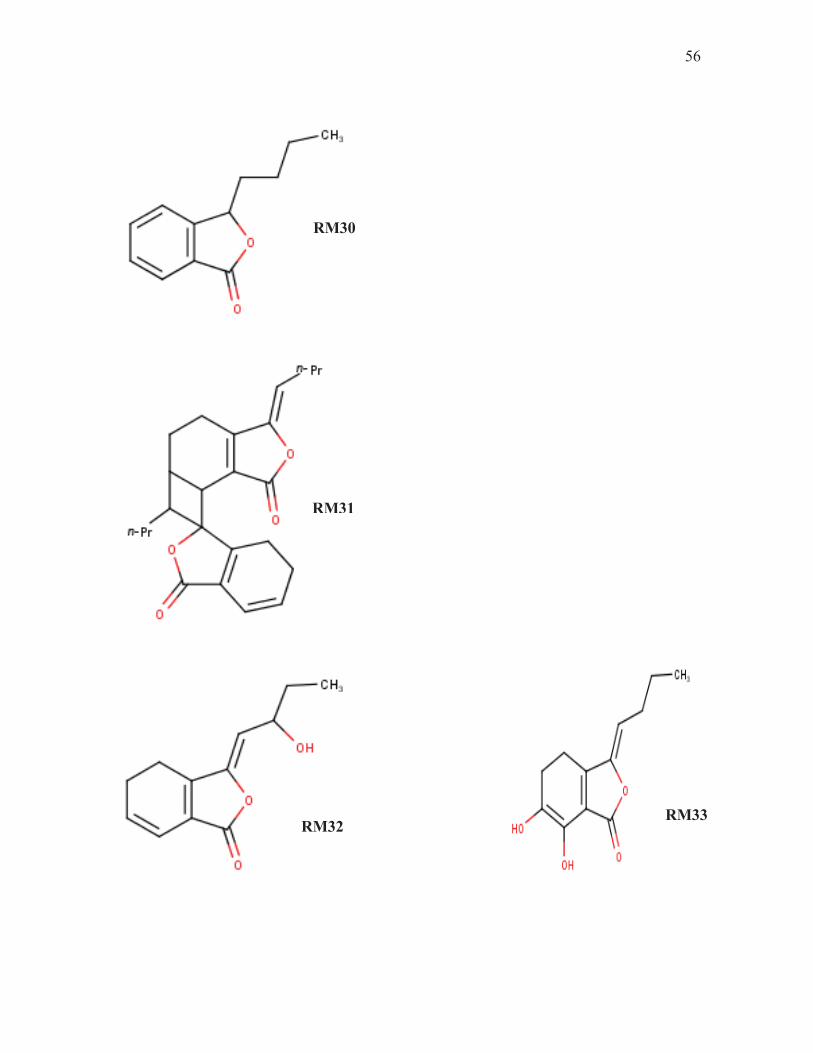
ESQUEMA 2
RM24 RM25
RM26
RM27
RM28 RM29
56
RM32 RM33
RM30
RM31
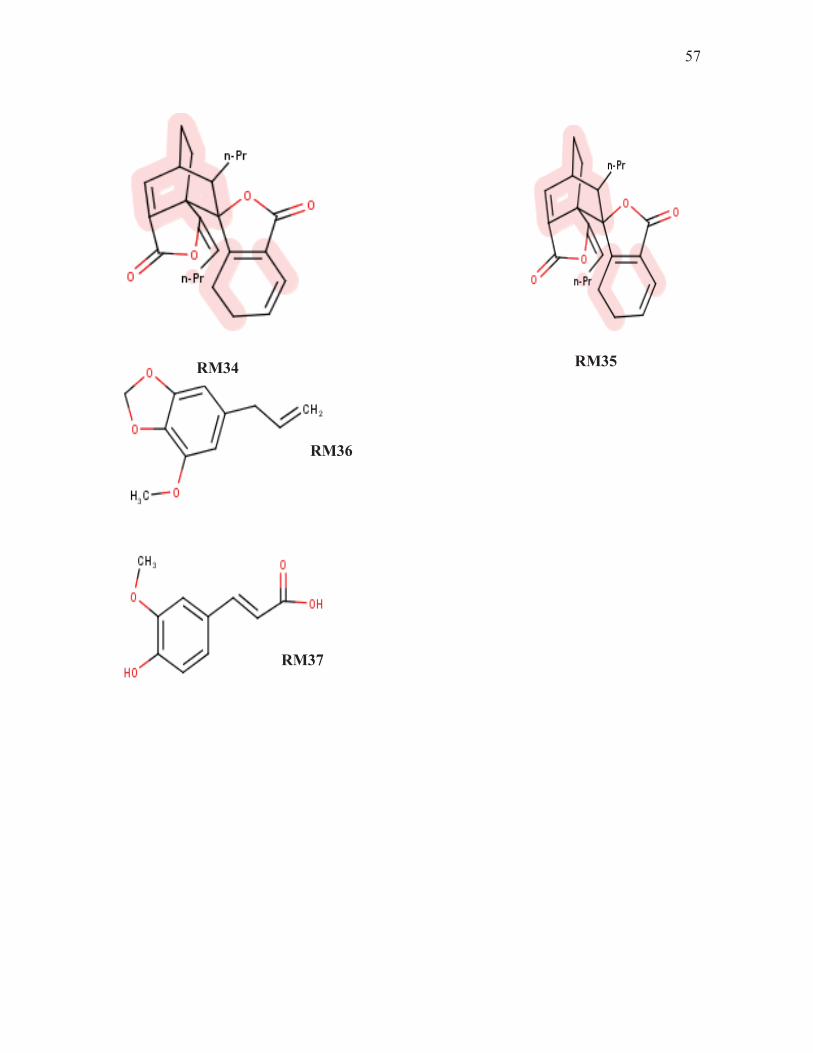
57
RM34 RM35
RM36
RM37
58
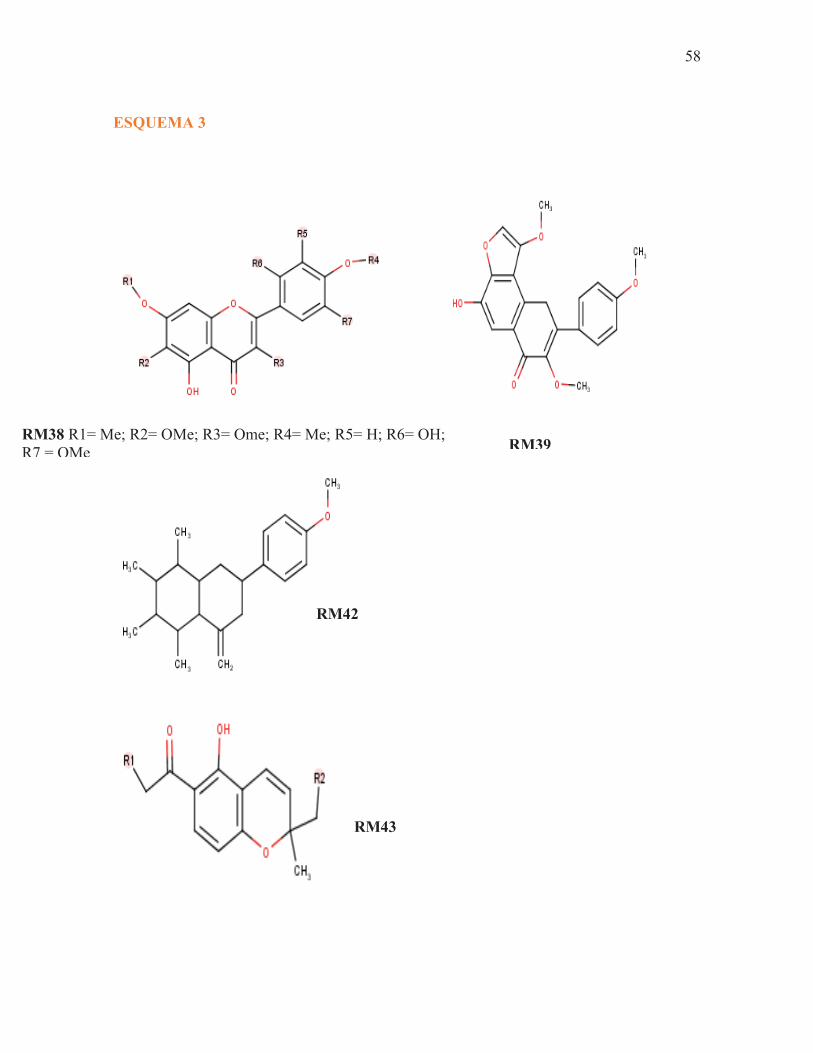
ESQUEMA 3
RM38 R1= Me; R2= OMe; R3= Ome; R4= Me; R5= H; R6= OH; R7 = OMe RM39
RM42
RM43

59
RM47 R1= tioglioxi; R2= Ac RM53
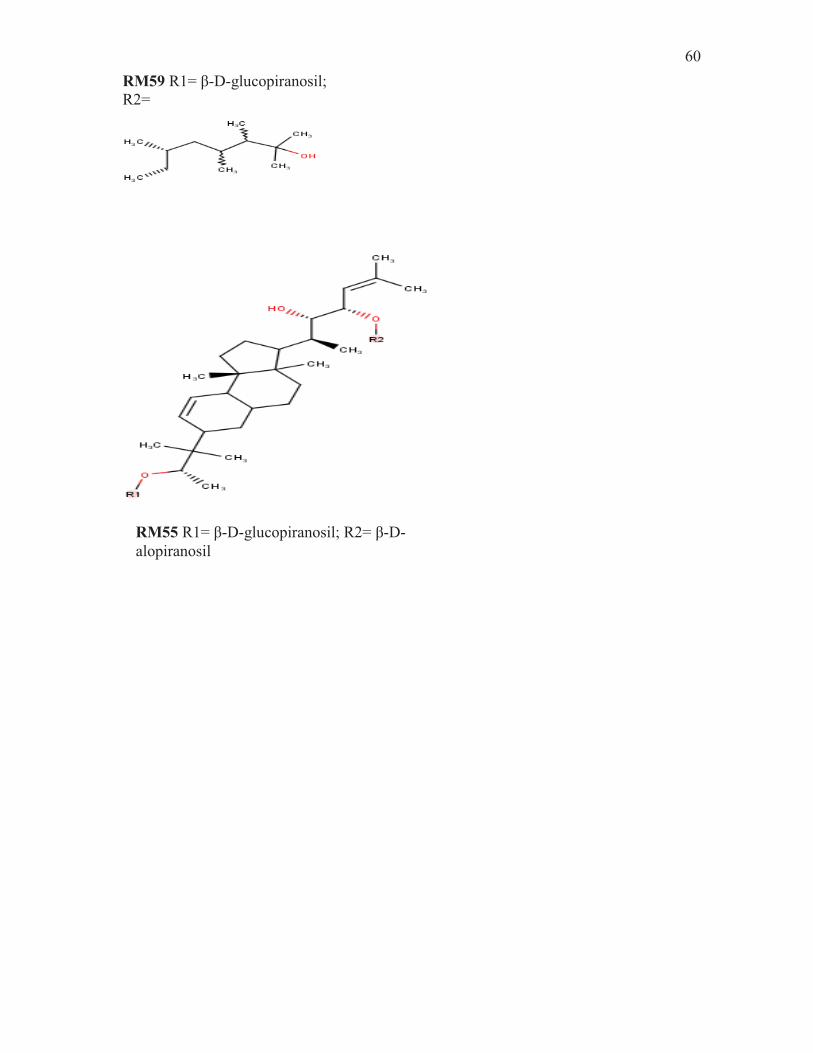
RM54 R1= β-D-glucopiranosil; R2=
60
RM55 R1= β-D-glucopiranosil; R2= β-D-alopiranosil
RM59 R1= β-D-glucopiranosil; R2=

61
ESQUEMA 4
RM56 R1= β-D-l i il
RM57 R1= β-D-alopiranosil; R2= O; R3= Me
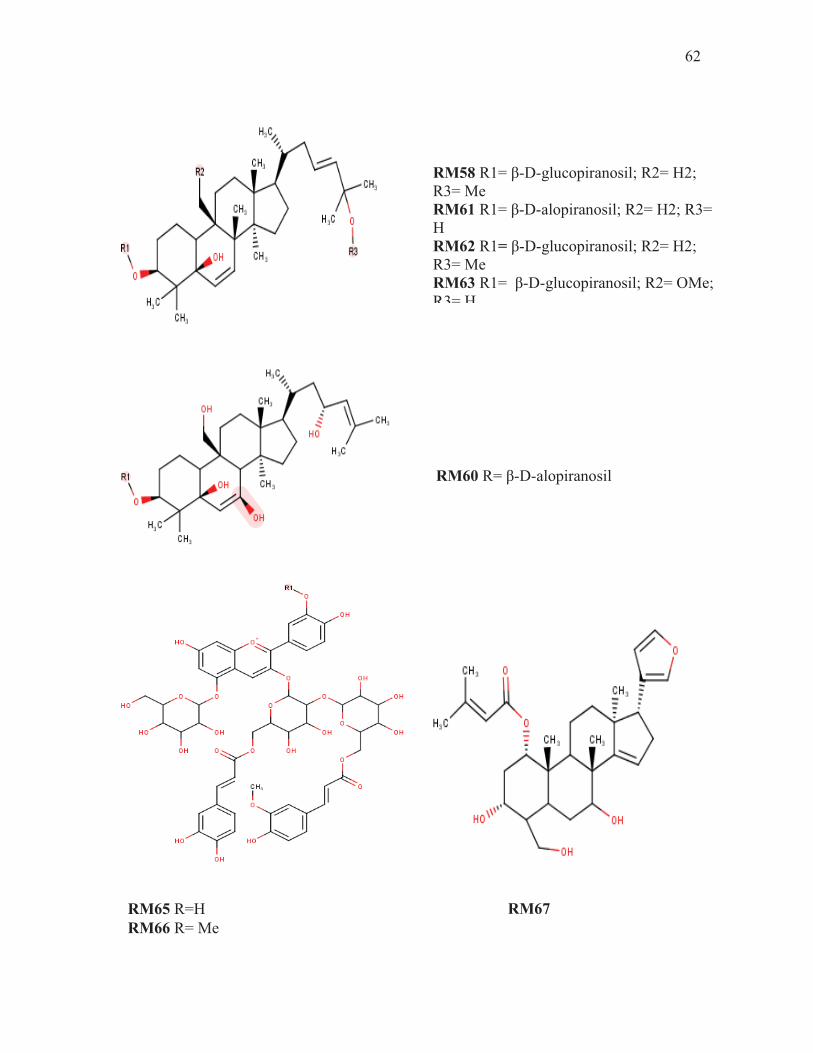
62
RM58 R1= β-D-glucopiranosil; R2= H2; R3= Me RM61 R1= β-D-alopiranosil; R2= H2; R3= H RM62 R1= β-D-glucopiranosil; R2= H2; R3= Me RM63 R1= β-D-glucopiranosil; R2= OMe; R3= H
RM60 R= β-D-alopiranosil
RM65 R=H RM66 R= Me
RM67

63
CONTROL
REFERENCIAS:
Berman, H. M. (2000). The Protein Data Bank. Nucleic Acids Research, 28(1), 235-242.
https://doi.org/10.1093/nar/28.1.235
Bursulaya, B. D., Totrov, M., Abagyan, R., & Brooks, C. L. (2003). Comparative study of
several algorithms for flexible ligand docking. Journal of Computer-Aided
Molecular Design, 17(11), 755-763.
Miglitol Acarbosa

64
Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J.,
… Richardson, D. C. (2010). MolProbity: all-atom structure validation for
macromolecular crystallography. Acta Crystallographica Section D: Biological
Crystallography, 66(Pt 1), 12-21. https://doi.org/10.1107/S0907444909042073
Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., …
Richardson, D. C. (2007). MolProbity: all-atom contacts and structure validation for
proteins and nucleic acids. Nucleic Acids Research, 35(Web Server), W375-W383.
https://doi.org/10.1093/nar/gkm216
Fox, T., & Kriegl, J. M. (2006). Machine learning techniques for in silico modeling of drug
metabolism. Current Topics in Medicinal Chemistry, 6(15), 1579-1591.
Hans Reinauer, & Philip D. Home. (2003). Diagnóstico y Monitorización de la Diabetes
Mellitus desde el Laboratorio. World Health Organization.
Houston, D. R., & Walkinshaw, M. D. (2013). Consensus Docking: Improving the
Reliability of Docking in a Virtual Screening Context. Journal of Chemical
Information and Modeling, 53(2), 384-390. https://doi.org/10.1021/ci300399w
Irwin, J. J., Sterling, T., Mysinger, M. M., Bolstad, E. S., & Coleman, R. G. (2012). ZINC:
A Free Tool to Discover Chemistry for Biology. Journal of Chemical Information
and Modeling, 52(7), 1757-1768. https://doi.org/10.1021/ci3001277

65
Kim, J.-S., Kwon, C.-S., & Son, K. H. (2000). Inhibition of Alpha-glucosidase and
Amylase by Luteolin, a Flavonoid. Bioscience, Biotechnology, and Biochemistry,
64(11), 2458-2461. https://doi.org/10.1271/bbb.64.2458
Laskowski, R. A. (2004). PDBsum more: new summaries and analyses of the known 3D
structures of proteins and nucleic acids. Nucleic Acids Research, 33(Database
issue), D266-D268. https://doi.org/10.1093/nar/gki001
LUO, F., GAO, J., CHENG, Y.-H., CUI, W., & JI, M.-J. (2012). Interaction Mechanisms of
Inhibitors of Glucoamylase by Molecular Dynamics Simulations and Free Energy
Calculations. Recuperado de
https://www.ingentaconnect.com/content/apcs/apcs/2012/00000028/00000009/art00
028#
Martínez, J. J. A., Antón, A. S., & Romero, F. B. (2000). Tratamiento de la diabetes
mellitus. Información terapéutica del Sistema Nacional de Salud, 24(2), 33-43.
Mata, R., Cristians, S., Escandón-Rivera, S., Juárez-Reyes, K., & Rivero-Cruz, I. (2013).
Mexican Antidiabetic Herbs: Valuable Sources of Inhibitors of α-Glucosidases.
Journal of Natural Products, 76(3), 468-483. https://doi.org/10.1021/np300869g

66
Mensch, J., Oyarzabal, J., Mackie, C., & Augustijns, P. (2009). In vivo, in vitro and in
silico methods for small molecule transfer across the BBB. Journal of
Pharmaceutical Sciences, 98(12), 4429-4468. https://doi.org/10.1002/jps.21745
Montero, J. M. S. (2016). Metodologías de modelado molecular en el diseño, sintesis y
explicación racional de resultados. Anales de la Real Academia Nacional de
Farmacia, 82(2). Recuperado de
http://www.analesranf.com/index.php/aranf/article/view/1708
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., &
Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with
selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785-
2791. https://doi.org/10.1002/jcc.21256
Park, H., Lee, J., & Lee, S. (2006). Critical assessment of the automated AutoDock as a
new docking tool for virtual screening. Proteins: Structure, Function, and
Bioinformatics, 65(3), 549-554. https://doi.org/10.1002/prot.21183
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E.
C., & Ferrin, T. E. (2004). UCSF Chimera?A visualization system for exploratory
research and analysis. Journal of Computational Chemistry, 25(13), 1605-1612.
https://doi.org/10.1002/jcc.20084

67
Prism - graphpad.com. (s. f.). Recuperado 19 de junio de 2018, de
https://www.graphpad.com/scientific-software/prism/
Ren, L., Qin, X., Cao, X., Wang, L., Bai, F., Bai, G., & Shen, Y. (2011). Structural insight
into substrate specificity of human intestinal maltase-glucoamylase. Protein & Cell,
2(10), 827-836. https://doi.org/10.1007/s13238-011-1105-3
Sim, L., Jayakanthan, K., Mohan, S., Nasi, R., Johnston, B. D., Pinto, B. M., & Rose, D. R.
(2010). New glucosidase inhibitors from an ayurvedic herbal treatment for type 2
diabetes: structures and inhibition of human intestinal maltase-glucoamylase with
compounds from Salacia reticulata. Biochemistry, 49(3), 443-451.
https://doi.org/10.1021/bi9016457
Sim, L., Quezada-Calvillo, R., Sterchi, E. E., Nichols, B. L., & Rose, D. R. (2008). Human
Intestinal Maltase–Glucoamylase: Crystal Structure of the N-Terminal Catalytic
Subunit and Basis of Inhibition and Substrate Specificity. Journal of Molecular
Biology, 375(3), 782-792. https://doi.org/10.1016/j.jmb.2007.10.069
The UniProt Consortium. (2017). UniProt: the universal protein knowledgebase. Nucleic
Acids Research, 45(D1), D158-D169. https://doi.org/10.1093/nar/gkw1099

68
Thomas Scior Eduardo Salinas Stefanón, & Evelyn Martinez. (2007). Los modelos in
silico. Una herramienta para el conocimiento farmacológico. Revista Elementos,
14(68), 755-763.
Warren, G. L., Andrews, C. W., Capelli, A.-M., Clarke, B., LaLonde, J., Lambert, M. H.,
… Head, M. S. (2006). A Critical Assessment of Docking Programs and Scoring
Functions. Journal of Medicinal Chemistry, 49(20), 5912-5931.
https://doi.org/10.1021/jm050362n
Wishart, D. S., Feunang, Y. D., Guo, A. C., Lo, E. J., Marcu, A., Grant, J. R., Wilson, M.
(2018). DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic
Acids Research, 46(D1), D1074-D1082. https://doi.org/10.1093/nar/gkx1037


















