
Vicente Arnau Llombart - uv.esvarnau/Bio_Inf_02.pdf · Técnicas Avanzadas de Inteligencia...
25
1 BIOINFORMÁTICA Vicente Arnau Llombart Técnicas Avanzadas de Inteligencia Artificial. http://www.uv.es/~varnau/TAIA_2011-12.htm E-mail: [email protected] Clase 2ª: Análisis de secuencias de DNA. BIOINFORMÁTICA Secuenciación del ADN (Celera Genomics). Análisis de una secuencia de DNA. Análisis comparativo entre dos secuencias. Secuencias Múltiples. Árboles Filogenéticos.
Transcript of Vicente Arnau Llombart - uv.esvarnau/Bio_Inf_02.pdf · Técnicas Avanzadas de Inteligencia...

1
BIOINFORMÁTICA
Vicente Arnau Llombart
Técnicas Avanzadas de Inteligencia Artificial.
http://www.uv.es/~varnau/TAIA_2011-12.htm
E-mail: [email protected]
Clase 2ª: Análisis de secuencias de DNA.
BIOINFORMÁTICA
Secuenciación del ADN (Celera Genomics).
Análisis de una secuencia de DNA.
Análisis comparativo entre dos secuencias.
Secuencias Múltiples. Árboles Filogenéticos.

2
Secuenciación del ADN (Celera Genomics).
•Celera Genomics es el nombre de una empresa estadounidense fundada en
mayo de 1998 por Applera Corporation y J. Craig Venter, con el objetivo primario
de secuenciar y ensamblar el genoma humano en plazo de tres años.
http://www.celera.com/
•En el año 2001 presentaron en la revista Science su primer esbozo, de 5
genomas de diferentes etnias, entre ellos, se encontraba el de su director, Craig
Venter.
Secuenciación del ADN (Celera Genomics).
Para ello utilizaron el método llamado: Shotgun
basado en la rotura del DNA en múltiples trozos, su clonación, y
búsqueda de solapamientos con aplicaciones bioinformáticas.

3
Se generan secuencias de entre 2000 y 50000 bases nucleicas.
Para poderlas solapar y reproducir la secuencia original se debe sobre-muestrear el original.
Se genera un programa con más de 500000 líneas de código para ensamblar el Genoma.
En 1998 Celera utilizó 700 ordenador conectados entre si para realizar esta tarea.
Aumentando posteriormente el poder computaciones, el Genoma Humano acabo secuenciándose 9 meses antes de la previsto.
Secuenciación del ADN (Celera Genomics).
Análisis de una secuencia de DNA
4
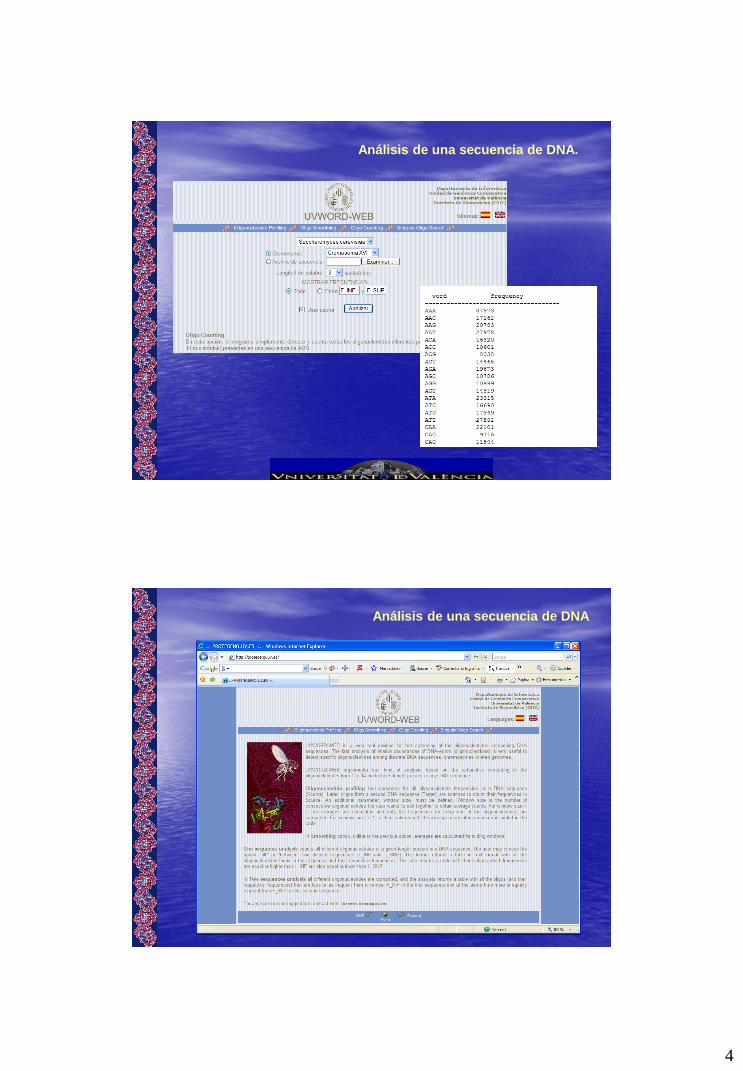
Análisis de una secuencia de DNA.
Análisis de una secuencia de DNA

5
Análisis de una secuencia de DNA.
Análisis de una secuencia de DNA.
Realizamos el análisis de cual es la frecuencia de aparición de cada palabra de k nucleótidos en una secuencia de entrada Source.

6
Análisis de una secuencia de DNA.
Esta estrategia nos permite 4 posibles análisis:
1) Secuencia larga sobre si misma.
2) Secuencia larga contra otras secuencias larga.
3) Secuencia corta sobre secuencia larga.
4) Secuencia larga sobre secuencias cortas.
Lo cual nos permite cosas como:
1) Analizar una secuencia determinada: localizar genes, regiones repetitivas, código único (codificador), …
2) Comparación de secuencias.
3) Identificar localización de secuencia en secuencia larga. Analizar densidades.
4) Analizar presencia de secuencia corta en diferentes genomas.
Análisis de una secuencia de DNA.
Ejemplos de uso con Drosophila melanogaster, cromosoma X: http://protegeno.uv.es/
Trozo de 5k bases de cromosoma X de la mosca: “trozo_mosca_X.fa” Presencia de dinucleótido CG en cromosomas humanos: “CG.TXT”

7
Análisis de una secuencia de DNA.
Trabajos publicados con el uso de esta herramienta de análisis:
"Comparative sequence analysis of the chimpanzee chromosomes 12 and 13 and its fused human counterpart, chromosome 2 " . Miguel Gallach, Vicente Arnau, and Ignacio Marín. ECCB05, 4th European Conference on Computational Biology 2005. JBI, VI Jornadas de Bioinformática. Octubre de 2005. "Global patterns of sequence evolution in Drosophila" Miguel Gallach, Vicente Arnau, Ignacio Marin. BMC Genomics 2007, 8:408.
Esta estrategia a sido publicada en:
"Fast comparison of DNA sequences by oligonucleotide profiling". Vicente Arnau, Miguel Gallach, Ignacio Marin. BMC Research Notes 2008. "UVWORD-WEB: a web-based tool for DNA word analysis" . Vicente Arnau, Jordi Llorens, Miguel Gallach and Ignacio Marín. ECCB05, 4th European Conference on Computational Biology 2005. Octubre de 2006.
Análisis de una secuencia de DNA.
“Discovery of human inversion polymorphisms by comparative analysis of human and chimpanzee DNA sequence assemblies”. Feuk L, et al. PloS Genetics, 2005.

8
Análisis de una secuencia de DNA.
Problema: En un artículo publicado recientemene en PloS Genetics* se mostraba las siguientes equivalencias:
1. El Chr_10_humano equivale al Chr_8_Chimpa.
2. El Chr_11_humano equivale al Chr_9_Chimpa.
Utilizando UVWORD-WEB realizar estudios estadísticos para demostrar esta afirmación, comparando estos 4 cromosomas entre sí dos a dos.
(*) “Discovery of human inversion polymorphisms by comparative analysis of human and chimpanzee DNA sequence assemblies”. Feuk L, et al. PloS Genetics, 2005.
Análisis de una secuencia de DNA.

9
Análisis de una secuencia de DNA.
Análisis comparativo entre dos secuencias.
José R. Valverde: “Comparar secuencias consiste en buscar todas las zonas de similitud significativa entre dos o más secuencias”.
Alex Sánchez: “El alineamiento de secuencias es probablemente la herramienta más utilizada en
bioinformática. Al determinar si una secuencia
desconocida es similar, en algún sentido, a secuencias
conocidas (e idealmente de estructura y función
conocidas) podremos identificarla y predecir su
estructura y función”.

10
SIMILITUD: es el resultado del análisis comparativo de dos o más secuencias.
Estas secuencia pueden ser de ácidos nucleicos o proteínas.
Es solo una medida matemática.
HOMOLOGÍA: es la medida cualitativa entre las secuencias cuando la similitud
que estas tienen es atribuible a razones evolutivas y no al azar.
La homología establece regiones entre las secuencias que se han conservado
con el tiempo.
Podemos medir la similitud entre cualquier par de secuencias, pero dos
secuencias son homologas o no lo son.
Análisis comparativo entre dos secuencias.
¿POR QUE CAMBIAN NUESTRAS SECUENCIAS DE UN INDIVIDUO O ESPECIE A OTRO?:
A lo largo de la evolución las secuencias descendientes de otra ancestral van
acumulando diversos tipos de mutaciones. Estas son mutaciones puntuales o
reorganizaciones genómicas, que pueden involucrar inserciones, delecciones,
inversiones, translocaciones o duplicaciones, mediados por distintos mecanismos
de recombinación (homóloga e ilegítima).
Análisis comparativo entre dos secuencias.
Cualquier análisis filogenético y/o evolutivo de secuencias requiere de un
alineamiento. Para ello se escriben las secuencias en filas una sobre la otra, de
modo que los sitios homólogos quedan alineados por columnas.
Pero veamos primero como se alinean dos secuencias:

11
Métodos de alineamiento.
Existen muchos programas disponibles en WWW para alinear secuencias y buscarlas en las BD.
La correcta elección del programa ( método) y de sus parámetros es muy
importante para la detección de similitudes. •Alineamiento de dos secuencias
•Métodos gráficos: Dotplot. Es intuitivo, pero difícil de cuantificar. •Algoritmos óptimos de alineamiento global (NW) o local (SW).
Obtienen el mejor alineamiento posible con programación dinámica Son demasiado exigentes para ser prácticos en búsquedas extensivas
•Alineamientos múltiples
•Algoritmos heurísticos para búsqueda en bases de datos FASTA, BLAST Dan soluciones buenas, no necesariamente óptimas Pueden ser mucho más rápidos
Análisis comparativo entre dos secuencias.
OBJETIVO: encontrar la posición relativa entre dos cadenas que haga que el número de coincidencias sea el máximo.
Veamos un ejemplo:
X: TCAGACGATTG (n=11) Y: ATCGGAGCTG (m=10)
Análisis comparativo entre dos secuencias.

12
Análisis comparativo entre dos secuencias.
El problema de comparar secuencias:
Cuando deseamos comparar dos secuencias, lo que queremos es hallar áreas de
similitud significativa. El problema es definir lo que quiere decir el término
significativo en biología. Para entenderlo mejor, veamos unos ejemplos.
Empezaremos con un sencillo par de secuencias:
ATGCATGCATGCATGCATATATATATATATATATGCATGCATGCATG
CGATCGATCGATCGATATATATATATGCATATATATGCATGCATGCATGCAT
No podemos limitarnos a poner una secuencia sobre la otra, sino que debemos
compararlas en todos los desplazamientos posibles en busca del mejor
alineamiento. Este podría ser el siguiente:
ATGCATGCATGCATGCATATATATATATATATATGCATGCATGCATG
|| || || |||||||||||| |||| |||||||||||
CGATCGATCGATCGATATATATATATGCATATATATGCATGCATGCATGCAT
Análisis comparativo entre dos secuencias.
Pero los organismos biológicos usan muchos mecanismos para cambiar. Podemos encontrarnos con cámbios en un nucleódito, con inserciones e incluso con delecciones. ¿Podemos mejorar el alineamiento a la luz de estos datos?
ATGCATGCATGCATGCATATATATATAT--ATATATGCATGCATGCATGCATGC
|| || || |||||||||||| |||||| ||||||||||||||||
CGATCGATCGATCGATATATATATATGCATATAT--ATGCATGCATGCATGCAT
Usando discontinuidades podemos aumentar dramáticamente el número de residuos coincidentes:
--AT-GCAT-GCATGC-ATGCATATATATATAT--ATATAT—GCATGCATGCATGCATGC
|| | || | || | |||||||||||| |||||| ||||||||||||||||
CGATCG-ATCG-AT-CG----ATATATATATATGCATATATATGCATGCATGCATGCAT
La pregunta es: ¿hasta qué punto sigue siendo significativo este alineamiento?

13
Análisis comparativo entre dos secuencias.
Si desplazamos las secuencias permitiendo mutaciones e introduciendo huecos podemos encontrar un alineamiento bastante bueno, quizás el mejor. Pero, ¿hemos extraído todos los hallazgos significativos? Una mirada de cerca muestra que hemos ignorado una propiedad importante que podemos destacar usando colores:
En efecto, hay una secuencia repetitiva distribuida por las secuencias. Ningún alineamiento único podría mostrarla puesto que solo podemos alinear una instancia de la repetición con otra instancia en la otra secuencia, pero no con todas las instancias.
Necesitamos un modo de inspeccionar la similitud entre secuencias que sea mejor que los meros alineamientos.
Análisis comparativo entre dos secuencias.
Sin embargo, la historia no acaba aquí todavía. Miremos las siguientes secuencias de cerca en busca de alguna similitud llamativa:
ATGCGACATTATATGGACGCCGACAATATGCATGACTAGCATAGCATGCGAT
||| | | | | | |||
TAGCGTACGAGACGTTCAGTAGGTATAACAGTCGCAGGTATCTTACAGCGTA
No parecen muy similares, ¿verdad?.
Y sin embargo ambas secuencias presentan un grado de similitud impresionante, solo que no es obvio a simple vista. Para verlo basta con dar la vuelta a una de las secuencias y cambiar sus extremos 5' y 3':
ATGCGACATTATATGGACGCCGACAATATGCATGACTAGCATAGCATGCGAT
|||||||||| ||||||||| ||||||||| |||||| ||| ||||||||||
ATGCGACATTCTATGGACGCTGACAATATGGATGACTTGCAGAGCATGCGAT

14
Análisis comparativo entre dos secuencias.
DotPlot: Método Gráfico de comparación de secuencias.
Con este método comparamos dos secuencias generando una tabla cartesiana de doble entrada (una por secuencia), colocando un punto en cada casilla con coincidencia.
El mejor alineamiento lo determina la diagonal con mayor número de puntos.
Pero:
¿Qué pasa si existen delecciones, mutaciones o inserciones?.
Análisis comparativo entre dos secuencias.
El siguiente es un ejemplo de un DotPlot entre las secuencias de aminoácidos de la rhodopsina del gallo (Gallus gallus) y la del cocodrilo (Alligator mississippiensis):

15
Análisis comparativo entre dos secuencias.
Una forma de mejorar los resultados es realizar una “limpieza” de la matriz de puntos, de la siguiente forma:
Recorrer la matriz por diagonales y eliminar todos aquellos puntos que no estén dentro de una sub-diagonal de al menos K=6 puntos, por ejemplo.
Veamos el resultado de compara una secuencia consigo misma.
Análisis comparativo entre dos secuencias.

16
Análisis comparativo entre dos secuencias.
Los alineamientos globales, que intentan alinear cada residuo en cada secuencia, son más útiles cuando las secuencias inicicales son similares y aproximadamente del mismo tamaño . Una estrategia general de alineamiento global es el algoritmo Needleman-Wunsch basado en programación dinámica.
Los alineamientos locales son más útiles para secuencias diferenciadas en las que
se sospecha que existen regiones muy similares o motivos de secuencias similares dentro de un contexto mayor. El algoritmo Smith-Waterman es un método general de alineamiento local basado en programación dinámica.
Análisis comparativo entre dos secuencias.
ALINEAMIENTO GLOBAL. ALGORITMO DE NEEDLEMAN & WUNSCH Procedimiento: Dadas dos secuencias A y B: A=a1a2...an y B=b1b2...bm Se define:
* Una función de similitud (coincidencias) S(ai,bj). * Las inserciones o delecciones se penalizan con un peso W.
Se construye una matriz H de i+1 filas y j+1 columnas. 1. Se inicializa con ceros la primera fila y la primera columna de Hij. 2. Llenado de Matriz Hij siguiendo la regla:

17
Análisis comparativo entre dos secuencias.
Ejemplo: Alinear las siguientes secuencias: A = GAATTCAGTTA
B = GGATCGA
Parámetros:
• Coincidencias = 1
• No coincidencias = 0
• Huecos = 0
Es decir:
W = 0
Análisis comparativo entre dos secuencias.

18
Análisis comparativo entre dos secuencias.
Recuperación de la solución (Backtracking) :
GGA-T-C-G--A
| | | | |
G-AATTCAGTTA
Análisis comparativo entre dos secuencias.
ALINEAMIENTO LOCAL. ALGORITMO DE SMITH & WATERMAN. (1981). Descripción:
Dadas dos secuencias SA y SB: SA=a1a2...an y SB=b1b2...bm Se define: • Una función de similitud (coincidencias) S(ai,bj). • Las inserciones o delecciones se penalizan con un peso W. Modelos para las penalizaciones de los gaps (Wk):
• Penalización logarítmica de la forma: W = A + B * log(N). • Una penalización de la forma: W = A + B * N. • O una ley de penalización de la forma: W = A + B * NC
donde A y B son definidos por el usuario y N representa la longitud del hueco
Se construye una matriz H de i+1 filas y j+1 columnas. Se inicializa con ceros la primera fila y la primera columna de Hij. Llenado de Matriz Hij siguiendo la regla:

19
Análisis comparativo entre dos secuencias.
Ejemplo: se tienen las siguientes secuencias: A: CAGCCUCGCUUAG, m= 13 B: AAUGCCAUUGACGG, n= 14 Parámetros:
S(ai,bj) = 1 si ai = bj S(ai,bj) = -1/3 si ai ≠ bj. Wk = 1 + 1/3 * k Wl = 1 + 1/3 * l
Análisis comparativo entre dos secuencias.
GCCAUUG GCC_UCG

20
Análisis comparativo entre dos secuencias.
Este algoritmo fue modificado mas tarde por Waterman y Eggert en 1987.
La modificación fue la siguiente:
Hij = max { Hi-1,j-1 + S(ai,bj) , Eij , Fij , 0 }
Eij = max {Hi,j-l - (u+v), Ei,j-l - v }
Fij = max {H i-l,j - (u+v), Fi-l,j - v }
Donde l es el tamaño posible del indels.
REFERENCIAS:
IDENTIFICATION FOR COMMON MOLECULAR SUBSEQUENCES. Smith & Waterman. 1981. Journal of
Molecular Biology 147: 195-197.
RAPID AND SENSITIVE PROTEIN SIMILARITY SEARCHES. Lipman and Pearson. 1985. Science 127:
1435-1441.
Análisis comparativo entre dos secuencias.
MATRICES DE SUSTITUCIÓN
Las matrices de sustitución son utilizadas en los análisis comparativos de
secuencias de aminoácidos.
Una matriz de sustitución se elabora bajo una teoría de evolución.
El resultado de la comparación de dos o más secuencias depende fuertemente
de la matriz de sustitución que se haya seleccionado.
Los algoritmos de alineamiento (comparación ) funcionan igual con una matriz
de distancias o con una matriz de sustitución (aunque se pueden obtener
diferentes resultados).
Una matriz de distancias es muy útil en la reconstrucción de un árbol
filogenético, mientras que una matriz de sustitución es utilizada para
realizar búsqueda en bases de datos.

21
Análisis comparativo entre dos secuencias.
MATRICES DE SUSTITUCIÓN PARA ÁCIDOS NUCLEICOS:
Análisis comparativo entre dos secuencias.
MATRICES DE SUSTITUCIÓN PARA PROTEÍNAS:
MATRIZ PAM
Las matrices PAM (“Percent Accepted Mutation”) fueron descritas por
Daykoff et al. (1978) están basadas en el alineamiento global de secuencias
de proteínas estrechamente relacionadas y asumen que una modificación en
algún sitio depende solamente del aminoácido presente en ese sitio.
MATRIZ BLOSUM
La matriz BLOSUM ("BLOcks SUbstitution“) fue propuesta por Steven
Henikoff and Jorja G. Henikoff en el año de 1992, fue creada a partir de un
estudio sobre bloques conservados.

22
Análisis comparativo entre dos secuencias.
PAM250
Análisis comparativo entre dos secuencias.

23
Análisis comparativo entre dos secuencias.
BLAST (“Basic Local Alignment Search Tool”).
BLAST es un conjunto de programas de búsqueda de similitud diseñados para explorar
todos las bases de datos de secuencias.
Programas de BLAST:
blastp: Compara una secuencia de aminoácidos contra una base de datos de secuencias de proteínas
blastn: Compara una secuencia de nucleótidos contra una base de datos de secuencias de nucleótidos.
blastx: Compara una secuencia de nucleótidos traducida en sus seis posibles marcos de lectura contra una base de datos de secuencias de proteínas.
tblastn:
Compara una secuencia de aminoácidos contra toda la base de datos de nucleótidos traducida en sus seis posibles marcos de lectura. Si se necesitara realizar este cálculo con FASTA sería necesario realizar las traducciones de las secuencias en los distintos marcos de lectura y ejecutar la búsqueda para cada uno de los seis marcos.
tblastx:
Compara las seis traducciones en sus marcos de lectura de la secuencia de nucleotidos, contra las seis traducciones en sus marcos de lectura de toda la base de datos de nucleótidos.
Análisis comparativo entre dos secuencias.
FASTA.
FASTA fue el primer algoritmo ampliamente utilizado para búsqueda de similitud en una
base de datos. FASTA busca alineamientos locales óptimos buscando coincidencias de
pequeñas subsecuencias denominadas palabras ("words o k-tuplas"), el score del primer
segmento en el que se aparean varias palabras se denomina "init1" y la suma de todos
los score de los segmentos se denomina "initn". La sensibilidad y velocidad del algoritmo
es inversamente proporcional a la longitud de la palabra utilizada en la búsqueda.
Desarrollado por David Lipman y William Pearson en el año de 1985.
Es empleado principalmente por el EMBL - EBI (“European Molecular Biology
Laboratories - European Bioinformatics Institute”), si se compara su velocidad con BLAST
se notará que es mucho más lento, incluso llegá hasta a emplear varias horas para
obtener los resultados, es por está razón que el EMBL envía los cálculos al usuario por
correo electróncio.
FASTA compara una secuencia de DNA o de proteínas contra todas las secuencias de
una base de datos y devuelve los mejores segmentos alineados.

24
Análisis comparativo entre dos secuencias.
ALINEAMIENTO MÚLTIPLE DE SECUENCIAS: Un alineamiento múltiple de secuencias es un alineamiento de tres o más secuencias biológicas, generalmente proteínas, ADN o ARN. Las secuencias a estudio poseen generalmente una relación evolutiva por la cual comparten un linaje y descienden de un ancestro común.
ÁRBOLES FILOGENÉTICOS:
• Definen subfamilias
– Relevantes para definir relaciones filogenéticas
S1 GIFTDIDMHFYVKKPGLDEFFTLVLRTLCMAA
S2 ALTTGIDMWTTAKRPDMDDYYTIIIPGLMNCI
S3 AVTTGLNMWTTAKRPGMDDFYTILLPGLMNCI
S4 GVTTGLNLYFTARRP--DEFYS-VLRTLCMCL
S5 GIFTDIDLHFYVKKP--DEFFSLVLRTLCMAA
S6 AVTTGLNLWTTAKRP--DDFYSILLPGLMNCI
S7 GLFTALNLHFFGRKP--EEYFSLVVDGLCNCI
Análisis comparativo entre dos secuencias.

25
BIOINFORMÁTICA
Vicente Arnau Llombart
Técnicas Avanzadas de Inteligencia Artificial.
http://www.uv.es/~varnau/TAIA_2011-12.htm
E-mail: [email protected]
Análisis comparativo entre dos secuencias.
![ARNAU - agora.xtec.cat · arnau i\/Án edurne] jairo aaron nÁyade gala . ann(s enzo dafne](https://static.fdocumento.com/doc/165x107/5f9c54edac215e48657ad01b/arnau-agoraxteccat-arnau-in-edurne-jairo-aaron-nyade-gala-anns-enzo.jpg)

















