







Idiomas
Páginas
Jurídico


ÍNDICE
Tema 1. Introducción. Concepto de ADN recombinante. Aplicaciones.
1
Tema 2. Restricción y modificación del ADN. Enzimas de restricción. Tipos
y utilización.
2
Tema 3.
Otras enzimas utilizadas en la producción de moléculas de ADN
recombinante. Enzimas de degradación. Enzimas de polimerización.
Enzimas de modificación. Marcaje de moléculas de ADN.
Producción de ADN recombinante.
5
Tema 4.
Amplificación enzimática de fragmentos de ADN y ARN.
Reacción en cadena de la polimerasa. Componentes y parámetros de
la reacción. PCR de ARN.
11
Tema 5.
Vectores de clonación en bacterias. Clonación. Tipos y
características de los vectores de clonación. Plásmidos. Vectores
basados en el genoma del fago λ. Cósmidos. BACs. YACs.
16
Tema 6.
Clonación en bacterias. Concepto de genoteca. Construcción de
genotecas de ADN genómico y de ADN copia. Representatividad de
una genoteca.
29
Tema 7.
Rastreo de genotecas. Detección e identificación de clones
individuales. Detección directa por complementación. Detección
indirecta por hibridación de ácidos nucleicos. Vectores de expresión
y detección por métodos inmunoquímicos.
33
Tema 8.
Caracterización estructural de moléculas de ADN recombinante.
Mapas de restricción. Secuenciación de ácidos nucleicos. Hibridación
de ácidos nucleicos en filtro. Localización del fragmento clonado en
el genoma. Determinación del número de copias de una secuencia en
el genoma.
39
Tema 9.
Caracterización funcional de moléculas de ADN recombinante.
Caracterización funcional in vitro. Caracterización funcional in vivo.
Introducción de genes en células eucariotas: métodos, vectores,
sistemas de selección.
50
Tema 10.
Mutagénesis in vitro. Genética reversa y tipos de mutagénesis.
Mutagénesis aleatoria por deleción, inserción y sustitución.
Mutagénesis dirigida. Inactivación de genes endógenos.
59
Tema 11.
Aplicaciones en genética humana. Aplicaciones al diagnóstico
clínico. Terapia génica. Medicina forense. Proyectos genoma.
69
Tema 12.
Aplicaciones en biología animal y vegetal. Obtención de plantas
transgénicas. Clonación animal. Transgénesis en animales. 80


Fundamentos de Genética Aplicada 1
Introducción
Hasta hace poco tiempo el ADN era una molécula que presentaba amplias dificultades para
su análisis bioquímico. Su gran longitud y monotonía hacían que la secuencia de
nucleótidos pudiese sólo ser estudiada mediante mecanismos indirectos. Para esto se
recurría a la determinación de la secuencia de las proteínas o del ARN. A partir de los años
70 se desarrollaron las herramientas de la ingeniería genética, o lo que se ha denominado
técnicas del ADN recombinante. Hoy en día se pueden separar regiones determinadas del
ADN, obtenerlas en grandes cantidades y determinar su secuencia de nucleótidos. También
es posible alterar un gen y transferirlo a células en cultivo o a la línea germinal de animales,
donde el gen modificado se incorpora como parte funcional y permanente del genoma.
Concepto de ADN recombinante
El ADN recombinante es una molécula de ADN formada por la unión de dos moléculas de
diferente origen. Generalmente se aplica este nombre a moléculas producidas por la unión
in vitro de ADN proveniente de dos
organismos diferentes que no se encuentran
juntos. Normalmente se trata de ADN
cromosómico que se une a un vector,
comúnmente un plásmido bacteriano.
Al introducirse este ADN recombinante en un
organismo se produce una modificación
genética que permite la adición de un nuevo
ADN al organismo conllevando a la
modificación de rasgos existentes o a la
expresión de nuevos rasgos.
Son necesarias herramientas que
linearicen los vectores y que corten el ADN
cromosómico, es decir, se necesitan enzimas
que tienen como sustrato el ADN. Después de
obtenerse el ADN recombinante debe
multiplicarse, para ello, se introduce en una
bacteria, que se dividirá formando un clon en
el que cada célula tendrá una copia del
plásmido recombinante.
Aplicaciones
Industria farmacéutica y química: se usan bacterias para producir sustancias de
interés para el hombre, como por ejemplo insulina.
Industria alimentaria: se usan bacterias para producir colorantes alimenticios.
Biorremediación: por ejemplo, degradación bacteriana de hidrocarburos.
Agricultura y ganadería: producción de plantas y animales transgénicos.
Medicina: técnicas de diagnostico molecular y terapia génica.
11

2 Alberto Fonte Polo
Restricción y modificación del ADN
En las técnicas de ingeniería genética se necesita fragmentar, modificar y multiplicar las
moléculas de ADN, y para ello se utilizan enzimas de diferentes tipos:
Enzimas nucleasas.
Enzimas de modificación.
Enzimas de polimerización.
Además se utilizan procedimientos químicos y físicos para fragmentar el ADN. Como
método químico cabe destacar la hidrólisis del ADN por pH ácido. Los ultrasonidos y el
cizallamiento son métodos físicos que fragmentan el ADN. Aunque a diferencia de los
métodos enzimáticos, los procedimientos químicos y físicos son inespecíficos.
Enzimas de restricción
Las nucleasas son enzimas que producen la rotura de los enlaces fosfodiéster de la cadena
polinucleotídica de los ácidos nucleicos. Tipos de nucleasas:
Exonucleasas: escinden el último nucleótido del extremo 5' o 3' de un
polinucleótido. Pueden degradar por completo un ácido nucleico lineal.
Endonucleasas: cortan los enlaces fosfodiéster situados en el interior de los
polinucleótidos. Estas enzimas no requieren un extremo libre, por lo que pueden
cortar ácidos nucleicos circulares.
Las nucleasas pueden ser enzimas inespecíficas, es decir, que cortan el ácido nucleico
independientemente del nucleótido que haya. Pero también existen endonucleasas (de
restricción) que sólo cortan cuando reconocen una determinada secuencia nucleotídica.
Las enzimas de restricción (o restrictasas) pueden reconocer una secuencia característica
de nucleótidos dentro de una molécula de ADN y cortar el ADN en ese punto en concreto,
llamado sitio o diana de restricción, o en un sitio no muy lejano a éste, dependiendo de la
enzima. Los sitios de restricción cuentan con entre 4 y 12 pares de bases, con las que son
reconocidos.
Existen tres tipos de enzimas de restricción (I, II, y III), forman parte de sistemas de
restricción-modificación, estos sistemas hacen la función del sistema inmune en las
bacterias. Cada enzima de restricción reconoce una diana, que también es reconocida por la
enzima de modificación. Las enzimas de modificación son metiltransferasas que añaden
grupos metilo a los nucleótidos de las dianas de restricción. Las restrictasas sólo reconocen
la diana cuando ésta no está metilada.
Cuando el ADN doble metilado en la
diana de restricción se replica, se originan dos
moléculas de ADN hemimetiladas, es decir,
que sólo tienen metilados los nucleótidos de
una cadena. En este caso las metiltransferasas
metilan la cadena de nueva síntesis. En el
caso de que un fago infecte a la bacteria, el
ADN que introducirá no estará metilado, y
por lo tanto será reconocido por las enzimas
de restricción y será degradado. Por otro lado,
22

Fundamentos de Genética Aplicada 3
existen fagos que no poseen dianas de restricción en su material genético, estos virus son
por lo tanto resistentes a la acción de las enzimas de restricción.
Se han descubierto más de 2.500 sistemas de restricción diferentes, y hay aproximadamente
300 enzimas de restricción comercializadas.
El nombre de cada enzima de restricción está asignado según el origen bacteriano de la
misma. La nomenclatura utilizada para denominar estas enzimas consiste en:
1. Tres letras que corresponden al nombre científico del microorganismo (ej.
Escherichia coli (Eco); Haemophilus influenzae (Hin)); y por ello las tres primeras
letras del nombre se escriben en cursiva.
2. La cepa, si la hubiese (ej. EcoR, aislada de la cepa "RY13" de E. coli).
3. En números romanos, un número para distinguir si hay más de una endonucleasa
aislada de esa misma especie. No confundir con el tipo de enzima de restricción.
De esta manera, el nombre de la enzima de restricción EcoRI se debe a:
Nomenclatura Ejemplo Corresponde a:
E Escherichia Género de la bacteria
co coli Especie de la bacteria
R RY13 Cepa de la bacteria
I La primera enzima identificada Orden de identificación de la enzima
Tipos y utilización
Tipos de enzimas de restricción:
Tipo I: estas restrictasas reconocen dianas largas (de 16 nucleótidos). Al reconocer
la secuencia específica de ADN (no metilado) corta al azar en sitios distintos al sitio
de reconocimiento, ya sea aguas arriba o aguas abajo. Corta las dos hebras en
puntos cercanos entre sí, poco específicos, a unos 1.000 pares de bases
aproximadamente después de la diana. Deja extremos cohesivos.
Tipo III: el sitio de reconocimiento es más corto. Corta las dos hebras en puntos
distantes entre sí, es por lo tanto un corte desplazado y deja extremos cohesivos. El
punto de corte se sitúa a 24-26 pares de bases de la diana de restricción.
Tipo II: estas son las enzimas de restricción que comúnmente se usan en el
laboratorio. Las enzimas de restricción de tipo II más frecuentes reconocen dianas
de 4-6 pares de bases, pero existen otras menos frecuentes que reconocen dianas de
8, 10 o 12 pb. Cortan las dos hebras en puntos muy específicos, dentro de la
secuencia de reconocimiento.
Las dianas pueden ser secuencias fijas de nucleótidos, o pueden ser flexibles como
por ejemplo:
5'GGNNCC 5'GGPyrPyrCC 5'GGPurPurCC
3'CCNNGG 3'CCPurPurGG 3'CCPyrPyrGG
N representa cualquier base, Pyr representa una base pirimidínica y Pur una base púrica.

4 Alberto Fonte Polo
En cualquier caso, las dianas son siempre secuencias palindrómicas, esto es,
secuencias de nucleótidos que son idénticas a su cadena complementaria cuando
cada una es leída en la misma dirección química.
5'-GTATAC-3'
||||||
3'-CATATG-5'
Secuencia de reconocimiento palindrómica
El corte puede generar extremos romos o extremos protuberantes, según la enzima.
Extremos romos: la enzima corta las dos hebras en el mismo punto.
5'---CAG CTG---3'
3'---GTC GAC---5'
Extremos protuberantes: cuando el corte ocurre en puntos distintos en cada
hebra. Pueden ser extremos protuberantes 5' o 3', según esté la región simplexa
en el extremo 5' o en el 3' respectivamente.
5'---G AATTC---3' 5'---CTTAA G---3'
3'---CTTAA G---5' 3'---G AATTC---5'
Extremos protuberantes 5’ Extremos protuberantes 3’
Los extremos protuberantes son cohesivos, es decir, pueden volver a ligarse si
se restablecen los puentes de hidrógeno entres las bases nitrogenadas.
Dos sistemas de restricción distintos pueden ser isoesquizómeros cuando ambos
reconocen la misma diana de restricción o dianas muy parecidas, pero producen los
mismos extremos cohesivos.
Cuando se pretende obtener un alto grado de fragmentamiento del ADN se emplean
enzimas de corte frecuente, que son aquellas enzimas que reconocen dianas de
pocos pares de bases. Puesto que, una secuencia corta de nucleótidos aparecerá con
más frecuencia en la molécula de ADN que una secuencia con muchos pares de
bases. Pero las dianas de restricción no están distribuidas uniformemente en el
ADN, sino que puede haber tramos con dianas muy próximas, y otros tramos que
por azar casi no presenten dianas. Por ello, mediante estas técnicas no se puede
obtener fragmentos de ADN del mismo tamaño.

Fundamentos de Genética Aplicada 5
Otras enzimas utilizadas en la producción de moléculas de ADN recombinante
Enzimas de degradación
Las enzimas de degradación que se usan en el laboratorio son nucleasas (exonucleasas y
endonucleasas), son enzimas que catalizan la ruptura de los enlaces fosfodiéster.
Es necesario valorar la enzima para determinar la concentración adecuada de enzima que
hay que utilizar, ya que un exceso podría conllevar la degradación total del ADN que se
esté estudiando.
Las principales enzimas son:
Exo λ: se trata de una enzima sintetizada por Escherichia coli cuando está infectada por el
fago λ. Es una exonucleasa que degrada el ADN en dirección 5'→3', lo que da lugar a
moléculas con extremos protuberantes 3'.
Exo III de E. coli: es una exonucleasa presente en E. coli que degrada en sentido 3'→5',
por lo que produce extremos protuberantes 5'. Actúa sobre moléculas de ADN intactas y
también sobre sustratos mellados, es decir, sobre moléculas de ADN que presentan un
espacio donde falta un enlace covalente, y por lo tanto tienen un extremo 3' sobre el que
puede actuar la enzima.
Exo VII de E. coli: enzima exonucleasa que utiliza moléculas simplexas como sustrato, es
capaz de reconocer ambos extremos. Degrada el ADN simplexo en dirección 3'→5' y
5'→3', por lo tanto produce moléculas progresivamente más cortas. También es capaz de
reconocer ADN semisimplexo, pero en tal caso sólo degradaría la parte simplexa de la
molécula.
Bal 31: es una enzima nucleasa que tiene actividad
exonucleásica 3'→5' y origina extremos
protuberantes 5'. También actúa como endonucleasa
realizando cortes en el interior de la molécula de
ADN. En el laboratorio esta enzima se usa para
mutagenizar el ADN, ya que produce la deleción de
los genes, esto es útil para determinar la parte
codificante de dicho gen. Dependiendo del tiempo
que esté actuando la enzima los fragmentos estarán
más o menos delecionados.
S1: enzima endonucleasa que corta extremos protuberantes en ambos sentidos.
33

6 Alberto Fonte Polo
ADNasa I de E. coli: esta enzima puede actuar como endonucleasa tanto de cadena simple
como de cadena doble. Si se trabaja con un tampón con Mg2+
la enzima corta solamente
una de las dos cadenas, introduce mellas en la molécula de ADN. Pero ante la presencia de
Mn2+
corta las dos cadenas en el mismo punto. En cualquier caso, siempre actúa sobre
ADN duplexo.
ARNasas: son enzimas endonucleasas que actúan sobre ARN monocatenario. Hay varios
tipos:
ARNasa A: rompe enlaces entre una pirimidina y otra base cualquiera.
ARNasa T: rompe enlaces entre una guanina y otro nucleótido.
ARNasa H: rompe sustratos híbridos ADN-ARN, por lo tanto, origina moléculas de
ADN monocatenarias.
Enzimas de polimerización
Son las ADN polimerasas y las ARN polimerasas. La mayoría de las enzimas de
polimerización necesitan un molde para poder copiar, el molde puede ser una molécula de
ADN o de ARN. Por ejemplo, las ADN polimerasas dependientes de ADN utilizan un
molde de ADN, y las ADN polimerasas dependientes de ARN utilizan un molde de ARN.
Además, estas enzimas también necesitan una cadena que proporcione un extremo 3' OH
libre para formar el enlace fosfodiéster, esta cadena actúa como cebador o primer. También
se requieren los desoxirribonucleótidos trifosfato correspondientes para la síntesis de la
nueva hebra.
ADN polimerasa I de E. coli: participa en la replicación del cromosoma bacteriano
corrigiendo los errores producidos. In vitro esta enzima tiene actividad polimerásica 5'→3',
actividad exonucleásica 3'→5' (actividad correctora de pruebas) y actividad exonucleásica
5'→3'. La actividad correctora de pruebas permite a la enzima eliminar los nucleótidos mal
apareados del extremo 3' de la cadena, y de este modo puede corregir sus propios errores. Y
mediante la actividad polimerasa 5'→3' puede rellenar los huecos con los nucleótidos
correspondientes.
Al tratar la ADN polimerasa I con una proteasa específica se obtienen dos
fragmentos de distinto tamaño. El polipéptido de mayor tamaño mantiene las actividades
polimerásica 5'→3' y exonucleásica 3'→5', se denomina fragmento Klenow. Es útil para la
investigación porque la actividad exonucleasa 5'→3' puede interferir en algunas
aplicaciones experimentales al acortar los extremos 5' de las moléculas de ADN. El gen que
codifica para el fragmento
Klenow ha sido
mutagenizado y se ha podido
obtener un fragmento con
actividad polimerasa 5'→3'
solamente.

Fundamentos de Genética Aplicada 7
ADN polimerasa del fago T7: esta enzima aparece en E. coli cuando está infectada por el
fago T7. En presencia de la enzima de E. coli tiorredoxina se une fuertemente al ADN
molde y no se separa fácilmente, de este modo se pueden sintetizar fragmentos muy largos
de ADN con poca cantidad de enzima. También se han introducido mutaciones en el gen
que codifica para esta ADN polimerasa, y así se ha conseguido la enzima sequenasa que
posee sólo la actividad polimerásica 5'→3'. Ésta ha sido la enzima utilizada hasta hace poco
para la secuenciación del ADN.
Taq polimerasa: es una enzima que resiste altas temperaturas, ha sido asilada de Thermus
aquaticus, una bacteria termófila que vive en las proximidades de las fuentes de agua
caliente. La Taq polimerasa es ampliamente utilizada por sus propiedades de
termorresistencia en las reacciones de PCR. En la reacción en cadena de la polimerasa se
separan las cadenas de la doble hélice del ADN mediante desnaturalización térmica a
temperaturas en torno a los 95°C, por lo que no pueden emplearse las ADN polimerasas
ordinarias. Pero la Taq polimerasa tiene una temperatura óptima de funcionamiento de
72°C, y su termoestabilidad es tal que su vida media a 95°C es de 40 minutos, lo que la
hace idónea para este tipo de procesos. Se trata de una ADN polimerasa que no tiene
actividad exonucleásica 3'→5', por lo que no corrige los errores que comete.
Pfu: es una ADN polimerasa que posee actividad correctora de pruebas, ha sido obtenida
de la arquea hipertermófila Pyrococcus furiosus. Es también usada en las técnicas de PCR.
ADN polimerasa ARN dependiente, también denominada transcriptasa inversa,
transcriptasa reversa o retrotranscriptasa: es una enzima de tipo ADN polimerasa, que
tiene como función sintetizar ADN de doble cadena utilizando como molde ARN
monocatenario. Esta enzima es específica de los retrovirus, un tipo de virus que presentan
un genoma de ARN monocatenario y se replican de manera inusual a través de una forma
intermedia de ADN bicatenario.
La transcriptasa inversa actúa del siguiente modo: a
partir de la molécula de ARN se sintetiza una cadena de
ADN, ambas moléculas permanecen unidas formando un
híbrido ADN-ARN. Esta enzima, además de poseer la
actividad polimerasa 5'→3', actúa también como
endonucleasa ARNasa H, por lo que puede romper la
molécula de ARN para dar lugar a ADN simplexo. A partir
del cual puede formarse ADN duplexo, denominado ADNc
(copia o complementario).
In vivo la retrotranscriptasa utiliza como cebador el ADN de la célula huésped, pero
in vitro se pueden usar varios tipos de cebadores:
1. El ARNm de las células eucariotas presenta una
cola poli-A en el extremo 3', en este caso se
utilizan cebadores de timina. Para ello se
sintetiza un oligonucleótido de aproximadamente 20 timinas, que actuará como
primer al proporcionar el extremo 3' sobre el que actuará la polimerasa.
2. Si el ARN no posee cola de poliadenilato, se usa un conjunto de cebadores de 4 o 6
nucleótidos con todas las combinaciones posibles. Al menos alguno de los
oligonucleótidos reconocerá alguna secuencia complementaria en el ARN y podrá
actuar como cebador.
3. En el caso de que se conozca la secuencia nucleotídica del ARN se puede sintetizar
un cebador específico.

8 Alberto Fonte Polo
ARN polimerasas: son un conjunto de enzimas capaces de polimerizar los ribonucleótidos
para sintetizar ARN a partir de una secuencia de ADN que sirve como molde, es decir, las
ARN polimerasas son dependientes de ADN. In vivo estas enzimas realizan la transcripción
del ADN, y para ello deben reconocer unas regiones promotoras situadas al comienzo del
gen. In vitro, hay que incorporar estas regiones promotoras al vector, para que la enzima se
pueda unir a la cadena de ADN. Hay que tener en cuenta que cada enzima reconoce un
promotor específico. Las ARN polimerasas que se suelen utilizar son las de los fagos T7,
T3 y SP6 de E. coli.
Enzimas “polimerasas” que no requieren molde:
Desoxirribonucleotidil transferasa terminal: es una enzima que sintetiza cadenas
de ADN sin la necesidad de un molde, y por lo tanto añade nucleótidos de forma
aleatoria. Requiere extremos 3' OH simplexos para poder polimerizar. Dependiendo
de los dNTPs que se añadan, la enzima creará una secuencia de nucleótidos u otra.
Poli A polimerasa: esta enzima añade nucleótidos de adenina en el extremo 3' de
moléculas de ARN simplexo. No necesita cadena patrón o molde, pues sólo añade
adenina. En condiciones naturales es la enzima de poliadenilación que añade la cola
poli-A en el ARN mensajero de los eucariotas.
Enzimas de modificación
Las enzimas de modificación alteran los extremos de las moléculas, normalmente, de ADN.
Las principales son:
Ligasa: es una enzima que forma enlaces covalentes entre el extremo 5' de una cadena
polinucleotídica y el extremo 3' de otra cadena adyacente. Ésta es probablemente la enzima
más usada en ingeniería genética.
Fosfatasa: enzima que elimina grupos fosfato del extremo 5', y origina por lo tanto
moléculas con grupos –OH en todos los extremos (en los 5' y en los 3'). De este modo se
evita que moléculas fragmentadas puedan unirse de nuevo, ya que la ligasa no reconoce los
extremos 5' OH.
Nucleotidil quinasa: enzima que añade grupos fosfato en los extremos de moléculas
desfosforiladas en el extremo 5'.

Fundamentos de Genética Aplicada 9
Marcaje de moléculas de ADN
Una de las aplicaciones de las enzimas que anteriormente se han estudiado es el marcaje de
las moléculas de ADN. Se realiza incorporando a la molécula de ADN un fluorocromo, es
decir, un componente que hace que la molécula sea fluorescente, un átomo radioactivo
como el 32
P o el 33
P, o un hapteno, que es la parte de un antígeno que por sí sola no dispara
respuesta inmune, pero sí posee especificidad. De este modo se puede realizar el
seguimiento de los procesos en los que está implicada dicha molécula de ADN. Se
distinguen dos tipos principales de marcaje, según quede marcada toda la molécula o sólo
se marquen los extremos:
Marcaje global: se usa para marcar fragmentos de más de 1.000 pares de bases de
longitud, existen dos técnicas diferentes:
Nick translation (traslación de mellas): se emplea
una enzima ADNasa que rompe en el interior de la
molécula de ADN, se hace en presencia de Mg2+
,
en este caso sólo corta una de las dos cadenas en
puntos aleatorios e introduce mellas. A
continuación se usa la ADN polimerasa I de E.
coli, sólo se requieren las actividades polimerasa
5'→3' y exonucleasa 5'→3'. La enzima se
introduce en la mella, y mediante la actividad
exonucleásica 5'→3' elimina los nucleótidos del
extremo 5' y mediante la actividad polimerásica
añade nucleótidos en el extremo 3' libre, es decir,
ocurre un traslado de la mella. Además se
necesitan dNTPs, se añaden nucleótidos fríos, es
decir, sin marcar, y nucleótidos calientes o
marcados, normalmente en una proporción de 1:4.
Así, el ADN de nueva síntesis quedará marcado.
Random priming (cebado al azar): como en
el caso anterior se parte de ADN duplexo,
pero se aumenta la temperatura hasta los
94ºC para generar cadenas simples por
desnaturalización. En este caso la enzima
que se emplea es el fragmento de Klenow
modificado (sólo con actividad polimerasa
5'→3'). Es necesario añadir cebadores que
proporcionen el extremo 3' OH, los
cebadores son secuencias simplexas cortas
de seis nucleótidos (oligonucleótidos
hexámeros). Hay que añadir una mezcla de
cebadores con todas las combinaciones
posibles de seis nucleótidos, alguno de ellos
reconocerá una secuencia complementaria y
se unirá en algún punto de las dos cadenas.
Al igual que en la técnica anterior se añaden
desoxirribonucleótidos trifosfato, algunos
de ellos marcados.

10 Alberto Fonte Polo
Marcaje en los extremos: se usa para marcar fragmentos de ADN muy pequeños, de entre
100 y 200 pares de bases.
Marcaje del extremo 5': se emplea una fosfatasa para eliminar el grupo fosfato de
los extremos 5'. Después, se añaden nucleótidos, normalmente dATP, que tienen
marcado con 32
P el grupo fosfato en posición γ (el más externo). La enzima
nucleotidil quinasa añadirá el grupo fosfato marcado a los extremos 5' OH.
Marcaje del extremo 3': se usa la enzima exo λ, que con su actividad exonucleásica
5'→3' elimina los nucleótidos de los extremos 5' dejando extremos protuberantes 3'.
Se añaden nucleótidos fríos y calientes (en menor proporción) y la
desoxirribonucleotidil transferasa terminal los incorporará a los extremos 3' de la
molécula de ADN, de este modo se genera una cola marcada.
Producción de ADN recombinante
Para generar ADN recombinante se requiere un vector de clonación, generalmente un
plásmido bacteriano, y un fragmento de ADN. En primer lugar, hay que cortar el plásmido
para obtener una molécula de ADN lineal, para ello se usa una enzima de restricción que
deja extremos romos. En segundo lugar, se tratan ambas moléculas de ADN por separado
con la enzima exo λ. Se generarán moléculas con extremos protuberantes 3'. Después se usa
la desoxirribonucleotidil transferasa terminal para que añada una cola de nucleótidos en los
extremos 3'. En cada preparación se añadirá un tipo de nucleótido diferente pero
complementario, por ejemplo, si a la preparación del plásmido se le añade guanina, al ADN
cromosómico se le añadirá citosina. Posteriormente se reúnen sendas preparaciones y en un
alto porcentaje ambas moléculas de ADN se unirán formando una molécula recombinante.
Por último, se necesita que una ADN polimerasa rellene los huecos que quedan en la
molécula con nucleótidos complementarios, y que una ligasa una los fragmentos para que la
molécula quede sellada.

Fundamentos de Genética Aplicada 11
Amplificación enzimática de fragmentos de ADN y ARN
Reacción en cadena de la polimerasa
La reacción en cadena de la polimerasa, conocida como PCR por sus siglas en inglés
(Polymerase Chain Reaction), es una técnica de biología molecular descrita en 1985 por
Kary Mullis, cuyo objetivo es obtener un gran número de copias de un fragmento de ADN
particular, en teoría, basta partir de una única copia de ese fragmento original, o molde.
Esta técnica sirve para amplificar un fragmento de ADN, su utilidad reside en que, tras la
amplificación resulta mucho más fácil estudiar e identificar el ADN.
Inicialmente la técnica era lenta, ya que las polimerasas que se usaban (fragmento Klenow)
se desnaturalizaban al realizar los cambios de temperatura y era necesario agregar nuevas
polimerasas en cada ciclo. Puesto que las temperaturas del ciclo (95 ºC en las fases de
desnaturalización del ADN) suponen la inmediata desnaturalización de toda proteína,
actualmente se emplean ADN polimerasas termoestables, extraídas de microorganismos,
generalmente arqueas, adaptados a vivir a esas temperaturas, por ejemplo: Thermus
aquaticus (polimerasa Taq) y Pyrococcus furiosus (Pfu). Hoy, todo el proceso de la PCR
está automatizado mediante un aparato llamado termociclador, que permite grandes
cambios de temperatura en pocos segundos.
Para realizar la técnica se necesitan:
ADN polimerasa termorresistente, la más común es la Taq polimerasa.
ADN molde, que contiene la región que se va a amplificar, debe estar
desnaturalizado.
Cebadores (o primers), son cadenas simplexas de oligonucleótidos complementarias
a una región de las dos hebras del ADN. Estas secuencias son reconocidas por la
polimerasa permitiendo iniciar la reacción. Delimitan la zona de ADN a amplificar.
Desoxinucleótidos trifosfato (dNTPs), el sustrato para polimerizar un nuevo ADN.
Una solución tampón que mantiene el pH adecuado para el funcionamiento de la
ADN polimerasa. Se usan tampones de Tris-HCl con un pH en torno a 7,5.
Cationes magnesio (Mg2+
), agregado comúnmente como cloruro de magnesio
(MgCl2), que actúan como cofactores de la polimerasa.
In vivo el origen de replicación marca el inicio de la síntesis de ADN, y la molécula molde
sólo es leída una vez, mientras que in vitro el inicio de la síntesis de ADN está marcado por
los cebadores, y ocurren varias rondas de replicación. In vivo los cebadores son secuencias
de ARN, mientras que in vitro son oligonucleótidos de ADN.
Componentes y parámetros de la reacción
El proceso de PCR por lo general consiste en una serie de 35 o 36 cambios repetidos de
temperatura llamados ciclos, este proceso tiene lugar en dos horas aproximadamente. De
este modo, a partir de una sola molécula de ADN se pueden llegar a conseguir 68×109
copias idénticas. Las temperaturas usadas y el tiempo aplicado en cada ciclo dependen de la
enzima y de los cebadores empleados.
44

12 Alberto Fonte Polo
Las etapas de un ciclo de replicación son las siguientes:
Desnaturalización: el primer paso consiste en desnaturalizar el ADN, es decir, en
separar las dos hebras de las cuales está constituido. In vivo las hebras de ADN se
separan por acción de las enzimas topoisomerasas que rompen los puentes de
hidrógeno. Pero in vitro el ADN se desnaturaliza por calentamiento a 94-95ºC.
Hibridación: el segundo paso de la PCR es la unión del cebador a la región
complementaria de la molécula de ADN molde. Para ello es necesario bajar la
temperatura a 50ºC aproximadamente, permitiendo así la hibridación. Los cebadores
actuarán como límites de la región de la molécula que va a ser amplificada.
Polimerización: fase de alargamiento o de síntesis. La ADN polimerasa sintetiza
una nueva hebra de ADN complementaria a la hebra molde añadiendo los dNTPs
complementarios en dirección 5'→3', uniendo el grupo 5'-fosfato de los dNTPs con
el grupo 3'-OH del cebador. La temperatura para este paso depende de la ADN
polimerasa que usemos. Para la Taq polimerasa, la temperatura de máxima actividad
está en 72°C.

Fundamentos de Genética Aplicada 13
Las moléculas de ADN obtenidas tras el tercer ciclo son de dos tipos: producto corto y
producto largo. El producto corto tiene una longitud perfectamente definida por los
extremos 5' de los cebadores y contiene exactamente la secuencia que se desea amplificar.
El producto largo es el que incorpora las cadenas de ADN originales de la muestra y cuyos
extremos 3' no están definidos. Con cada nuevo ciclo se incrementa el número de moléculas
de ADN delimitadas por los cebadores.
En la PCR hay una amplificación exponencial:
En la reacción en cadena de la polimerasa se parte, normalmente, de una cantidad de ADN
de 50 ng, pero actualmente se puede amplificar incluso una sola molécula de 3,3 pg. Y
usualmente se trabaja con ADN no deteriorado, pero se ha llegado a amplificar ADN de
herbarios antiguos e incluso de fósiles.
Para llevar a cabo la PCR se añaden dNTPs en exceso y cebadores en exceso. Cuando la
reacción se acaba no es por falta de dNTPs o cebadores, sino porque la ADN polimerasa se
ha soltado de la cadena.

14 Alberto Fonte Polo
Normalmente la Taq polimerasa usa moldes de 100 pb – 2 kb, por encima de 2 kb la
enzima en muy poco procesiva. La Taq polimerasa tiene actividad polimerásica 5'→3' y
exonucleásica 5'→3', pero no tiene actividad exonucleasa 3'→5', y por ello no puede
corregir sus propios errores, y tiene una alta tasa de error (10–4
, es decir, comete un error
cada 10.000 bases incorporadas). Hay que tener en cuenta que estos errores son
acumulativos. La Pfu polimerasa es más lenta, pero posee actividad correctora de pruebas,
y por lo tanto tiene menor tasa de error.
Los cebadores usados en PCR tienen un tamaño que va desde 15 hasta 30 pares de bases,
aunque lo más normal es que tengan un tamaño de 20-24 pb. Tienen que ser secuencias
totalmente específicas de la secuencia que se quiere amplificar. Normalmente tienen una
composición de guanina-citosina del 50% aproximadamente. La temperatura de hibridación
está determinada por la composición de nucleótidos del primer, si la cantidad de G-C es
elevada la temperatura de hibridación será alta, 50-60ºC (55ºC Tª óptima). Pero si hay más
cantidad de A-T se requerirá menor temperatura de hibridación, y el cebador
complementará regiones inespecíficas del ADN. Al sintetizar los cebadores hay que evitar
que haya tramos que contengan bases (3-4) repetidas, para que no se formen apareamientos
incorrectos. Además, el cebador no debe tener secuencias complementarias, pues se podrían
formar bucles, y tampoco deben existir secuencias complementarias entre un cebador y
otro, para que no se formen dímeros.
Es necesario conocer las secuencias nucleotídicas laterales a la región que se quiere copiar
del ADN molde, para poder sintetizar los cebadores complementarios. Si se desconocen
estas secuencias hay que recurrir a otros métodos:
Si se conoce la secuencia de aminoácidos codificada por la secuencia de ADN que
se pretende replicar, se puede predecir la secuencia nucleotídica de dicho ADN.
Pero hay que tener en cuenta que el código genético es degenerado, y que un mismo
aminoácido puede estar codificado por más de un triplete. Por ello, se elabora una
mezcla (oligonucleótido degenerado) de todos los posibles cebadores, según la
secuencia aminoacídica.
Si se conocen secuencias homólogas al ADN que se quiere amplificar se pueden
sintetizar los cebadores específicos. Por ejemplo, conociendo el gen de la insulina
de rata se pueden diseñar cebadores para el gen de la insulina humana, puesto que
son genes homólogos. Normalmente se trabaja con genes de varias especies
distintas, y se buscan las regiones más conservadas como posibles promotores.
También se puede elaborar una mezcla de oligonucleótidos con variaciones en la
tercera base de los codones.
PCR de ARN
La reacción en cadena de la polimerasa con transcripción reversa (RT-PCR del inglés
reverse transcription polymerase chain reaction) es una variante de la PCR, en la que una
hebra de ARN es retrotranscrita en ADN complementario (ADNc) usando una enzima
denominada transcriptasa reversa, y el resultado, se amplifica en un PCR tradicional.
En este proceso se parte de una población de ARNm que se extrae de una preparación de
tejido o de un cultivo celular, la reacción será distinta según se quiera amplificar toda la
población de ARNm, o por el contrario, sólo un tipo concreto de ARNm. En cualquier caso,
la reacción comienza siempre del mismo modo: se añade un oligonucleótido poli-T de 20 o

Fundamentos de Genética Aplicada 15
24 nucleótidos que actuará como cebador, este poli-T complementa con la cola poli-A del
ARNm y proporcionará el extremo 3' OH.
La enzima retrotranscriptasa sintetizará una cadena de ADN a partir de la cadena de ARN,
y posteriormente, usando su actividad endonucleasa H degradará la cadena de ARN. La
transcriptasa inversa actúa a una temperatura de 37ºC. La reacción puede continuar de dos
modo distintos:
En el caso de que se quiera amplificar todas
las moléculas de ARNm sin distinción, se
usará la desoxirribonucleotidil transferasa
terminal, esta enzima sintetiza cadenas de
ADN sin molde. Si se añaden, por ejemplo,
desoxirribonucleótidos trifosfato de guanina
la enzima añadirá una cola poli-G en el
extremo 3' del ADN. A partir de este punto
se procede con una reacción PCR normal. Se
añade la enzima Taq polimerasa, una mezcla
de dNTPs y dos cebadores de 20-25
nucleótidos: un oligonucleótido poli-C y un
oligonucleótido poli-T, que complementarán
con las regiones poli-G y poli-A del ADN.
Si se quiere amplificar sólo un tipo de ARNm producido por un gen concreto, es
necesario conocer la secuencia nucleotídica del ARNm, y en consecuencia la del
ADN, para poder diseñar los cebadores específicos. Se añaden los dNTPs, la Taq
polimerasa y los dos primers específicos para que tenga lugar la reacción en cadena
de la polimerasa estándar.
La reacción RT-PCR es útil para buscar genes concretos, y probar su expresión en ciertos
tejidos.

16 Alberto Fonte Polo
Vectores de clonación en bacterias
Clonación
Un clon es un conjunto de células genéticamente idénticas, que proceden de un mismo
progenitor por división celular.
El proceso de clonación de genes implica la construcción de moléculas de ADN
recombinante, en estas moléculas se incluye el gen, o fragmento de ADN, a clonar. La
molécula de ADN recombinante es posteriormente introducida en células hospedadoras,
para que se replique usando las enzimas de la célula, de modo que todas las células
descendientes poseerán, al menos, una copia de la molécula recombinante. La obtención de
las moléculas de ADN recombinante es un proceso que se realiza in vitro, pero la
replicación en el interior de la célula hospedadora es un proceso totalmente in vivo.
Las células hospedadoras son normalmente células bacterianas, aunque ocasionalmente
también puede transferirse el ADN recombinante a células eucariotas, esto ocurre, sobre
todo, cuando se quiere estudiar la expresión de genes eucariotas. La bacteria E. coli es la
más usada para este fin. Se trabaja siempre con cepas modificadas genéticamente, a las que
se les ha eliminado el sistema de restricción-modificación, para, de este modo, invalidar los
sistemas que tiene la bacteria para evitar la intromisión de ADN extraño.
Una molécula de ADN recombinante se compone de:
ADN exógeno (o pasajero): es el gen, o fragmento de ADN, que se quiere clonar.
Éste puede tener distintos orígenes:
– Puede proceder de un genoma fragmentado.
– Puede ser un fragmento específico de ADN, amplificado mediante PCR.
– Puede ser ADNc (copia) procedente de ARNm.
– O puede ser ADN simplexo, que deberá pasarse a duplexo para clonarlo.
ADN vector: molécula que porta el fragmento de ADN exógeno. Se clasifican en:
– Vectores de clonación: aquellos que permiten mantener el ADN exógeno en
la célula hospedadora.
– Vectores de expresión: permiten transcribir y traducir la información
genética dentro de la célula hospedadora.
Tipos y características de los vectores de clonación
Los distintos tipos de vectores de clonación difieren en la especificidad de la célula
huésped, el tamaño de los insertos que pueden transportar y en características como el
número de copias que producen y el número y tipo de genes marcadores que contienen.
Plásmidos: fueron los primeros vectores que se desarrollaron, y aún son
ampliamente usados. Estos vectores proceden de moléculas de ADN de doble
cadena extracromosómicas que se encuentran de manera natural y que se replican
autónomamente dentro de las células bacterianas. Portan fragmentos pequeños de
ADN pasajero, de entre 0,1 y 8 kb (kilobases). Algunos plásmidos usados como
vectores son: pBR322, pUC18, pBluescript…
55

Fundamentos de Genética Aplicada 17
Vectores λ de inserción: son vectores basados en el genoma del fago lambda, que
pueden portar fragmentos de ADN de 0,1–8 kb.
Vectores λ de sustitución o de reemplazamiento: son también vectores basados en
el genoma del fago λ, pero pueden portar fragmento mayores (8–23 kb).
Cósmidos: son vectores híbridos utilizando partes del cromosoma de lambda y de
plásmidos. Pueden transportar entre 20 y 45 kb de ADN insertado.
BAC (bacterial artificial chromosome): es un cromosoma artificial bacteriano que
puede transportar insertos de 300 kb a 1,5 Mb (megabases).
YAC (yeast artificial chromosome): son cromosomas artificiales de levadura,
permiten clonar grandes trozos de ADN, de 2–3 Mb de tamaño. Al ser fragmentos
tan grandes, muy frecuentemente se producen alteraciones de la molécula de ADN.
Características generales de los vectores de clonación:
El ADN no esencial debe ser mantenido al mínimo. Para que estos vectores se
reproduzcan eficazmente en las bacterias es necesario que tengan el tamaño más
pequeño posible.
Tienen que ser moléculas autorreplicativas, y por lo tanto, deben tener su propio
origen de replicación.
Deben poseer genes marcadores, que sirven para identificar las células que
contienen el vector de clonación. Se suelen utilizar como marcadores, genes de
resistencia a antibióticos y genes de bioluminiscencia.
Deben tener un sitio de clonación, es decir, un sitio que permita introducir el ADN
pasajero. Estos sitios son dianas de restricción, puede haber una sola diana que sea
reconocida por una sola enzima, o varias dianas reconocidas por varias enzimas de
restricción. Pero, cada diana de restricción debe estar presente sólo una vez en el
vector, ya que si no, éste se fragmentaría.
Plásmidos
Características de los plásmidos naturales:
Son moléculas circulares de ADN duplexo de tamaños muy variables, desde 1 kb
hasta 8 kb.
Son moléculas de ADN extracromosómico que portan información adicional, no
esencial, para la bacteria. En muchas ocasiones las bacterias con plásmidos
presentan algún tipo de ventaja respecto de las que no tienen.
Son moléculas autorreplicativas, tienen un origen de replicación (ORI). Se sirven de
las enzimas de la célula bacteriana para replicarse, pero contienen genes
reguladores de la replicación, si éstos son muy estrictos la bacteria sólo tendrá dos
o tres copias del plásmido, pero si los genes reguladores de la transcripción son
relajados puede haber hasta 800 copias del plásmido.
Pueden transferirse por conjugación de
una bacteria a otra, puesto que poseen
genes tra (transferencia) y genes mob
(movilidad). Éstos no están presentes en
los plásmidos usados como vectores,
pues no interesa que se transfieran de
una célula a otra.
Plásmido
ORI
Genes reguladores
de la replicación
mob
tra

18 Alberto Fonte Polo
En el caso de los plásmidos usados como vectores, sólo es necesario que posean el origen
de replicación. Y tienen genes reguladores relajados, para conseguir el mayor número de
copias posible. Los plásmidos vectores que se usan en el laboratorio son totalmente
artificiales, no existen en la naturaleza. Se les ha incluido genes marcadores procedentes de
genomas bacterianos, y fragmentos de ADN artificiales con dianas de restricción.
Plásmido pBR322: es el primer plásmido utilizado en biotecnología, fue diseñado en 1977
por Bolívar y Rodríguez (de ahí su nombre pBR). A este plásmido se le han añadido dos
genes marcadores: un gen de resistencia a la ampicilina (ampr) y otro de resistencia a la
tetraciclina (tetr); y tres dianas de restricción: EcoRI, PstI y BamHI. La inserción de ADN
foráneo puede hacerse en cualquiera de las tres secuencias.
La diana PstI se encuentra inserta en el gen que confiere resistencia a ampicilina, y la diana
BamHI está dentro del gen de resistencia a la tetraciclina. Por ejemplo, si la enzima PstI
reconoce la diana PstI cortará el plásmido por ese punto, e introducirá el ADN exógeno
interrumpiendo e invalidado el gen ampr. Por lo tanto, esta bacteria no podrá crecer en un
medio con el antibiótico ampicilina, pero sí en un medio con tetraciclina. Si el ADN
exógeno se introduce en la diana BamHI la bacteria será sensible a la tetraciclina y
resistente a la ampicilina. Pero si el ADN exógeno se introduce en la diana EcoRI, la
bacteria presentará resistencia a los dos antibióticos. De este modo puede distinguirse entre
bacterias sin plásmido, con plásmido salvaje, o con plásmido recombinante, y además se
puede saber el lugar donde se ha insertado el ADN pasajero.
Plásmidos pUC (plasmid of University of California, plásmido de la Universidad de
California): son un conjunto de plásmidos (por ej. pUC18, pUC19) muy utilizados como
vectores, ya que presentan unas características que son muy beneficiosas:
Son pequeños, de modo que pueden transportar fragmentos de ADN relativamente
grandes (de 8 a 10 kb).
Tienen un origen de replicación y pueden producir hasta 500 copias de los
fragmentos de ADN insertado por célula (tienen control relajado de la replicación).
Se han modificado para que presenten muchas secuencias de reconocimiento para
enzimas de restricción que se han agrupado en una región denominada polylinker o
sitio de clonación múltiple (MCS: multiple cloning site).

Fundamentos de Genética Aplicada 19
Permiten la identificación sencilla de los plásmidos recombinantes, ya que
contienen un fragmento del gen bacteriano lacZ que produce colonias de color azul.
Si se inserta un fragmento de ADN en el polylinker, se inactiva este gen, y da lugar
a colonias de color blanco.
El plásmido pBluescript es un vector comercializado de la serie pUC. Está formado por una
molécula circular de ADN duplexo, de tamaño inferior a 3 kb, que comprende:
1. Un fragmento lacZ + polylinker + 2 promotores: el fragmento del gen lacZ procede
del operón lactosa de E. coli, y contiene el minigen de la β-galactosidasa (también
contiene el promotor lac). En el extremo 5' terminal de este fragmento lacZ se ha
introducido el polylinker. El polylinker es rico en sitios de restricción únicos, es
importante que las dianas de restricción estén presentes sólo una vez en el plásmido,
pues si no, se fragmentaría la molécula, y lo que se pretende es linealizarla. Los
promotores T3 y T7 también interrumpen el gen lacZ, se sitúan flanqueando el
polylinker. Estas regiones promotoras proceden del fago T3 y T7 respectivamente, y
son reconocidas por las enzimas ARN polimerasas de dichos fagos.
2. Un origen de replicación del fago M13 o F1: estos bacteriófagos son similares al
fago ΦX174 de E. coli, su material genético es una molécula de ADN circular
simplexo, pero cuando infecta a la bacteria se convierte en ADN circular duplexo, y
por medio del mecanismo del circulo rodante se originan moléculas simplexas
nuevamente. Esta región f1 origin se emplea cuando se quiere replicar un ADN en
forma de monohebra. No obstante, la replicación sólo se puede hacer por
coinfección con un virus auxiliar (helper) que aporte los genes que codifican las
enzimas necesarias para la replicación. Este procedimiento ha sido muy utilizado
para secuenciar el ADN, puesto que para esta tarea debe pasarse de ADN duplexo a
ADN simplexo.
3. Un gen de resistencia a la ampicilina: este gen permite, gracias al cultivo en
presencia de ampicilina, seleccionar todas las bacterias que han incorporado
Bluescript, ya sea con o sin el inserto. (La selección de los Bluescripts
recombinantes se realiza según si la β-galactosidasa que se obtiene es, o no,
funcional).
4. Una secuencia origen de replicación (ColE1 ori): esta secuencia permite la
multiplicación del plásmido en E. coli.

20 Alberto Fonte Polo
El gen lacZ del operón lactosa codifica la
enzima β-galactosidasa, que cataliza la reacción
de hidrólisis de la lactosa en glucosa más
galactosa. Esta enzima debe estar en forma
tetramérica para ser efectiva. Para que se
produzca la tetramerización de la β-
galactosidasa se requiere la información del
minigen (fragmento de la región 5' de lacZ), que codifica los 149 aminoácidos del extremo
N-terminal del péptido. Los restantes aminoácidos constituyen un monómero de β-
galactosidasa.
El plásmido pBluescript contiene el minigen de β-galactosidasa con capacidad de
tetramerizar. Este plásmido se introduce en cepas modificadas de E. coli que contienen un
gen lacZ defectivo que sólo codifica los monómeros de la β-galactosidasa. De este modo,
ocurre una complementación: el péptido que codifica el minigen hace que los cuatro
monómeros se ensamblen para formar la β-galactosidasa funcional.
El gen lacZ está reprimido de forma natural, y sólo se traduce en presencia de un inductor,
el galactósido IPTG. In vivo, la lactosa es la molécula inductora. IPTG induce por
desactivación de la represión la síntesis de la β-galactosidasa.
La presencia de la enzima β-galactosidasa se detecta añadiendo al cultivo celular un
sustrato de ésta, denominado X-Gal (5-bromo-4-cloro-3-indolil-β-D-galactósido). X-Gal es
un β-galactósido que, al igual que la lactosa, puede ser hidrolizado por la β-galactosidasa.
La ventaja de este sustrato es que su hidrólisis libera, además de galactosa, una sustancia X
(5-bromo-4-cloro-3-hidroxi-indol) coloreada en azul.
5′ 3′
NH2 COOH
minigen β-galactosidasa
LacZ

Fundamentos de Genética Aplicada 21
Pero, cuando se inserta una molécula de ADN pasajero dentro del plásmido no se forma el
tetrámero β-galactosidasa. Y esto es porque el ADN se introduce dentro del minigen, y la
proteína que se genera no tiene la capacidad de tetramerizar. Al no producirse β-
galactosidasa funcional X-Gal no se metaboliza y no cambia a color azul.
En resumen, en presencia de ampicilina sólo sobreviven las bacterias transformadas.
Aquellas colonias de color blanco corresponden a bacterias con plásmido recombinante,
mientras que las colonias de color azul corresponden a bacterias con plásmido pBluescript
salvaje, es decir, sin inserto de ADN foráneo.
Vectores basados en el genoma del fago λ
El bacteriófago lambda es un virus que infecta a la bacteria Escherichia coli. Se trata de un
virus complejo de ADN lineal bicatenario. Los extremos de su material genético son
cohesivos y complementarios (extremos cos) y ello hace que tras la infección su genoma se
circularice, comportándose, en caso de seguir un ciclo lisogénico, como un plásmido y
aprovechando las enzimas
recombinantes de la bacteria para
integrarse en el genoma de ésta. El
fago no tiene por qué integrarse, y
de hecho es más habitual que se
comporte como un virus de ciclo
lítico.
En el ciclo lisogénico, el
virus se inserta en un punto
concreto del genoma de la
bacteria. En este estado, el
virus se replica cuando lo
hace la bacteria, pasando
su genoma a las réplicas de
E. coli.
El ciclo lítico es la forma más habitual de actuación del virus al infectar la célula, y
también es la vía que sigue al final del ciclo lisogénico. En ella se producen
partículas virales que son liberadas al medio una vez que la bacteria hospedadora es
lisada, matando a la célula en el proceso.
El virus replica su genoma circular empezando por un punto de éste, y desenrollando sólo
una de las dos hebras. El resultado es un genoma lineal muy largo, consistente en una gran
cantidad de repeticiones del genoma original de forma seguida, lo que se conoce como
concatámero (o concatémero). La enzima terminasa es una endonucleasa que corta el
concatámero mediante sucesivos cortes en escalera (en las regiones cos). El virus también
sintetiza las proteínas de su cápside, y se ensambla en el citoplasma de la bacteria. Dentro
de la cápside sólo cabe, prácticamente, el genoma de un virus. Finalmente, la célula es
lisada, liberando los viriones al medio.

22 Alberto Fonte Polo
El genoma del fago lambda se ha cartografiado y secuenciado completamente. Para ser
utilizado como vector, el tercio central de su cromosoma puede ser reemplazado por ADN
foráneo, sin que ello afecte a la capacidad del fago de infectar células y formar calvas.
Antes de introducir el ADN exógeno hay que eliminar partes no útiles del genoma del
virus, para, de este modo, dejar espacio, pues en la cápside sólo hay hueco para 50 kb, y el
genoma del virus tiene 48.502 pb. Las regiones prescindibles corresponden a los genes que
controlan el ciclo lisogénico.
Dependiendo de la cantidad de ADN vírico que se elimine, para insertar el ADN pasajero,
existen dos tipos de vectores: vectores de inserción y vectores de sustitución.
En los vectores de inserción se ha eliminado sólo una parte de los genes que controlan la
lisogenia. Los vectores de inserción sólo presentan un sitio de restricción, donde se
introduce el inserto. Sirven para la clonación de pequeños fragmentos (hasta 8 kb).
Por ejemplo, el vector de inserción λgt10 presenta sólo la diana de restricción EcoRI, que
está situada en el gen cI que codifica el represor λ, éste impide el ciclo lítico. Esto hace que
la inserción de ADN extraño en este sitio inactive el gen cI. El represor no se sintetiza y el
ciclo del virus orienta hacia la vía lítica. En cambio, si el gen cI está activo el fago entra en
ciclo lisogénico. El vector λgt11 utiliza como sistema de selección el gen lacZ del operón
lactosa.
Regiones prescindibles del genoma del fago λ

Fundamentos de Genética Aplicada 23
En los vectores de sustitución (o de reemplazamiento) se sustituye toda la región
prescindible del genoma del fago por el ADN de interés. De este modo se pueden insertar
moléculas exógenas de mayor tamaño (de hasta 20-25 kb). Los vectores de sustitución
tienen dos sitios de restricción que flanquean el segmento de ADN que es sustituido por el
ADN que se va a clonar. Al tratar el genoma del fago con una enzima de restricción se
obtendrán tres fragmentos: brazo izquierdo, brazo derecho y región stuffer (región
prescindible).
En el ADN stuffer se han introducido varias dianas de restricción para poder fragmentarlo
en pequeños trozos que, por centrifugación, pueden separarse fácilmente de los fragmentos
de ADN mayores. El ADN pasajero ocupará el lugar del ADN stuffer, aunque también
pueden ligarse los dos brazos sin incorporar el ADN exógeno. Por eso, es necesario
seleccionar las moléculas recombinantes.
Los vectores λ recombinantes se empaquetan in vitro en las cápsides proteicas del fago λ, y
se pasan a un césped bacteriano. Se debe añadir la cantidad adecuada de fagos, para que
cada célula huésped bacteriana sea infectada por uno sólo de estos virus. Dentro de las
bacterias, los vectores se replican y forman múltiples copias del fago infectivo, llevando
todas ellas el fragmento de ADN de interés en la clonación. Cada fago infectará una célula,
se replicará y la lisará. A continuación, la descendencia de viriones infectará y, en
consecuencia, lisará las bacterias adyacentes al lugar de la primera célula infectada, creando
entonces una región clara o calva en el campo bacteriano opaco. Los fagos contenidos en
cada calva, o placa, de lisis pueden recogerse para ser analizados.
Proceso de empaquetamiento in vitro

24 Alberto Fonte Polo
Para conseguir cápsides víricas vacías se usan cepas mutantes del fago λ. Una cepa formará
colas pero no cápsides, debido a una mutación, otra cepa no produce colas pero sí cápsides.
Cuando se reúnen las colas, las cápsides y el ADN recombinante ocurre el
empaquetamiento in vitro de éste. Los vectores λ tienen extremos cohesivos 5', esto hace
que se forme un concatémero de vectores, algunos de ellos tendrán inserta la región de
ADN exógeno y otros no. Junto al extracto de empaquetamiento (proteínas fágicas) se
añade la enzima terminasa, que corta e individualiza cada molécula. Dentro de la cápside
sólo se introducen moléculas de ADN de 50 kb aproximadamente, por lo tanto, las
moléculas brazo izquierdo–brazo derecho no forman partículas infectivas, pues al tener un
tamaño menor no entran en la cápside. Todos los virus que se formen contienen en la
cápside moléculas del tipo brazo izquierdo–ADN exógeno–brazo derecho porque son las
que tienen el tamaño adecuado. De este modo se ha conseguido seleccionar las moléculas
recombinantes.
Cósmidos
Los cósmidos son vectores de clonación que permiten la introducción de insertos de ADN
de hasta 45 kb. Realmente un cósmido consiste en un plásmido al cual se le han adicionado
unos segmentos del genoma de un bacteriófago como el fago λ, por lo tanto, presenta
características intermedias entre los plásmidos y los vectores λ de reemplazamiento. Un
cósmido contiene:
Del plásmido, el origen de replicación y genes marcadores de resistencia a
antibióticos, esto depende del tipo de plásmido del cual derive y, por tanto, puede
contener más segmentos como por ejemplo el gen lacZ (que permite la selección
blanco/azul de las colonias) u orígenes de replicación para fagos como el M13 (que
permiten la obtención de ADN simplexo, usado para aplicaciones como la
secuenciación del inserto).
Del fago λ, los extremos cos, extremos complementarios del genoma del fago y que
se emplean para la recircularización del mismo.
Estos vectores facilitan el trabajo porque permiten el empaquetamiento in vitro, gracias a
los extremos cos, de los cósmidos en las partículas fágicas λ. Los fagos empaquetados se
usarán para infectar cepas de E. coli, consiguiendo un plásmido en la bacteria, ya que el
cósmido se recirculariza, al entrar a ésta, gracias a los extremos cos.
Uno de los cósmidos más usados es el SuperCos, contiene:
– Dos sitios cos, entre los cuales se sitúa la diana de restricción Xba I.
– Un origen de replicación (ori).
– Un sitio de clonaje múltiple
(MCS), es un polylinker que no
interrumpe ningún gen
marcador, incluye la diana de
restricción BamH I.
– Dos genes marcadores: uno de
resistencia a ampicilina y otro
de resistencia a neomicina.
– SV40 ori: origen de replicación
del virus animal SV40, usado
para replicar el vector cuando se
introduce en células animales.

Fundamentos de Genética Aplicada 25
Para producir cósmidos recombinantes, en primer lugar, se usa la enzima de restricción
BamHI para cortar el vector y linealizarlo. Después, se emplea la enzima fosfatasa alcalina
que hidroliza los grupos P de los extremos 5' y los transforma en extremos 5' OH y, de este
modo, se evita que el plásmido se circularice de nuevo.
Posteriormente se inserta el ADN exógeno, previamente cortado con la restrictasa Sau3AI.
La enzima Sau3AI genera extremos compatibles con la enzima BamHI, es decir, son
enzimas isoesquizómeras. Para ello, se hace una digestión parcial del ADN que se quiere
introducir en el cósmido. En una digestión parcial las condiciones de la reacción impiden
que sean reconocidas todas las dianas, y ocurren menos cortes de los que se esperarían en
condiciones normales. Como resultado se obtienen fragmentos de ADN de un tamaño
mayor. Se usa la enzima Sau3AI en lugar de la enzima BamHI, porque la primera reconoce
una diana de sólo 4 pares de bases, y la segunda tiene una diana de 6 pares de bases. Es
decir, si se cortara el ADN usando una diana de 6 pb los fragmentos que se obtendrían
serían demasiado grandes, ya que esta diana aparece con menos frecuencia que una de 4 pb.
La ADN ligasa une los extremos 3'-OH sólo con los 5'-P, y no con los 5'-OH, por eso queda
una mella entre el ADN exógeno y el ADN del cósmido. Hay que recordar que los
extremos que ha dejado la enzima BamHI en el cósmido tienen grupos –OH en el extremo
3' y en el 5', debido a la acción de la fosfatasa.
En segundo lugar, se digiere el cósmido con la enzima de restricción XbaI, que corta el
vector entre las regiones cos. Los cósmidos linealizados pueden unirse, por los extremos
cos, y formar un concatámero.
Por último, se añaden los extractos de empaquetamiento del fago λ junto con la enzima
terminasa, para empaquetar in vitro el cósmido con el ADN que se quiere clonar. Del
mismo modo que ocurría con los vectores de sustitución, dentro de las cápsides sólo se
empaquetan las moléculas que tienen ADN recombinante, pues son las que tienen el
tamaño adecuado.
Estos fagos no entran en ciclo lítico (tampoco en ciclo lisogénico), puesto que, los únicos
genes que hay en el cósmido procedentes del genoma del fago λ son los extremos cos. Y
por lo tanto, no producen la lisis de las bacterias que infectan, ni se reproducen dentro de
éstas. El fago sólo se utiliza para introducir el cósmido, que se comporta como un plásmido,
en el interior de las bacterias, este proceso se denomina transducción. Las colonias de
bacterias que contienen en su interior ADN recombinante, se seleccionan gracias a que el
cósmido lleva genes de resistencia a antibióticos.

26 Alberto Fonte Polo
BACs
El cromosoma artificial bacteriano, o BAC (bacterial artificial chromosome), es un vector
usado para clonar fragmentos de ADN de 300 kb de tamaño medio. Se distinguen dos tipos:
Los BACs propiamente dichos, están basados en el plásmido factor-F (factor de
fertilidad) encontrado de modo natural en la bacteria Escherichia coli. Este factor es
un episoma que puede integrarse en el cromosoma bacteriano y es responsable de la
conjugación bacteriana.
Los PACs (P1-derived artificial chromosome), son cromosomas artificiales
bacterianos derivados del fago P1, alojan menos cantidad de ADN pasajero que los
BACs.
Un PAC típico consta de:
– Dos secuencias pac, son secuencias repetidas protuberantes que permiten el
empaquetamiento in vitro.
– Dos secuencias lox que intervienen en la circularización del vector.
– Un origen de replicación (ori).
– Un gen marcador de resistencia a la canamicina (kanr).
– Un gen marcador sac implicado en el metabolismo de la sacarosa, que está
interrumpido con un sitio de clonaje múltiple (MCS).
De forma natural el bacteriófago P1 sólo presenta un sitio lox, pero en el PAC se han
insertado dos regiones lox. En el interior de E. coli, la enzima recombinasa reconoce los
sitios lox y produce la circularización del ADN situado entre éstos.
Para la selección de las moléculas PAC recombinantes se usan cepas de E. coli sac–, es
decir, cepas que no pueden crecer en presencia de sacarosa. Esto es debido a que en el
metabolismo de la sacarosa se produce un levano, que resulta tóxico para estas bacterias.
Dentro del gen sac se encuentra un polylinker con dianas de restricción. Cuando se
introduce una molécula de ADN recombinante el gen sac queda interrumpido, las bacterias
no sintetizan el levano tóxico y pueden crecer con normalidad. Pero aquellas bacterias que
no poseen vectores recombinantes mueren, puesto que, el gen sac se expresa con
normalidad y se produce el compuesto tóxico.
Para el empaquetamiento in vitro de los PACs se usan extractos de empaquetamiento del
fago P1 y enzima pacasa. La pacasa reconoce los extremos pac y permite el
empaquetamiento del vector en las partículas fágicas. Dentro de la cápside del fago el
vector se encuentra en forma lineal, pero cuando se introduce en la bacteria, ésta sintetiza
recombinasa, y se circulariza la molécula.
Un BAC propiamente dicho incorpora insertos, normalmente, de 300 kb, pero se ha
conseguido clonar fragmentos de hasta 1 Mb. Presenta genes procedentes del plásmido F:
– oriS: origen de replicación.
– rep: control de la replicación del plásmido.
– parA, parB, parC: genes implicados en el mantenimiento de un número bajo de
copias (1 o 2) del plásmido en la bacteria.

Fundamentos de Genética Aplicada 27
Además, tiene otros genes que no proceden del factor-F:
– cmr: gen de resistencia al cloranfenicol.
– Minigen lacZ con un polylinker (MCS) en su
interior. Se usa para el ensayo de alfa-
complementación, del mismo modo que ocurre
en el plásmido pBluescript. A ambos lados del
minigen lacZ hay una diana de restricción de la
enzima NotI, esta restrictasa se usa para
desinsertar el ADN pasajero.
– lox: sitio reconocido por la enzima recombinasa,
permite linealizar el vector.
Procedimiento de clonación: en primer lugar, se corta el
vector con BamHI para linealizarlo, y se añade fosfatasa
alcalina para desfosforilar los extremos 5' del vector, y evitar autoligación. Después, se
hace una digestión parcial del ADN que se quiere clonar con la enzima Sau3A, la cual
genera extremos compatibles a los de la enzima BamHI, y se lleva a cabo la reacción de
ligación de ambas moléculas de ADN. Para la transformación de las bacterias se emplea la
electroporación. Este método consiste en la aplicación de un campo eléctrico que produce
poros transitorios en la membrana citoplasmática de la bacteria, a través de los cuales puede
introducirse el BAC.
La selección de los clones se hace en un medio con cloranfenicol, con IPTG y con X-gal.
Las bacterias que resisten el antibiótico son las que han incorporado el BAC, las colonias
que presentan color azul son aquellas cuyo vector no tiene ADN exógeno, y las colonias
blancas tienen BACs recombinantes.
YACs
El cromosoma artificial de levaduras o YAC (yeast artificial chromosome) es el vector de
clonación de mayor capacidad (hasta 2 Mb). Es un vector que pretende imitar las
características de un cromosoma normal de una levadura, portando un centrómero y
telómeros terminales. Esto permite clonar en levaduras grandes secuencias de ADN, al
comportarse como un cromosoma propio de este microorganismo eucariota. Son utilizados
en construcción de genotecas genómicas, sin embargo, son más inestables que otros
vectores como los BACs, que han acabado imponiéndose. Un problema que presentan los
YACs es que el ADN que se inserta puede proceder de dos regiones distintas del genoma.
Los YACs contienen características de bacterias:
– Origen de replicación (ori) de tipo plasmídico.
– Gen marcador de resistencia a ampicilina (ampr).
Y características de levaduras:
– Secuencia de replicación de levaduras (SRA).
– Genes marcadores de selección: un gen TRP relacionado con el metabolismo del
triptófano, y un gen URA relacionado con el metabolismo del uracilo.
– Gen marcador de inserción sup, relacionado con el metabolismo de la adenina,
interrumpido con un sitio de clonaje múltiple (MCS). Este gen identifica las células
de levadura que incorporan el vector, ya que las células huésped que expresan

28 Alberto Fonte Polo
normalmente el gen sup muestran un color rojizo (sup+), mientras que las que
presentan ADN recombinante adquieren un color blanquecino (sup–).
– Centrómero (CEN), permite la segregación durante la división celular.
– Dos secuencias teloméricas (TEL), procedentes de un ciliado del género
Tetrahymena, pero que funcionan con las levaduras. Entre estas dos secuencias se
sitúa una diana de restricción para la enzima BamHI.
Procedimiento de clonación: se digiere el vector y el ADN que se quiere clonar con la
misma restrictasa, o con restrictasas isoesquizómeras, para que ocurra el ligamiento entre
ambas moléculas. Para que el vector se comporte como un cromosoma más de la levadura
hay que linealizarlo, mediante el uso de la enzima de restricción que reconoce la diana que
se encuentra entre las regiones TEL, dejando, de este modo, funcionales las secuencias
teloméricas. Los YACs se introducen en el interior de levaduras para que ocurra la
clonación del ADN recombinante, pero también se pueden introducir en bacterias, pues
poseen un origen de replicación de procariotas.

Fundamentos de Genética Aplicada 29
Clonación en bacterias
Concepto de genoteca
Una genoteca (genomic library en inglés) es un conjunto de clones en los que está
contenido el genoma de un organismo.
La construcción de una genoteca implica: aislar el genoma completo de la célula (obtención
de ADN genómico), fraccionar el genoma en fragmentos de un tamaño apropiado, clonar
cada fragmento en un vector, e introducir cada molécula recombinante en una célula
hospedadora.
Las genotecas se usan para aislar y estudiar genes, o secuencias genómicas. Cuando se
quiere clonar un fragmento de ADN previamente amplificado mediante PCR no se
construye una genoteca, ya que se parte de una muestra pura.
Existen dos tipos de genotecas según contengan ADN genómico o ADNc:
Genoteca genómica: contiene el genoma de un organismo. Se emplea para aislar un
gen determinado completo (con intrones y exones, en el caso de eucariotas) o para
clonar regiones reguladoras que no se transcriben (por ejemplo, promotores).
Genoteca de ADN copia: está constituida por ADN copia, sintetizado, por acción
de la transcriptasa inversa, a partir de los ARNm expresados en una célula, tejido u
organismo. Esta genoteca es útil cuando se quiere clonar un gen procesado, para ser
expresado directamente a ARN. Cuando se quiere expresar un gen eucariota en
bacterias, éste debe estar procesado, ya que las enzimas procarióticas no reconocen
las regiones intrónicas.
Parámetros de una genoteca:
Tamaño de la genoteca, es decir, el número de clones que constituye una genoteca.
Tamaño medio de los insertos, condiciona el tamaño global de la genoteca.
Representatividad, cuanto mayor sea la probabilidad de encontrar un clon
determinado más representativa será la genoteca.
Hay que tener en cuenta que, la clonación es un proceso aleatorio, puede haber fragmentos
del genoma que, por azar, estén más representados en la genoteca, y otros que no lo estén
en absoluto. Lo importante es que todos los fragmentos estén presente en al menos un
vector, pero existen ciertas regiones del genoma, con secuencias repetitivas, que son
difíciles de clonar, pues presentan gran capacidad para alterarse. Para que una librería
genómica sea representativa, el número de clones debe ser superior al número de
fragmentos generados.
Construcción de genotecas de ADN genómico y de ADN copia
Construcción de genotecas genómicas: el número de clones componentes debe ser, en la
práctica, suficiente para abarcar todo el genoma, incluyendo tanto el ADN codificante
como el no codificante. La preparación del ADN de partida se realiza por escisión del
genoma con enzimas de restricción bajo condiciones de rotura parcial, es decir, que no
66

30 Alberto Fonte Polo
permitan la actuación exhaustiva de la enzima sobre todos los sitios de restricción presentes
en el ADN. La rotura se produce en cada molécula aleatoriamente sobre el conjunto de
sitios de restricción, de forma que aparecen un número menor de fragmentos, y estos son de
mayor tamaño, respecto a lo teóricamente posible, y diferentes de una a otra molécula.
Construcción de genotecas de ADN complementario: a diferencia del caso anterior, las
genotecas de ADNc representan exclusivamente a la parte codificante del genoma. La
clonación del ADN copia está basada en la acción de la transcriptasa inversa, una ADN
polimerasa dependiente de ARN derivada de retrovirus, que puede sintetizar una cadena de
ADNc complementaria de un molde de ARN. La retrotranscriptasa requiere un cebador
para iniciar la síntesis de ADN, tal como un oligonucleótido constituido por timidinas
(oligo-dT); este fragmento se une a la cola poli-A en el extremo 3' de las moléculas de
ARNm y proporciona un cebador que es ampliado por la transcriptasa inversa para la
síntesis de una copia complementaria. Después se elimina el ARNm, y la cadena única de
ADNc se usa como plantilla para la ADN polimerasa, y se origina una molécula de doble
cadena.

Fundamentos de Genética Aplicada 31
A las moléculas de ADNc de doble cadena, se le añaden en ambos extremos unas
moléculas cortas de ADN, denominadas adaptadores (o ligadores). Los adaptadores
presentan en su interior una diana de restricción, y cuando se digiere el ADNc con la
enzima de restricción específica se generan extremos cohesivos. Pero, la diana de los
adaptadores también puede estar presente dentro de la molécula de ADNc original, en este
caso la molécula se fragmentaria al hacer la digestión con la enzima de restricción. Para
evitar esto, hay que metilar el ADNc antes de ligar los adaptadores, ya que cuando una
diana está metilada no es reconocida por la enzima. Por último, las moléculas de ADNc con
los extremos cohesivos se pueden ligar a un vector apropiado para crear una genoteca
representativa de todos los transcritos originales de ARNm que se encuentran en la célula o
en el tejido original.
Representatividad de una genoteca
A partir del tamaño del genoma completo y del tamaño medio de los insertos se puede
calcular el número mínimo de clones necesario para que la genoteca represente al genoma
completo. Sin embargo, el carácter aleatorio de la preparación de fragmentos y del proceso
de clonación hace que, para tener certeza de que una genoteca contiene cualquier región del
genoma, se requiera en la práctica un número de clones muy superior al teórico. Para
calcular el número de clones mínimo para que una genoteca sea representativa se utiliza la
siguiente fórmula:
N = ln 1–P
ln 1– 1n
N: número de clones; P: probabilidad de que un clon en la genoteca tenga una secuencia
determinada; n: número de fragmentos distintos que se pueden generar, depende del tamaño
del genoma y del tamaño medio del inserto.
n = tamaño medio del inserto (pb)
tamaño del genoma (pb)

32 Alberto Fonte Polo
Por ejemplo: el tamaño del genoma humano es de aproximadamente 3∙109 pb, si se quieren
hacer insertos de alrededor de 20∙103 pb, ¿qué cantidad de clones hay que usar para trabajar
con una probabilidad del 95%, y para una probabilidad del 99%?
Se pueden generar 1,5∙105 fragmentos distintos (3∙10
9 pb 2∙10
4 pb ). Si todos los
fragmentos se clonaran se obtendrían 1,5∙105 clones, pero como el proceso es
aleatorio hay que trabajar con un número de clones mayor:
P = 95% N = ln 1–0,95
ln 1– 11,5∙10
5 = 450.000 clones
P = 99% N = ln 1–0,99
ln 1– 11,5∙10
5 = 690.000 clones

Fundamentos de Genética Aplicada 33
Rastreo de genotecas
Detección e identificación de clones individuales
El rastreo de genotecas incluye una serie de procedimientos que permiten detectar, de
entre los centenares de miles de clones que componen una genoteca, aquel clon que es
portador de la secuencia de interés.
Las estrategias que se pueden llevar a cabo para el rastreo de las genotecas dependerán del
conocimiento previo, que posea el investigador, de la secuencia de interés. Pueden darse
dos situaciones:
Que se conozca la secuencia de nucleótidos que componen el fragmento de ADN
buscado, es decir, que se conozca su estructura.
O que se conozca el producto de expresión (proteína) del fragmento, esto es, que se
conozca su función.
Algunos de los métodos usados para la identificación de clones recombinantes de interés en
una genoteca son: la hibridación de ADN usando alguna sonda marcada, y el rastreo
inmunológico (uso de anticuerpos frente al producto de interés).
Detección directa por complementación
El método más usado, cuando se conoce la estructura del fragmento de interés, es la
hibridación de ácidos nucleicos con una sonda específica.
Su fundamento estriba en la posibilidad de formación de ADN de cadena doble entre
cadenas sencillas del ADN a sondear y cadenas sencillas de ADN marcado (ADN sonda).
Una sonda es un fragmento de ácido nucleico que se usa para el reconocimiento específico
de una molécula determinada de ácido nucleico de cadena sencilla, en un proceso de
hibridación. Existen varios tipos fundamentales de sondas de ácidos nucleicos: ADN
genómico, ADN copia, ARN o un oligonucleótido sintético. La sonda se puede marcar de
varias maneras, mediante nick translation, random priming, o por PCR.
Ahora bien, ¿cuál es la fuente de la sonda? Lógicamente, el usar una sonda de ADN
determinada se justifica porque sepamos o sospechemos que dicho ADN puede tener
parecido con el ADN que estamos buscando en la genoteca. Los principales orígenes de las
sondas moleculares son los siguientes:
Una secuencia de ADN procedente de otro organismo y de la que tenemos indicios
de que puede tener parecido con lo que estamos buscando. A este tipo de sondas se
las suele denominar como sondas heterólogas.
Una sonda sintética elaborada a partir de la información disponible sobre la
secuencia de aminoácidos de la proteína correspondiente al gen que estamos
buscando. Con esta información, y teniendo en cuenta que el código genético es
degenerado, podemos sintetizar todos los posibles oligonucleótidos sinónimos que
codificarían dicha porción de proteína. Marcando esa mezcla de oligonucleótidos,
los podemos usar como sonda para localizar el gen en una genoteca. Pero, de entre
todos los oligonucleótidos, sólo unos pocos serán homólogos a la secuencia de
77

34 Alberto Fonte Polo
interés, y por lo tanto, habrá una señal de hibridación muy débil. Por otro lado, hay
que tener en cuenta que puede haber falsos positivos cuando algunos de los
oligonucleótidos hibriden con regiones del ADN que no son las buscadas.
El procedimiento para el rastreo por hibridación de ADN es el siguiente: las genotecas se
construyen en placas de Petri con medio de cultivo selectivo. Las miles de colonias de una
genoteca se replican a filtros de nylon o de nitrocelulosa, las colonias se fijan a la
membrana de nylon mediante alta temperatura (80ºC) o por luz ultravioleta. Posteriormente
se usa una solución alcalina (NaOH) para lisar las bacterias y desnaturalizar el ADN
recombinante.
El ADN sonda se desnaturaliza también para convertirlo a cadenas sencillas. Y se
mezcla el ADN sonda desnaturalizado y marcado con los filtros que contienen el ADN de
la genoteca (igualmente desnaturalizado), esta reacción se realiza a una temperatura (60-
65ºC) adecuada para que la sonda se una a la región especifica, pero no renaturalicen ambas
cadenas. Si el ADN sonda tiene cierto grado de homología con el ADN de alguno de los
clones, se emparejará con dicho ADN para generar doble hélice. Después, hay que lavar la
muestra a alta temperatura para eliminar el exceso de sonda, y para eliminar las sondas que
se hayan unido en regiones inespecíficas.
Finalmente revelamos el resultado, el procedimiento de detección dependerá del
tipo de marcaje realizado. En la película fotográfica de autorradiografía, encontraremos
unas manchas peculiares correspondientes a las colonias que hayan dado la hibridación con
la sonda. Con esta información vamos a las placas matrices donde están las colonias
originales de células vivas, y recuperamos los clones, que podemos seguir estudiando.
Detección indirecta por hibridación de ácidos nucleicos
Cuando no se dispone de una sonda específica se puede recurrir a la hibridación diferencial
o a la hibridación sustractiva de ácidos nucleicos.
Hibridación diferencial: este método permite la detección de clones que se expresan
abundantemente en un tejido, y muy poco, o nada, en otro tejido diferente. Se extrae el
ARNm de cada uno de los tejidos, y se retrotranscribe a ADNc de doble cadena, mediante
la transcriptasa inversa. Parte del ADNc del tejido 1 se marca y se usa como sonda, y parte
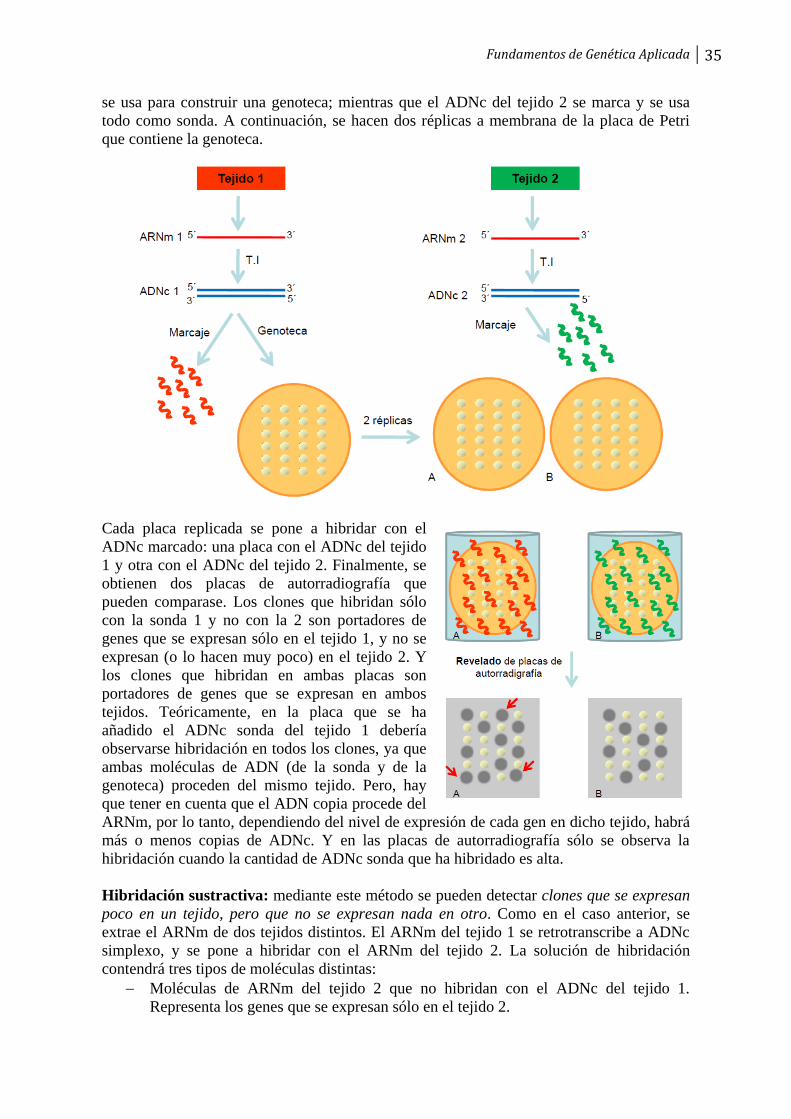
Fundamentos de Genética Aplicada 35
se usa para construir una genoteca; mientras que el ADNc del tejido 2 se marca y se usa
todo como sonda. A continuación, se hacen dos réplicas a membrana de la placa de Petri
que contiene la genoteca.
Cada placa replicada se pone a hibridar con el
ADNc marcado: una placa con el ADNc del tejido
1 y otra con el ADNc del tejido 2. Finalmente, se
obtienen dos placas de autorradiografía que
pueden comparase. Los clones que hibridan sólo
con la sonda 1 y no con la 2 son portadores de
genes que se expresan sólo en el tejido 1, y no se
expresan (o lo hacen muy poco) en el tejido 2. Y
los clones que hibridan en ambas placas son
portadores de genes que se expresan en ambos
tejidos. Teóricamente, en la placa que se ha
añadido el ADNc sonda del tejido 1 debería
observarse hibridación en todos los clones, ya que
ambas moléculas de ADN (de la sonda y de la
genoteca) proceden del mismo tejido. Pero, hay
que tener en cuenta que el ADN copia procede del
ARNm, por lo tanto, dependiendo del nivel de expresión de cada gen en dicho tejido, habrá
más o menos copias de ADNc. Y en las placas de autorradiografía sólo se observa la
hibridación cuando la cantidad de ADNc sonda que ha hibridado es alta.
Hibridación sustractiva: mediante este método se pueden detectar clones que se expresan
poco en un tejido, pero que no se expresan nada en otro. Como en el caso anterior, se
extrae el ARNm de dos tejidos distintos. El ARNm del tejido 1 se retrotranscribe a ADNc
simplexo, y se pone a hibridar con el ARNm del tejido 2. La solución de hibridación
contendrá tres tipos de moléculas distintas:
Moléculas de ARNm del tejido 2 que no hibridan con el ADNc del tejido 1.
Representa los genes que se expresan sólo en el tejido 2.

36 Alberto Fonte Polo
Moléculas de ADNc del tejido 1 que no han encontrado secuencias homólogas en el
ARNm del tejido 2. Son los genes exclusivos del tejido 1.
Moléculas híbridas de ADN-ARN. Son los genes que se expresan en ambos tejidos.
La mezcla de las tres moléculas se hace pasar por una columna de hidroxiapatito, que
retiene las moléculas duplexas. Se recoge la fracción eluida de la columna, que contiene las
moléculas simplexas (ADNc 1 y ARNm 2), y se pone en presencia de una ADN polimerasa
y de nucleótidos (dNTPs). La enzima ADN polimerasa no usará como sustrato el ARNm,
sólo trabajará con el ADNc del tejido 1, el cual pasará a ser ADNc de doble cadena. Éste
será usado para construir una genoteca enriquecida con clones que se expresan poco en el
tejido 1 y nada en el tejido 2.
Vectores de expresión y detección por métodos inmunoquímicos
Para el rastreo mediante métodos inmunológicos es necesario construir genotecas obtenidas
con vectores de expresión.
Los vectores de expresión son vectores de clonación que permiten la expresión del inserto.
Por lo tanto, tienen que permitir la transcripción y traducción del ADN exógeno. Estos
vectores presentan las siguientes características:
Un origen de replicación (ori).
Un gen marcador.

Fundamentos de Genética Aplicada 37
Un promotor que permita
la transcripción del ADN
insertado. Esta región
promotora está situada
aguas arriba (región 5')
del gen, y es reconocida
por la maquinaria de
replicación. El promotor
tiene que ser fuerte, es
decir, tiene que permitir
una elevada expresión del
gen, el 50% de la proteína
total celular debe ser la
codificada por el inserto;
y también tiene que ser
inducible, es decir, debe
permitir el control del momento de la expresión mediante inductores y/o represores.
Un sitio de unión al ribosoma (RBS) situado a continuación del promotor. Es una
región que se transcribe pero que no se traduce, y permite la traducción eficaz del
fragmento clonado. Las regiones no traducidas de los genes se denominan UTR (del
inglés untranslated region), en este caso se dice que es una región 5'-UTR. Existen
dos tipos:
o Secuencia Shine-Dalgarno: se usa para expresar fragmentos de ADN
procarióticos. Se trata de una secuencia consenso situada unos nucleótidos
antes del codón de inicio de la traducción, que es complementaria a una zona
de la región 3' del ARN ribosomal 16S.
o Secuencia Kozak: se usa cuando se quiere expresar fragmentos eucarióticos.
Permite la unión del mensajero al ribosoma.
Un sitio de inicio de la traducción, en eucariotas el codón de inicio más empleado
es AUG.
Un sitio de clonación múltiple (MCS), o polylinker, con dianas de restricción únicas.
Una señal de terminación que interviene en la finalización de la transcripción, y que
permite la liberación del ARN mensajero.
Antes y después del MCS, se suelen insertar unos genes que codifican para
proteínas de fusión. Las proteínas de fusión aparecen normalmente en la región C- y
N-terminal de la proteína diana, están en fase con ella, y no debe haber codones de
paro entre ambas. Las proteínas de fusión tienen distintas funciones: aumentan la
expresión, mejoran la solubilidad, favorecen la purificación, y ayudan a la
localización de la proteína de interés. Por ejemplo, el gen ompF produce una
proteína de membrana externa en E. coli, y se incluye como proteína de fusión en
algunos vectores. Este gen presenta un promotor propio y una región RBS.
Seguidamente al gen ompF se encuentra el ADN insertado, y a continuación de éste
se encuentra, por ejemplo, el gen lacZ, que permite localizar los clones
recombinantes. Cuando se expresan estos genes se obtiene una proteína trihíbrida,
formada por la proteína ompF, la proteína diana, y la enzima β-galactosidasa. El
producto del gen ompF hace que la proteína trihíbrida se localice en la membrana
citoplasmática de la bacteria hospedadora, y la actividad β-galactosidasa hace que
las colonias portadoras de ADN recombinante aparezcan de color azul si se les
añade X-Gal.

38 Alberto Fonte Polo
Siempre que se incluyen proteínas de fusión en el vector, se añaden también sitios
de reconocimiento de proteasas entre los genes que codifican para las proteínas de
fusión y el sitio de clonación múltiple. De este modo, es posible aislar de forma
específica la proteína diana de las proteínas de fusión.
Si no disponemos de una sonda de ADN, y si el gen que estamos buscando en la genoteca
se expresa hasta nivel de proteína, podemos recurrir al rastreo inmunoquímico (siempre y
cuando contemos con anticuerpos frente a la proteína en cuestión).
Para buscar en la genoteca aquellos clones portadores de la proteína recombinante,
se procede del siguiente modo: se hacen réplicas de todas las colonias de la genoteca a
membranas de nylon o nitrocelulosa, y se fijan las bacterias a la membrana. Después, las
colonias de réplica se lisan, para dejar expuestas las proteínas producidas por estos clones.
El filtro con las proteínas se trata con un anticuerpo específico que reconoce la proteína que
estamos buscando (anticuerpo primario, que no está marcado). Y se lava la muestra para
quitar los productos en exceso y no unidos. A continuación, se trata el filtro con un segundo
anticuerpo. Este anticuerpo secundario está marcado y reconoce al anticuerpo primario, de
modo que varias moléculas de anticuerpo secundario se unirán a cada molécula de
anticuerpo primario, así se amplifica la señal. El resultado es que en la placa
autorradiográfica aparecerá una señal que se corresponde con aquel clon que ha expresado
la proteína recombinante, proteína que es el producto de expresión del ADN pasajero
insertado en el vector de expresión.

Fundamentos de Genética Aplicada 39
Caracterización estructural de moléculas de ADN recombinante
Mapas de restricción
Un mapa de restricción es un mapa físico que muestra las posiciones relativas de las
dianas, de un conjunto de enzimas de restricción, contenidas en una secuencia de ADN. Las
distancias entre las dianas de corte se indican en pares de bases. Antes de construir el mapa
de restricción hay que estimar el tamaño del fragmento clonado.
Estimación del tamaño del fragmento clonado en la molécula de ADN recombinante:
en primer lugar, mediante la técnica conocida como miniprep, se extrae del cultivo
bacteriano el ADN de naturaleza plasmídica. Una vez aislado el ADN recombinante se
procede a separar el inserto del vector, para lo cual hay dos métodos:
1. Digestión con la enzima de restricción usada en la clonación.
2. Amplificación del inserto por PCR, usando los cebadores adyacentes al sitio de
clonación múltiple (MCS). Por ejemplo, en el plásmido pBluescript se usan los
primers T3 y T7.
En segundo lugar, se realiza una electroforesis en gel de agarosa. A pHs cercanos a la
neutralidad, el ADN está cargado negativamente y migra del cátodo al ánodo con una
movilidad que depende de su tamaño. Normalmente, los fragmentos lineares de ADN
cortos migran a través del gel más rápidamente que los fragmentos largos. La tasa de
migración depende del tamaño de los fragmentos lineares y del gradiente de voltaje que se
establece entre los electrodos. Los productos de la
digestión con la enzima de restricción se cargan en un
pocillo del gel, para separarlos y poderlos visualizarlos.
Se observarán dos bandas, una se corresponde con el
ADN vector y otra con el ADN pasajero. En el caso del
ADN amplificado por PCR se observa una sola banda
(ADN pasajero). Los tamaños de los fragmentos que
han corrido en el gel, se determinan mediante
comparación con los tamaños de fragmentos conocidos
(los patrones) situados en otra calle del gel.
Construcción de un mapa de restricción: para construir el mapa hay que separar el
inserto del vector, para ello, se corta el gel de agarosa con un bisturí y se extrae la banda del
ADN exógeno. Después, el inserto purificado se digiere con diferentes enzimas de
restricción:
88
–
+
Patrón E.R. PCR
Vector
Inserto

40 Alberto Fonte Polo
Digestión con la enzima de restricción A.
Digestión con la enzima de restricción B.
Doble digestión con las enzimas de restricción A y B.
Los distintos fragmentos de ADN generados se separan por electroforesis. Los fragmentos
se organizan en forma de mapa, comparando los tamaños de fragmentos en las calles donde
se cortó con una sola enzima (digestiones simples) con los de las calles donde se cortó con
dos enzimas (digestiones dobles). El tamaño de cada molécula de ADN se estima
comparándolo con las bandas de ADN patrón de tamaño conocido.
Por ejemplo, la digestión de un determinado fragmento de ADN de 3300 pares de bases, da
lugar a los fragmentos que se muestran a continuación:
Enzima de restricción Tamaño de los fragmentos
generados (pb)
A
1500
1000
800
B
1700
900
700
A+B
1500
900
600
200
100
Posteriormente, se extraen del gel de agarosa los fragmentos obtenidos por la digestión con
la enzima A, y se digieren con la enzima B, y viceversa. En este caso es obtienen los
siguientes fragmentos:
1500 pb B 1500 pb 1700 pb
A 1500 pb + 200 pb
1000 pb B 900 pb + 100 pb 900 pb
A 900 pb
800 pb B 600 pb + 200 pb 700 pb
A 600 pb + 100 pb
Patrón Inserto A B A+B

Fundamentos de Genética Aplicada 41
Se observa que, cada fragmento producido por la digestión del fragmento original A con la
enzima B también se encuentra en una de las digestiones dobles en las que el fragmento
original B se ha cortado con la enzima A. Estos datos permiten colocar las dianas de corte y
elaborar un mapa inequívoco. La clave del mapeo de restricción es el uso de fragmentos
solapados.
Los fragmentos que son digeridos sólo por una de las dos enzimas se corresponden con los
extremos de la molécula de ADN exógeno, en este ejemplo serían los fragmentos de 1500
pb y de 900 pb. Estos fragmentos se buscan en la doble digestión y se ordenan. Cuando el
fragmento de 1700 pb se digiere con la enzima A se obtienen dos fragmentos, uno de 200
pb y otro de 1500 pb, el fragmento 1500 se dispone en un extremo y a continuación de éste
está el fragmento 200. Del mismo modo se puede determinar que el fragmento de 100 pb
está contiguo al fragmento de 900 pb. Y por lo tanto el fragmento de 600 pb está entre el
200 pb y el 100 pb. El mapa de restricción resultante sería el siguiente:
Éste es el método usado para hacer mapas de dianas (de 6 nucleótidos) de enzimas de corte
poco frecuente, pero no sirve si se quieren hacer mapas de restricción que muestren las
dianas (de 4 nucleótidos) de enzimas que cortan más frecuentemente el ADN. Por ejemplo,
asumiendo que en un ADN de gran longitud las dianas quedan distribuidas aleatoriamente,
cuanto más corta sea la secuencia del sitio de restricción, más probabilidad habrá de
encontrarla por puro azar, es decir, mayor será la frecuencia de reconocimiento. Esto hará
que se generen fragmentos de ADN más pequeños, y por lo tanto, será muy poco probable
que dentro de éstos haya una diana para una segunda enzima de restricción. En estos casos
hay que recurrir a otras estrategias.
Se usa una enzima fosfatasa para desfosforilar los
extremos 5' P de la molécula de ADN que se pretende
mapear. Se añade ATP marcado, y la enzima nucleotidil
quinasa regenerará los grupos fosfato en los extremos 5',
generándose una molécula de ADN marcada en sus
extremos. A continuación, se corta la molécula con una
enzima de restricción que genere grandes fragmentos, y
mediante electroforesis se aísla el fragmento de mayor
tamaño. Si se usara la enzima B del ejemplo, el fragmento que se seleccionaría sería el de
1700 pb, como es un fragmento de uno de los extremos presenta marcaje en 5' P. Este
fragmento se digiere parcialmente con una enzima de restricción de corte frecuente, bajo
condiciones de rotura parcial que no permiten que la enzima corte todas las dianas
presentes en el ADN. Y se obtiene una colección de fragmentos solapantes de diferentes
tamaños, según la diana que haya sido cortada en cada uno.
A
A+B
B
200
100
1500 800 1000
1700 700 900
1500 600 900
A A
A A
B B
B B

42 Alberto Fonte Polo
Estos fragmentos se separan mediante electroforesis y se detecta el marcaje
del extremo 5' en una placa de autorradiografía, de este modo se puede
conocer la posición de las dianas en el ADN. Hay que tener en cuenta que
los fragmentos de digestión internos no pueden ser detectados, ya que no
presentan marcaje.
Utilidades de los mapas de restricción:
Se usan para localizar dianas de enzimas de restricción, para un posterior análisis
genómico o una subclonación.
Sirven para ordenar clones solapantes, en la construcción de genotecas genómicas.
Secuenciación de ácidos nucleicos
El análisis más detallado de la estructura del ADN consiste en averiguar la secuencia de
nucleótidos. Los métodos más utilizados son el de secuenciación automática y el método
enzimático de terminación de cadena de Sanger, también conocido como método didesoxi.
Método enzimático de terminación de cadena o método didesoxi de Sanger: para
obtener la secuencia de bases nitrogenadas de un segmento de ADN por este método, se
necesitan los siguientes compuestos:
El ADN molde o segmento de ADN que se desea secuenciar. Para poder secuenciar
un segmento de ADN, previamente se necesita tener gran cantidad de ese
fragmento, y por tanto, hay que clonarlo en un vector apropiado. Además, debe estar
en estado de hélice sencilla.
Una enzima que replique el ADN, normalmente la ADN Polimerasa I del
bacteriófago T4. Ésta emplea como molde ADN de hélice sencilla y siguiendo las
reglas de complementariedad de bases va añadiendo nucleótidos a partir de un
cebador.
Un cebador, que suele ser un oligonucleótido corto de alrededor de 20 bases de
longitud necesario para que la ADN polimerasa I comience a añadir nucleótidos por
el extremo 3' OH. Este cebador debe poseer una secuencia de bases complementaria
a la del fragmento de ADN que se desea secuenciar. Debido a que la secuencia de
nucleótidos del segmento que se quiere secuenciar es desconocida, se emplea un
primer con secuencia complementaria al vector empleado para clonar el fragmento
de ADN, además, este cebador procede de una región del vector muy cercana al
punto de inserción del ADN problema cuya secuencia se conoce. El primer
utilizado suele marcarse radiactivamente.
Los cuatro nucleótidos trifosfato (dATP, dCTP, dGTP y dTTP). A veces en vez de
marcar radiactivamente el cebador, se marca radiactivamente uno de los cuatro
nucleótidos trifosfato en cada reacción.
Por último, se necesitan nucleótidos didesoxi (ddATP, ddTTP, ddCTP y ddGTP).
Los nucleótidos didesoxi son nucleótidos modificados que han perdido el grupo
hidroxilo de la posición 3' de la desoxirribosa. Estos nucleótidos pueden
incorporarse a la cadena de ADN naciente, pero no es posible que se una a ellos
ningún otro nucleótido por el extremo 3'. Por tanto, una vez incorporado un
nucleótido didesoxi se termina la síntesis de la cadena de ADN.
En primer lugar, deben realizarse en cuatro tubos diferentes, cuatro mezclas de reacción.
Cada mezcla de reacción contiene los cuatro nucleótidos trifosfato (dATP, dCTP, dTTP y

Fundamentos de Genética Aplicada 43
dGTP), ADN polimerasa I, un cebador marcado radiactivamente y un nucleótido didesoxi,
por ejemplo ddATP, a una concentración baja. El nucleótido didesoxi utilizado (ddATP en
este ejemplo) competirá con su homólogo (dATP) por incorporarse a la cadena de ADN
que se está sintetizando, produciendo la terminación de la síntesis en el momento y lugar
donde se incorpora. Por este sistema, en cada mezcla de reacción se producen una serie de
moléculas de ADN de nueva síntesis de diferente longitud que terminan todas en el mismo
nucleótido y marcadas todas radiactivamente por el extremo 5' (todas contienen en el
extremo 5' el cebador utilizado).
Los fragmentos de ADN de nueva síntesis obtenidos en cada mezcla de reacción se separan
por tamaños mediante electroforesis en geles verticales de acrilamida, muy finos y de gran
longitud, que permiten distinguir fragmentos de ADN que se diferencian en un sólo
nucleótido. Los productos de cada una de las cuatro mezclas de reacción se insertan en
cuatro calles o carriles diferentes del gel. Una vez terminada la electroforesis, el gel se pone
en contacto con una película fotográfica de autorradiografía. La aparición de una banda en
una posición concreta de la autorradiografía en una de las cuatro calles nos indica que en
ese punto de la secuencia del ADN de nueva síntesis (complementario al ADN molde) está
la base correspondiente al nucleótido didesoxi utilizado en la mezcla de reacción
correspondiente.
Teniendo en cuenta que el ADN de nueva síntesis crece en la dirección 5'→3', si
comenzamos a leer el gel por los fragmentos de menor tamaño (extremo 5') y avanzamos

44 Alberto Fonte Polo
aumentando el tamaño de los fragmentos (hacia 3'), obtendremos la secuencia del ADN de
nueva síntesis en la dirección 5'→3'.
Método automático de secuenciación: la principal diferencia entre el método enzimático
de terminación de cadena y el método automático de secuenciación radica, en primer lugar,
en el tipo de marcaje. En el método automático en vez de radiactividad se utiliza
fluorescencia y lo habitual es realizar cuatro mezclas de reacción, cada una con un
didesoxinucleótido trifosfato (ddNTP) marcado con un fluorocromo distinto. Este sistema
permite automatizar el proceso de manera que es posible leer al mismo tiempo las
moléculas de ADN de nueva síntesis producto de las cuatro mezclas de reacción. La
segunda diferencia radica en el sistema de detección de los fragmentos de ADN. La
detección del tipo de fluorescencia correspondiente a cada reacción se lleva a cabo al
mismo tiempo que la electroforesis, de manera que los fragmentos de ADN de menor
tamaño que ya han sido detectados se dejan escapar del gel, permitiendo este sistema
aumentar el número de nucleótidos que se pueden determinar en cada electroforesis y, por
consiguiente, en cada secuenciación.
Hibridación de ácidos nucleicos en filtro
El Southern-blot es un método, desarrollado por el biólogo inglés Edwin Southern, que
permite detectar la presencia de una secuencia de ADN en una mezcla compleja de este
ácido nucleico, mediante el uso de electroforesis en gel y de hibridación utilizando sondas
específicas.

Fundamentos de Genética Aplicada 45
Para analizar una muestra de ADN genómico, éste debe ser
previamente fragmentado, utilizando enzimas de restricción. Los
fragmentos obtenidos se separan, de mayor a menor tamaño,
mediante una electroforesis en gel de agarosa. Cuando se digiere una
molécula de ADN recombinante (menor número de dianas) y se carga
en un gel, se resuelve en bandas discretas, pero en el caso del ADN
genómico (más dianas) no se observan bandas definidas, sino todo un
rastro de fragmentos de ADN de distintos tamaños.
Una vez terminada la electroforesis y sin teñir el gel, los fragmentos de ADN se traspasan a
una membrana de nylon. Para ello, se dispone el gel boca abajo sobre un papel que está en
contacto con una solución desnaturalizante, y por encima del gel se coloca la membrana de
nylon. El líquido asciende por capilaridad, y el ADN es arrastrado desde el gel hasta la
membrana, donde queda retenido.
A continuación, se fija el ADN por medio de luz ultravioleta o mediante calor. El filtro se
incuba con una sonda marcada, específica para la secuencia que se desea identificar. Esta
sonda es una secuencia de ácido nucleico que reconocerá a la secuencia de ADN
inmovilizada en el filtro, a través del reconocimiento de secuencias complementarias de
ácidos nucleicos. De este modo, en la placa de autorradiografía se detectan aquellos
fragmentos del ADN genómico que son homólogos a la sonda empleada.
Si en lugar de transferir ADN a la membrana se transfiere ARN, la técnica se denomina
Northern blot, se usa en ensayos de expresión de fragmentos. La detección de proteínas, en
una muestra de un tejido homogeneizado o extracto, es denominada Western blot.
Patrón ADNr ADNg

46 Alberto Fonte Polo
Aplicaciones del Southern:
Obtención de RFLPs desde genomas de especies próximas, para buscar diferencias
estructurales entre genomas próximos que presentan mutaciones en dianas de
enzimas de restricción.
Elaboración de mapas genéticos.
Determinación del número de copias de un fragmento en el genoma.
El término RFLP (del inglés restriction fragment length polymorphism, es decir,
polimorfismo para la longitud de un fragmento de restricción) se refiere a secuencias
específicas de nucleótidos en el ADN que son reconocidas y cortadas por las enzimas de
restricción y que varían entre individuos. Las secuencias de restricción presentan
usualmente patrones de distancia, longitud y disposición diferentes en el ADN de diferentes
individuos de una población, por lo que se dice que la población es polimórfica para estos
fragmentos de restricción.
Una forma de detectar dicho polimorfismo es mediante Southern-blot. Para ello, se
digiere el ADN genómico de dos especies con una enzima de restricción y luego se hibrida
la mezcla de fragmentos resultante con una sonda marcada que hibrida en la zona de la
diana que muestra polimorfismo.
La especie 1 muestra en el Southern blot una única banda (el fragmento acotado por dos dianas de
restricción para la enzima empleada), la especie 2 muestra dos bandas, pues la sonda complementa con una
secuencia en ambos fragmentos.
Localización del fragmento clonado en el genoma
Se pretende asignar el fragmento clonado a un locus particular. Los genomas eucariotas
están fragmentados en cromosomas, por lo tanto, hay que asignar el fragmento a un
cromosoma particular.
Un mapa cromosómico representa el orden lineal de los genes en un cromosoma. El
conjunto de mapas cromosómicos constituye el mapa genómico del organismo.
Existen dos variantes fundamentales de mapas:
Los mapas genéticos o de ligamiento, definidos mediante unidades de frecuencia
de recombinación. Para construir los mapas genéticos es necesario analizar la
descendencia de un cruzamiento particular. Se basa en que la probabilidad de
recombinación entre dos genes es menor cuanto menor es la distancia entre ellos.
Los mapas físicos, en los que las distancias entre loci se expresan en pares de bases,
como ocurre, por ejemplo, en los mapas de restricción. Los mapas físicos se
obtienen ordenando fragmentos clonados. El mapa físico más completo es la
secuencia total de nucleótidos que conforman el genoma del organismo.
Especie 2
Especie 1
1 2
Sonda

Fundamentos de Genética Aplicada 47
Los mapas genéticos son la única forma de localizar genes en especies con genomas no
secuenciados. Y son el paso previo a la secuenciación del genoma.
Para hacer un mapa genético es necesario que existan, al menos, dos alelos distintos que
den lugar a un fenotipo distinto. La forma de análisis de los fenotipos depende del
conocimiento del gen y de las herramientas de las que dispongamos.
Ejemplo:
La anemia falciforme es una enfermedad autosómica recesiva con una alta prevalencia en
determinadas regiones geográficas. Los pacientes afectados por esta patología presentan en
homocigosis la sustitución de una glutamina por una valina en el tercer aminoácido de la β-
globina. Dicha mutación se corresponde con el cambio de una adenina por una timina en el
codón 6 de dicho gen, que al mismo tiempo altera la diana de restricción para la enzima
MstII. Esto da lugar a la existencia de RFLPs para el gen de la β-globina, entre individuos
sanos y enfermos.
Aprovechando esta circunstancia se ha diseñado un método muy sencillo para detectar la
mutación. Dicha diana se pierde con la mutación que causa anemia falciforme.
Flanqueando a esta diana existen dos dianas para la misma enzima: una a 1,2 kb y otra a 0,2
kb de distancia. La digestión del ADN genómico de un individuo con la enzima MstII, dará
lugar a miles de fragmentos entre los cuales estarán los correspondientes al locus de la β-
globina. Podemos ahora separar todos los fragmentos según su tamaño en un gel de agarosa
y transferir el resultado a un filtro de nitrocelulosa por la técnica de Southern. Utilizando
una sonda complementaria al fragmento de 1,2 kb, podremos detectar aquellos fragmentos
de ADN que contengan la secuencia complementaria a la sonda.
Si un individuo es homocigoto para el alelo normal de la β-globina, el único fragmento que
será detectado por la sonda será el de 1,2 kb. Si el individuo es portador del alelo de anemia
falciforme detectaremos dos fragmentos: el de 1,2 kb y el de 1,4 kb, uno proveniente del
cromosoma que tiene la secuencia normal (con diana) y el otro proveniente del cromosoma
con la secuencia de la anemia falciforme (sin diana). Finalmente, en un individuo
homocigoto para la anemia falciforme sólo detectaremos el fragmento de 1,4 kb.

48 Alberto Fonte Polo
Determinación del número de copias de una secuencia en el genoma
La mayor parte de los genes son de copia única, pero hay secuencias de más de una copia,
repetidas en tándem o dispersas. La determinación del número de copias de una secuencia
de ADN en el genoma depende de las regiones adyacentes al fragmento insertado.
Si las regiones adyacentes a las diferentes copias, del fragmento clonado, en el genoma son
distintas, se determina el número de copias por RFLPs. Para ello, se digiere el ADN
genómico con una enzima de restricción, se hace una electroforesis en gel de agarosa y se
realiza un Southern Blot. Por ejemplo, si hay dos copias del fragmento clonado en el
genoma, y las regiones adyacentes al inserto son distintas, las dianas para la restrictasa
usada en la digestión estarán en distinta posición en cada una de las copias. Por lo tanto, al
hibridar el fragmento clonado con la membrana de nylon, y después del revelado
autorradiográfico, se observarán dos bandas que corresponden a las dos copias del
fragmento en el genoma.
Si, por el contrario, las regiones adyacentes al fragmento clonado son iguales, se observará
una sola banda en la película de autorradiografía, y no se puede saber el número de copias.
Por lo tanto, hay que recurrir a los ensayos de Dot-Blot.
El dot blot consiste en colocar, sobre una membrana, una serie de diluciones de ADN en
forma de pequeños círculos o puntos, para su posterior hibridación con la sonda. En este
caso, se hacen diluciones del ADN genómico y diluciones del fragmento clonado, y se
hibridan ambos conjuntos de diluciones con el inserto marcado. Se revela el resultado, y se
buscan las diluciones que coincidan en intensidad de señal.
Copia 1 en la región X del genoma
Copia 2 en la región Y del genoma
E.R. A E.R. A
E.R. A E.R. A
Membrana de nylon Autorradiografía Gel de electroforesis
ADN + E.R. A
Sonda
marcada
Membrana de nylon Autorradiografía Gel de electroforesis
ADN + E.R. A
Sonda
marcada Copia 1 en la región X del genoma
Copia 2 en la región Y del genoma
E.R. A E.R. A
E.R. A E.R. A

Fundamentos de Genética Aplicada 49
Sabiendo el tamaño del genoma y el tamaño del inserto, se puede estimar el número de
copias del ADN clonado que hay en el genoma.
Por ejemplo: el tamaño del genoma es de 3,3∙109 pb, y el inserto mide 3∙10
3 pb. Y en el dot
blot se ha visto que, la intensidad de la dilución de 10 µg de ADN genómico coincide con
la dilución de 10 ng del inserto. Sabiendo que en un picogramo de ADN hay 109 pares de
bases, ¿cuántas copias del fragmento clonado hay en el genoma?
Moléculas de inserto en 10 ng:
10 ng = 104 pg = 10
13 pb
1013
/ 3∙103 = 3,3∙10
9 insertos
Número de genomas en 10 µg:
10 µg = 107 pg = 10
16 pb
1016
/ 3, 3∙109 = 3∙10
6 genomas
3,3∙10
9
3∙106 = 1.100 copias del fragmento clonado en el genoma
Diluciones de
ADNg
Diluciones de
ADN clonado

50 Alberto Fonte Polo
Caracterización funcional de moléculas de ADN recombinante
Para conocer la función del fragmento de ADN clonado hay que estudiar los productos que
codifica (ARN y proteínas), y saber en qué circunstancias celulares se expresan, es decir, en
que tipo celular y en qué momento del desarrollo.
En la caracterización funcional de moléculas de ADN recombinante se hacen estudios a dos
niveles:
Análisis funcional in vitro: se trabaja con el fragmento clonado.
Análisis funcional in vivo: hay que introducir el fragmento clonado en células
(bacterias o células eucariotas) o en organismos (transgénicos) para que se exprese.
Las herramientas de estudio serán distintas según se haya clonado un gen o un promotor.
Caracterización funcional in vitro
Si el fragmento clonado es un gen:
Si se ha clonado un gen, o un fragmento génico, éste puede ser usado para saber en qué tipo
celular y en qué estadio celular se está expresando. El Northern-blot y la RT-PCR son las
técnicas usadas para este fin.
El Northern-blot es una técnica de detección de moléculas de ácido ribonucleico de
una secuencia dada dentro de una mezcla compleja (por ejemplo, un ARN
mensajero para un péptido dado en una extracción de ARN total). Para ello, se
extrae el ARN de los distintos tejidos, se digiere con una enzima de restricción, y se
somete a una electroforesis en gel a fin de separar los fragmentos en base a su
tamaño. Tras esto, se transfiere por capilaridad el contenido del gel, ya resuelto, a
una membrana en la cual se efectúa la hibridación de una sonda (el fragmento
clonado) marcada radiactiva o químicamente.
La señal de hibridación que se observa por autorradiografía indica el tejido
donde se ha expresado el fragmento de ADN clonado, el momento de expresión, y
la intensidad con la que lo hace.
Membrana de nylon
Sonda
marcada
Gel de electroforesis
Pat
rón
T
ejid
o A
T
ejid
o B
T
ejid
o C
–
+
Película radiográfica
99

Fundamentos de Genética Aplicada 51
La RT-PCR es un procedimiento rápido y cuantitativo para el análisis del nivel de
expresión de genes. Esta técnica utiliza la capacidad de la transcriptasa reversa (RT)
de convertir el ARN en ADNc de una sola hebra y la acopla con la amplificación
por PCR de tipos específicos de moléculas de ADNc presentes en la reacción de
RT. Los ADNc que se producen durante la reacción de la retrotranscriptasa
representan una ventana en el patrón de genes que están siendo expresados en el
momento en que el ARN fue aislado.
El ARN celular total se extrae de tejidos, o de células, y se utiliza como
plantilla para la transcriptasa inversa. Una pequeña alícuota de la reacción de RT se
agrega entonces a una reacción PCR que contiene los primers específicos de las
secuencias que uno desea amplificar, que se han diseñado gracias a que se conoce la
secuencia de bases del fragmento clonado. Los productos del RT-PCR pueden
entonces ser visualizados en un gel de agarosa, de este modo, se demuestra que el
gen clonado se está expresando en el tejido en cuestión.
Si el fragmento clonado es un promotor:
El promotor de un gen es la región de ADN que controla la iniciación de la transcripción
de dicho gen a ARN. El promotor forma parte de la región reguladora del gen, es decir, no
tiene una función codificante y no se transcribe, pero es necesario para el inicio de la
transcripción. Dicha región -el promotor- se encuentra aguas arriba (extremo 5') del gen, y
es el lugar donde se une la ARN polimerasa, y en eucariotas, además, los factores
transcripcionales. Los promotores tienen secuencias de nucleótidos definidas (secuencias
consenso) como, por ejemplo, la caja TATA que aparece en los promotores eucariotas.
Para demostrar que en el fragmento de ADN clonado hay motivos de unión a proteína
(factores transcripcionales), se emplean dos técnicas: la huella de la DNasa y los geles de
retardo.
Geles de retardo: se basan en que la migración de un fragmento de ADN sobre un
gel de electroforesis es más lenta (está “retardada”) si el fragmento está unido a una
proteína.
En una primera etapa, el fragmento de ADN en estudio (marcado) se incuba
con las proteínas (los factores transcripcionales) y después se realiza una
electroforesis. Como en realidad hay muy pocas proteínas reguladoras, sólo un
escaso porcentaje de ADN será retardado. En el caso de que exista interacción
ADN-proteínas, se observa una banda débil encima de la banda de ADN principal.
Es necesario hacer un ensayo control, para
asegurarse de que no se trata de uniones
inespecíficas, realizando una electroforesis
del fragmento de ADN en ausencia de las
proteínas.
Lo más habitual es utilizar un
extracto completo de proteínas nucleares,
entre las cuales se incluyen los factores
transcripcionales. O pueden emplearse
diferentes fracciones proteicas purificadas a
partir del extracto nuclear.
Banda
retardada
–
+
Control
Fragmento clonado
+ extracto nuclear

52 Alberto Fonte Polo
Huella de la DNasa: esta técnica permite localizar de forma precisa la región de
ADN donde se unen las proteínas. Se basa en la capacidad de la enzima DNasa de
hacer cortes, al azar, en la molécula de ADN. La interacción de una proteína con un
ADN protege a este último de los ataques de la DNasa.
Se realizan dos ensayos en paralelo, en uno se digiere el fragmento clonado
(marcado) con la DNasa, éste servirá de control; y por otro lado, el fragmento
clonado se pone en presencia del extracto nuclear, y seguidamente se realiza la
digestión con la DNasa. Las condiciones de digestión de la DNasa se han adecuado
para que sólo se produzca un corte por molécula. Las roturas se producen al azar y
esto hace que, estadísticamente, aparezcan todas las bases de la secuencia de ADN
en el gel. El ADN incubado con la DNasa en ausencia de proteínas dará lugar, sobre
el gel de electroforesis, a toda una serie de bandas que corresponderán a los
diferentes fragmentos de ADN.
Por el contrario, si en el ADN, después de una incubación con proteínas,
ciertas regiones quedan protegidas del ataque enzimático, aparecerá en el gel una
zona desprovista de bandas. La proteína deja su “huella” sobre el ADN. Es posible
conocer exactamente la localización sobre el ADN de la zona protegida, donde hay,
por tanto, interacción con los factores de transcripción.
Sin embargo, aunque se ha demostrado que el fragmento de ADN tiene regiones de unión a
proteínas, no se sabe si esa región corresponde a un promotor. Es necesario recurrir a la
caracterización funcional in vivo, para lo cual, hay que introducir el fragmento de ADN
clonado en una célula hospedadora.
Caracterización funcional in vivo
El fragmento clonado se expresa en células, que pueden ser bacterias o células eucariotas, o
en organismos transgénicos.

Fundamentos de Genética Aplicada 53
La expresión de genes eucariotas en células bacterianas, normalmente en Escherichia
coli, es útil en experimentación básica:
Para obtener ARN que luego puede ser usado como sonda.
Para obtener proteínas y caracterizarlas funcional y estructuralmente.
Para obtener anticuerpos contra un producto génico específico, y usarlos en ensayos
inmunológicos.
También se utiliza en ensayos biotecnológicos, ya que las bacterias pueden usarse como
“fábricas de proteínas”, es decir, es posible usar el poder reproductivo de las bacterias para
expresar grandes cantidades de proteínas de interés terapéutico o industrial.
Para que el fragmento de ADN se exprese en el interior de las células debe estar
incluido en un vector de expresión, que cuente con los motivos necesarios para que el
inserto se traduzca y se exprese como proteína.
La expresión del fragmento clonado en E. coli conlleva algunos problemas:
Expresar genes eucariotas en células procariotas da problemas de estabilidad, esto
hace que la productividad sea menor.
En ocasiones, las proteínas sintetizadas forman cuerpos de inclusión, y para
romperlos hay que recurrir a tratamientos muy agresivos.
Otras veces, la proteína producida no es funcional, ya que no han ocurrido las
modificaciones postraduccionales (glicosilaciones, metilaciones, formación de
puentes disulfuro…) necesarias para que la proteína adquiera su conformación
nativa, puesto que E. coli carece de las enzimas precisas.
En estos casos, cuando no es posible expresar convenientemente el fragmento clonado en
bacterias, hay que recurrir a la expresión en células eucariotas.
Introducción de genes en células eucariotas: métodos, vectores, sistemas de selección
La introducción de material genético externo en células eucariotas se conoce con el nombre
de transfección. Por el contrario, la introducción de ADN exógeno en células procariotas
se conoce como transformación. El término transformación también se usa para referirse al
paso de una célula normal a una célula cancerígena.
Para la transfección se usan, normalmente, células de levadura. Aunque también es
común el uso de células de mamífero, en este caso se recurre a líneas celulares inmortales
procedentes de células tumorales.
Existen dos tipos de transfección: transitoria y estable.
Para la mayoría de aplicaciones de la transfección, es suficiente que el gen
transfectado se exprese transitoriamente. Como el ADN introducido en la
transfección normalmente no se inserta en el genoma nuclear, el ADN exógeno se
expresa durante 4 o 5 días, pero no se transmite a la descendencia.
Cuando se desea que el gen transfectado permanezca en el genoma de la célula y de
su progenie, se debe efectuar una transfección estable.
Hay varios métodos para introducir ADN en una célula eucariota:
Para la transfección en células de levadura se emplea el método de
permeabilización de la membrana con sales de litio y con polietilenglicol, y
seguidamente, se da un choque térmico.

54 Alberto Fonte Polo
La coprecipitación con fosfato cálcico se usa en levaduras, células animales y
protoplastos. El ADN se pone en presencia de cloruro de calcio (CaCl2) y ácido
fosfórico (H3PO4). Los iones fosfato cargados negativamente se combinan con los
cationes calcio y, de este modo, se forma un precipitado de ADN y fosfato cálcico
[ADN–Ca3(PO4)2]. Esta suspensión se añade a las células que se quieren transfectar,
éstas toman parte del precipitado por endocitosis, y junto con él, el ADN. Con este
método, del 90% de las células transfectadas sólo el 4% integran el fragmento de
ADN en su genoma.
Otro método es la electroporación, consiste en un aumento de la permeabilidad de
la membrana plasmática celular causado por un campo eléctrico aplicado
externamente. Cuando el voltaje (2-4 kV) que atraviesa una membrana plasmática
excede su rigidez dieléctrica se forman poros. Si la fuerza del campo eléctrico
aplicado y/o la duración de la exposición al mismo es la adecuada, los poros
formados por el pulso eléctrico se sellan tras un corto periodo de tiempo, durante el
cual la molécula de ADN tiene la oportunidad de entrar a la célula. Sin embargo,
una exposición excesiva de células vivas a campos eléctricos puede causar apoptosis
y/o necrosis, procesos que provocan la muerte celular. Por este método, sólo
sobreviven el 60-90% de las células, aunque el rendimiento es de 20-30 células
transfectadas de cada 105 supervivientes.
Un método muy eficiente es la inclusión del ADN en liposomas, pequeños cuerpos
formados por una membrana en cierto modo similar a la membrana plasmática de la
célula y que puede fusionarse con la misma, liberando el ADN al interior celular.
Los liposomas se forman por la interacción entre las cargas negativas del ADN, y
las cargas positivas de los lípidos catiónicos que envuelven a éste. La tasa de
transfección de este método es similar a la de la electroporación.
Otra técnica de transfección es el bombardeo con microesferas de metal (bolas de
oro o tungsteno de una micra de diámetro) recubiertas de ADN. Las microesferas
son proyectadas, con una pistola génica, a enorme velocidad sobre las células que
deben modificarse. Estas bolas se retrasarán progresivamente cruzando las distintas
capas celulares. Algunas de las células alcanzadas integrarán espontáneamente los
genes en su genoma.
La microinyección resulta ser una técnica muy efectiva, aunque laboriosa. Consiste
en la introducción mecánica de las moléculas de ADN en el interior de la célula,
utilizando para ello una microaguja. La microinyección es normalmente realizada
bajo un microscopio óptico llamado micromanipulador. Es el método que se emplea
en la introducción del ADN recombinante en las células embrionarias en el proceso
de obtención de animales transgénicos.
Mediante infección viral también se consigue introducir el ADN en el núcleo de la
célula eucariota, usando un virus como vector. En este caso, la técnica es
denominada transducción, y se dice que las células se transducen.
Para seleccionar aquellas células eucariotas que han incluido el gen de interés se usan genes
marcadores. Se entiende por marcadores aquellas secuencias que se incluyen en el vector y
permiten detectar las células que han sido transfectadas. Existen dos tipos de genes
marcadores:
Genes de selección: confieren una ventaja proliferativa a las células transfectadas.
Pueden ser, a su vez, de dos tipos:
o Genes de resistencia a antibióticos: los más usados son los genes puro, neo
e higro, que confieren resistencia a puromicina, neomicina e higromicina,
respectivamente.

Fundamentos de Genética Aplicada 55
o Genes implicados en el metabolismo de las bases nitrogenadas: son genes
que participan en la síntesis de novo de las bases, o bien, genes que permiten
el reciclado de las mismas. Dentro de los genes que permiten el reciclado de
las bases nitrogenadas, caben destacar los siguientes:
tk+: gen de la timidina quinasa, participa en el reciclado de los
nucleótidos de timina, fosforilándolos. Las células eucariotas que van
a ser transfectadas deben ser tk–, en presencia de aminopterina las
células tk– mueren. La aminopterina inhibe la síntesis de novo de
bases nitrogenadas. Sólo aquellas células que han incorporado un
vector con el gen marcador tk+ son capaces de vivir en un medio con
aminopterina, de este modo, se pueden detectar las células
transfectadas.
hprt+: gen de la hipoxantina fosforribosil transferasa, interviene en el
reciclado de la guanina. Como en el caso anterior, para la selección
de las células transfectadas se usan células que carecen del gen hprt.
Si se las pone en un medio con aminopterina, solamente sobrevivirán
las células recombinantes porque tienen la hipoxantina fosforribosil
transferasa necesaria para el reciclado de las bases.
tkhs+: gen de la timidina quinasa de herpes simple, permite la
selección negativa de las células que lo poseen. Las células que son
transfectadas con el gen tkhs+ mueren en presencia de ganciclovir, un
análogo de la guanina. El ganciclovir es fosforilado por el gen tkhs+,
y se incorpora en el ADN durante la replicación, en lugar de la
guanina, causando la muerte celular.
Genes señal: estos genes marcadores confieren a la célula una característica
distintiva visible, como por ejemplo, un color diferencial.
o Genes señal usados en transfección estable:
GFP: gen de la proteína fluorescente verde, obtenido de la medusa
Aequorea victoria. Es una proteína muy poco tóxica y estable,
absorbe luz azul y emite luz de color verde cuando es iluminada con
luz de 395 nm de longitud de onda.
lacZ: gen que codifica para la enzima β–galactosidasa, que en
presencia de X-Gal, colorea a las células de azul.
o Genes señal usados en transfección transitoria:
Gen de la β–glucuronidasa: en presencia de X-Glu origina un
producto de color azul.
Gen de la luciferasa: la luciferasa es una enzima presente en las
luciérnagas, que cataliza la degradación de la luciferina a un
producto final liberando luz (fotones). Mediante un luminómetro se
puede detectar este fugaz destello de luz amarilla.
CAT: gen de la cloranfenicol acetil transferasa, esta enzima acetila el
cloranfenicol, y el cloranfenicol acetilado migra de forma diferente
en cromatografía.
Vectores de transfección: existen distintos mecanismos vectoriales de transfección celular:
Transfección de ADN lineal con el inserto y otra molécula de ADN con el gen
marcador, que se añade a la célula eucariota diana, y se integra en el genoma de
ésta, al azar, y en un número de copias variable.
Plásmidos mixtos: son construcciones a partir de plásmidos de bacterias, a los que
se les añade los motivos de expresión necesarios para la replicación en el interior de

56 Alberto Fonte Polo
células eucariotas; tales como un origen de replicación eucariota, un sitio de unión
al ribosoma, un promotor, etc. Estos plásmidos se mantiene en bacterias, y son
posteriormente transfectados a células eucariotas.
Vectores virales: se aprovecha el mecanismo de infección viral para transducir
células eucariotas. Se usan siempre virus defectivos, es decir, carentes de parte del
genoma vírico original, y por lo tanto, no son capaces de replicarse de forma
autónoma.
Mecanismo general de transfección vírica usando virus defectivos:
1. Construcción del vector viral. Se elimina el mayor número posible de genes y
secuencias del genoma viral, para que pueda captar el ADN exógeno.
2. Empaquetamiento del vector viral, mediante células empaquetadoras o mediante
empaquetamiento in vitro, para formar partículas virales infectivas. Las células
empaquetadoras son células eucariotas que poseen los genes que han sido
eliminados del genoma viral, y aportan, por tanto, la composición proteica que
necesitan los virus para su funcionalidad.
3. Infección de las células eucariotas diana.
Algunos vectores virales usados en transfección:
Vectores derivados de SV40: el SV40 es un virus pequeño de ADN circular de
doble cadena, hallado tanto en monos como en humanos, y está implicado en
algunos tipos de cáncer. Parte del genoma de este virus se usa para construir un
vector viral mixto.
Es necesario recurrir al uso de células empaquetadoras, estas células son portadoras
de los genes para los que es defectivo el vector, o en otros casos, estas células son
cotransfectadas con virus auxiliares. De cualquier modo, dentro de las células
empaquetadoras se van a producir partículas recombinantes, con el vector portador
del gen clonado que se pretende expresar en las células diana. Estos virus,
defectivos pero con capacidad infectiva, se usan para transfectar células de una línea
celular de mono africano, con el fin de producir grandes cantidades de la proteína de
interés.
neoR
PBR322
Ori SV40
Promotor temprano
Promotor tardío
MCS
Células empaquetadoras Células diana
Proteína de interés
Partículas virales
Vector viral
Gen foráneo

Fundamentos de Genética Aplicada 57
Vectores derivados de retrovirus: los retrovirus son virus de ARN lineal. Los
vectores retrovirales pueden incluir insertos de hasta 8 Kb, e integran el ADN de
interés en el genoma de la célula diana. Sin embargo, únicamente sirven para
infectar células que se encuentran en división. Además, la tasa de transfección es
pequeña y se obtienen pocas partículas virales. En el proceso de infección el
genoma del retrovirus es retrotranscrito, mediante la transcriptasa inversa, en el
citoplasma a ADNc, que es trasladado al núcleo, donde se integra en el genoma de
la célula hospedadora en forma de provirus. En cada extremo del genoma del
provirus hay dos regiones largas y repetidas LTR (Long Terminal Repeats), que
incluyen secuencias promotoras, de integración y de empaquetamiento. Entre los
dos LTR se sitúan los genes virales gag, pol y env que codifican para proteínas
estructurales del virus: proteínas de la matriz, enzima polimerasa (retrotranscriptasa)
y proteínas de la cubierta del virus, respectivamente.
El vector retroviral se obtiene después de sustituir los genes gag, pol y env, por el
ADN de interés; no obstante, este vector contendrá los LTR virales, además de un
gen de resistencia a antibiótico y un sitio de clonación, donde se inserta el ADN de
interés, con un promotor para que se exprese.
Es necesaria la transfección del vector a células empaquetadoras, éstas tiene
incorporados los genes retrovirales gag, pol y env en el genoma. Sólo cuando entren
en contacto los genes retrovirales de las células empaquetadoras y las regiones LTR,
necesarias para la encapsidación del virus, presentes en el vector, será cuando se
producirán los retrovirus recombinantes que contiene el gen de interés. En el medio
extracelular se acumularán las partículas virales defectivas (carecen de los genes
gag, pol y env), que serán capaces de transducir las células eucariotas diana y
conseguir así que estas células contengan y expresen el gen de interés. En las células
diana no ocurre la complementación, y por lo tanto, en el interior de estas células no
se forman nuevas partículas virales.
Vectores derivados de adenovirus: los adenovirus son virus de ADN duplexo
lineal, son más grandes y complejos que los retrovirus, causan infecciones
respiratorias benignas en humanos. Los vectores adenovirales son defectivos, y es
necesario recurrir a células empaquetadoras. Se pueden llegar a insertar en ellos
hasta 7,5 Kb de ADN exógeno. La gran ventaja de usar un adenovirus como vector
es la alta eficacia de transfección, así como una producción de grandes títulos.
Infecta tanto células en división como quiescentes. Entre los principales
inconvenientes encontramos que el genoma vírico no llega a integrarse en la célula,
de modo que los genes de interés se expresan sólo transitoriamente. Además se ha
visto que, en terapia génica in vivo, los adenovirus producen respuesta inmune
celular e inflamatoria en los organismos tratados con estos vectores.
Promotor
MCS neoR
LTR LTR Vector
retroviral
LTR gag pol env LTR

58 Alberto Fonte Polo
Vectores derivados de adenoasociados: son parvovirus, contienen ADN simplexo
como material genético, y requieren la coinfección con un adenovirus para
multiplicarse. Son vectores que combinan las ventajas de los retrovirales y los
adenovirales. Su capacidad de integrar ADN exógeno es pequeña, sólo de 5 Kb. Las
principales ventajas son que los virus adenoasociados integran su ADN en el
genoma de la célula diana. Además, pueden infectar tanto a células en división
como a las que no lo están (muy útil en experimentos in vivo). El riesgo de una
respuesta inmune está minimizado ya que no producen proteínas víricas. La
desventaja de estos vectores es que para producir partículas virales es necesaria la
cotransfección de las células empaquetadoras con un adenovirus o un herpesvirus.
Para separar las partículas virales deseadas de los virus auxiliares que se producen
en las células empaquetadoras, se hace una centrifugación en gradiente de densidad.
Vectores derivados de herpesvirus: los herpesvirus son una familia de virus de
animales poco conocida, cuyo genoma se compone de ADN bicatenario y lineal.
Infectan de forma específica a las neuronas, tanto a células quiescentes como en
división. No se integran en el genoma, por lo tanto, el gen transfectado se expresa
transitoriamente. La gran ventaja de estos vectores es el gran tamaño de su ADN,
que les permite aceptar grandes fragmentos genómicos, incluso genes con sus
propias regiones reguladoras.
En resumen, ¿para qué clonar en células eucariotas?:
1. Análisis de función génica y regulación: para demostrar que el promotor clonado
funciona in vivo en la célula. Se hacen ensayos de expresión transitoria, que
permiten conocer en qué célula se expresa el gen que es regulado por dicho
promotor, y en qué condiciones induce la expresión. Para lo cual, se clona la
secuencia reguladora (posible promotor) junto con un gen señal, por ejemplo el gen
GFP de la proteína fluorescente verde. Luego, se transfecta a células donde se
sospeche que puede expresarse el gen regulado por el supuesto promotor. Si las
células adquieren coloración verde, significa que se expresa el gen GFP junto con el
promotor, ya que en estas células se está expresando el gen regulado por el
promotor clonado. En cambio, si las células no cambian de color quiere decir que no
hay factores que permitan la transcripción del gen, debido a que ese tipo celular no
es el adecuado, o no se ha inducido correctamente.
2. Producción de proteínas de interés a nivel industrial: es un proceso lento y
costoso, y presenta importantes limitaciones. La integración del gen de interés, en el
genoma, es rara y aleatoria. Tampoco se puede controlar el número de copias del
inserto que se integran. La mayor parte del genoma no se expresa, y si el gen se
inserta en una de estas regiones no se expresará, pues no hay promotores. Y si se
integran muchas copias en una misma región del genoma puede haber efectos de
dosis, e inhibirse la expresión del gen.

Fundamentos de Genética Aplicada 59
Mutagénesis in vitro
Genética reversa y tipos de mutagénesis
La genética reversa, o genética inversa, es una disciplina genética que, partiendo del
conocimiento de un fragmento concreto de ADN clonado y/o secuenciado investiga sobre
su función biológica alterando dicho ADN mediante mutación. El gen mutagenizado in
vitro se introduce en el organismo y se observa el fenotipo que produce, para poder
dilucidar la función de dicho gen.
En cambio, la genética clásica busca situar y clonar un determinado gen de interés
partiendo de individuos cuyo fenotipo es llamativo (generalmente mutantes). Para lo cual,
se producen mutaciones in vivo que conducen a fenotipos mutantes estables y heredables.
Estos fenotipos son analizados para llegar a conocer, como en el caso anterior, la función de
los genes que intervienen en la expresión de dichos caracteres fenotípicos.
En genética se denomina mutagénesis a la producción de mutaciones sobre ADN, clonado
o no. De realizarse in vitro, dicha alteración puede realizarse al azar (mutagénesis
aleatoria), sobre cualquier secuencia, o bien de forma dirigida (mutagénesis dirigida)
sobre una secuencia conocida y en la posición de interés. En el caso de realizarse in vivo,
sobre organismos y no sobre ADN clonado, se realiza a gran escala y sin conocimiento de
secuencia, empleando para ello sustancias denominadas mutágenos.
Para mutagenizar un fragmento de ADN clonado hay que conocer su secuencia, o al
menos tener un mapa de restricción detallado.
Mutagénesis aleatoria por deleción, inserción y sustitución
La mutagénesis aleatoria es una técnica que se utiliza en los laboratorios de investigación
para introducir cambio al azar en una secuencia de ADN. Las mutaciones que se generan
pueden ser por deleción, inserción y sustitución; y se pueden tratar de bases simples o de
agrupaciones de muchos nucleótidos.
Mutagénesis por deleción:
Se emplean dos enzimas de restricción distintas para digerir el fragmento clonado que está
incluido en el vector. El vector con el inserto delecionado se separa del otro fragmento
mediante electroforesis en gel de agarosa. El vector (con parte del inserto) corresponde a la
banda que menos ha migrado, y el fragmento cortado del gen clonado corresponde a la
banda que más ha corrido en el gel. Se extrae el vector del gel y se pone en presencia de
ADN ligasa para que se circularice de nuevo, generándose un vector que porta un gen
clonado que ha perdido parte de su secuencia. Hay que tener en cuenta que, si las
restrictasas empleadas dejan extremos cohesivos no compatibles hay que usar la polimerasa
T4 en ausencia de dNTPs para que genere extremos romos, después se usará la ADN ligasa
para ligar el vector.
Pero, si en lugar de emplear dos enzimas de restricción sólo se usa una, ésta tendrá
que generar siempre extremos cohesivos. Luego, se usará otra enzima para dejar los
extremos romos, y después la ligasa recircularizará el vector. En este caso se produce la
deleción de un número de bases menor.
1100
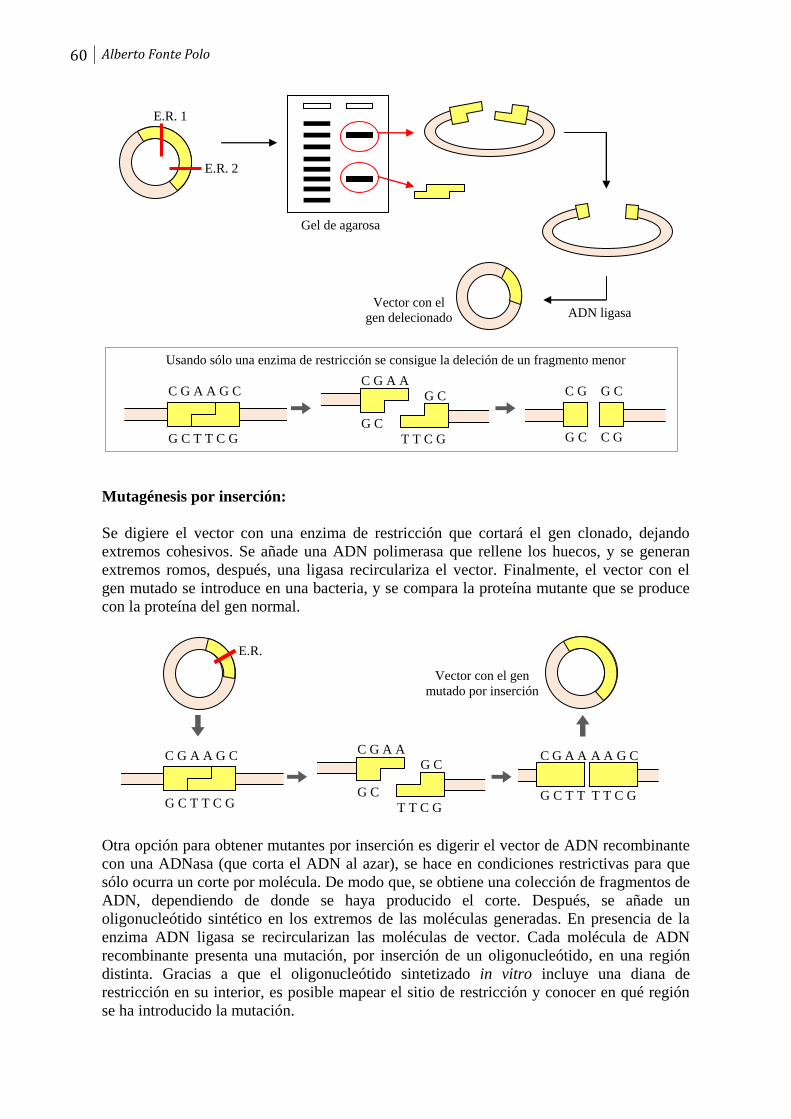
60 Alberto Fonte Polo
Mutagénesis por inserción:
Se digiere el vector con una enzima de restricción que cortará el gen clonado, dejando
extremos cohesivos. Se añade una ADN polimerasa que rellene los huecos, y se generan
extremos romos, después, una ligasa recirculariza el vector. Finalmente, el vector con el
gen mutado se introduce en una bacteria, y se compara la proteína mutante que se produce
con la proteína del gen normal.
Otra opción para obtener mutantes por inserción es digerir el vector de ADN recombinante
con una ADNasa (que corta el ADN al azar), se hace en condiciones restrictivas para que
sólo ocurra un corte por molécula. De modo que, se obtiene una colección de fragmentos de
ADN, dependiendo de donde se haya producido el corte. Después, se añade un
oligonucleótido sintético en los extremos de las moléculas generadas. En presencia de la
enzima ADN ligasa se recircularizan las moléculas de vector. Cada molécula de ADN
recombinante presenta una mutación, por inserción de un oligonucleótido, en una región
distinta. Gracias a que el oligonucleótido sintetizado in vitro incluye una diana de
restricción en su interior, es posible mapear el sitio de restricción y conocer en qué región
se ha introducido la mutación.
E.R.
Vector con el gen
mutado por inserción
C G A A A A G C
G C T T T T C G
C G A A G C
G C T T C G
C G A A
G C
G C
T T C G
C G G C
G C C G
C G A A G C
G C T T C G
C G A A
G C
G C
T T C G
Usando sólo una enzima de restricción se consigue la deleción de un fragmento menor
Gel de agarosa
E.R. 1
E.R. 2
Vector con el
gen delecionado ADN ligasa

Fundamentos de Genética Aplicada 61
Mutagénesis por sustitución en vectores de cadena simple:
Mediante agentes químicos y/o enzimáticos se consigue mutagenizar el ADN clonado,
mediante la sustitución puntual de una base por otra distinta.
Por ejemplo, el bisulfito sódico (NaHSO3) es un agente químico que desamina la
citosina y la transforma en uracilo. Para estos experimentos se utiliza un vector de cadena
simple, habitualmente la serie M13 de vectores de bacteriófagos recombinantes. El
bacteriófago M13 contiene ADN circular de cadena sencilla (forma replicativa) que se
replica a ADN de cadena doble dentro de las células infectadas, por el mecanismo del
círculo rodante. Si se trata el vector M13 monohebra con bisulfito sódico se consigue que
las citosinas se transformen en uracilos. Estos uracilos pueden aparear con un
oligonucleótido adecuado, el cual sirve de cebador para sintetizar la cadena de ADN
complementaria. De este modo, se generan moléculas de vector que incluyen una mutación.
Del mismo modo, pueden usarse análogos de bases como el 5-bromouracilo (5BU), que en
su forma ceto, es análogo a la timina y, por ello, se aparea con la adenina; mientras que en
su forma enol (es decir, en su forma tautomérica), es análogo de la citosina y se aparea con
la guanina.
Cuando el vector M13 de cadena sencilla se replica a ADN duplexo en presencia de
5-bromouracilo, éste se incorpora en el ADN en lugar de la timina. Si el 5-bromouracilo se
tautomeriza a la forma enol, después de dos rondas de replicación, se produce una
transición A–T → G–C.
Mutagénesis por sustitución en vectores de cadena doble:
El vector de doble cadena, se digiere con una enzima de restricción en presencia de
bromuro de etidio (BrEt). El bromuro de etidio es un agente intercalante usado
comúnmente como marcador de ácidos nucleicos para procesos como la electroforesis en
5BU 5BU*
A A A
5BU*
G
T
A
5BU*
G
C
G
T
A
T
A
C U
NH2
AAA
UUU UUU
M13
CCC
NaHSO3
AAA
1 2
4 3
1 2 3 4
2 3 4 1
3 4 1 2
4 1
3 2
1 2
4 3
2 3
1 4

62 Alberto Fonte Polo
gel de agarosa. Además, en presencia de bromuro de etidio la restrictasa corta sólo en una
de las dos cadenas, es decir, produce mellas en la molécula de ADN. Si después se trata la
preparación con una exonucleasa se consigue extender la mella, por ejemplo con la
exonucleasa III de E. coli que elimina nucleótidos uno por uno desde el extremo 3'–OH.
Seguidamente, se añade un agente mutágeno, como por ejemplo el bisulfito sódico, que
produce mutaciones por sustitución de la citosina por uracilo. Después, una ADN
polimerasa añade nucleótidos en sentido 5'→3', y rellena el hueco. Por último, una ligasa
sellará el vector mutagenizado.
Mutagénesis dirigida
Un enfoque muy empleado en la mutagénesis in vitro es la mutagénesis dirigida.
Utilizando este método, podemos generar mutaciones en cualquier posición de un gen de
secuencia conocida. A partir de la secuencia, se sintetizan químicamente segmentos cortos
de ADN denominados oligonucleótidos, correspondientes a cualquier sitio escogido del
gen, al que pueden unirse mediante la hibridación entre cadenas sencillas de ADN.
Una de las aplicaciones de la mutagénesis dirigida es el estudio de la relación estructura-
función de una proteína. Las mutaciones se introducen en un punto muy concreto del
correspondiente ADN clonado. Se induce la expresión de la proteína y después se
comparan las propiedades de la forma salvaje y de la forma mutante.
Mutagénesis dirigida usando un oligonucleótido mutado y un vector monohebra:
El gen de interés se inserta en un vector de ADN de cadena sencilla, como el fago M13. Se
diseña un oligonucleótido portador de la mutación deseada y se permite que hibride con la
región complementaria del ADN clonado. El oligonucleótido actúa como cebador para la
síntesis in vitro de la cadena complementaria insertada en el vector M13. La mutación que
deseamos generar se programa en la secuencia del cebador sintético. A pesar de que las
bases mutadas están mal emparejadas, la hibridación del oligonucleótido sintético con el
ADN complementario se produce si se realiza a baja temperatura y con concentraciones
elevadas de sal.
CCC UUU
AAA
UUU CCC CCC
3'
NaHSO3
E.R.
BrEt
ADN Pol
Exo III

Fundamentos de Genética Aplicada 63
Una vez que la ADN polimerasa completa la síntesis in vitro, se deja replicar el ADN de
M13 en E. coli, obteniéndose así un gran número de fagos portadores de la mutación
deseada. Para distinguir entre los fagos mutantes y los silvestres es necesario hacer una
selección restrictiva. Por ejemplo, se puede utilizar el oligonucleótido marcado como
sonda. A temperatura baja, la base mal emparejada no impide que el cebador hibride con
ambos tipos de fagos; sin embargo a temperatura elevada, el cebador sólo hibridará con el
fago mutante.
Mutagénesis dirigida por inserción de un cassette:
Otra forma de mutagénesis dirigida es la sustitución de un fragmento de doble cadena por
un oligonucleótido sintético (denominado cassette) que incluya el cambio. Este método
permite introducir más mutaciones que otros métodos como el de PCR.
Se usan dos enzimas de restricción distintas que van a hacer una digestión parcial,
cortando el gen clonado, pero no el vector en el que está incluido. Después de cortar el
vector con las dos enzimas de restricción se obtiene un plásmido abierto, que se separa del
fragmento del gen clonado mediante electroforesis en gel de agarosa. Se extrae el vector del
gel, y se inserta el fragmento de ADN sintetizado in vitro en el vector linearizado. El
oligonucleótido sintético (que lleva incorporada la mutación) tiene extremos
complementarios a los generados en el vector digerido con las dos enzimas de restricción,
de este modo se consigue el ligamiento entre ambas moléculas de ADN.
Mutagénesis dirigida por PCR:
Es posible sintetizar oligonucleótidos con mutaciones, que pueden ser usados como
cebadores en reacciones de amplificación por PCR, lo que permite el enriquecimiento en el
gen mutante por el propio proceso de amplificación. En una primera ronda de PCR, se
utiliza un cebador portador de la mutación deseada. El producto de esta reacción se emplea
como cebador en una segunda ronda de PCR, cuyo
producto es el alelo mutante.
Se puede también añadir secuencias útiles, que
contengan por ejemplo uno o más sitios de restricción,
a los extremos 5' de los fragmentos amplificados. Los
cambios no se pueden introducir en el lado 3' de los
oligonucleótidos elegidos para realizar la PCR, porque
al no hibridar correctamente no produciría el cebado de
la cadena.
Gel de agarosa
E.R. 1
E.R. 2
ADN ligasa
Fragmento de ADN sintético
que contiene el cambio deseado
Plásmido con el
gen mutado
Plásmido de ADN
recombinante
Gen clonado

64 Alberto Fonte Polo
Inactivación de genes endógenos
La inactivación de genes en el organismo nos da información sobre la función de dichos
genes. Se pueden inactivar genes por recombinación homóloga, o usando moléculas de
ARN o ADN antisentido.
Inactivación de genes por recombinación homóloga:
Cuando se transfecta un cultivo celular con una molécula de ADN determinada, ésta se
integra, al azar, en cualquier punto del genoma por recombinación heteróloga. Pero,
mediante recombinación homóloga se puede obtener una inserción precisa (gene
targeting) y no aleatoria. Es decir, es posible reemplazar una secuencia por una secuencia
homóloga que diferirá de la original porque procede de otra especie o porque presenta una
mutación. La recombinación homóloga es más frecuente en plásmidos y en levaduras
(ocurre en una de cada 106 células transfectadas) que en eucariotas superiores. Pero, puesto
que la recombinación homóloga ocurre muy excepcionalmente, es necesario recurrir a
sistemas para favorecer la selección de las células transfectadas. Se pueden distinguir dos
sistemas de enriquecimiento de células transfectadas por recombinación homóloga:
Sistemas de selección positiva: dentro del gen clonado, se introduce un gen de
resistencia al antibiótico neomicina (neoR) sin promotor. Lo más probable es que el
fragmento de ADN clonado se integre en el genoma en cualquier punto al azar, y
por lo tanto, al no integrarse en una zona con el promotor adecuado no se expresará
el gen neo. Pero, si el fragmento clonado se integra en la región homóloga del
genoma, con el promotor del gen original, el gen neo se podrá expresar. Por lo
tanto, en presencia del antibiótico sólo sobrevivirán las células transfectadas en las
que se ha producido recombinación homóloga.
Sistemas de doble selección positiva-negativa: en este caso, se incluye el gen de
resistencia a neomicina con promotor en el interior del gen clonado, y el gen de la
timidina quinasa viral del herpes simple (tkhs+), también con su promotor, en un
extremo del gen clonado. Gracias a estos dos genes, se puede proceder a una doble
selección:
Positiva: cultivando las células en presencia de neomicina. Las células que
no han incorporado el gen neo, es decir, que no han incorporado el gen
clonado, son destruidas.

Fundamentos de Genética Aplicada 65
Negativa: cultivando las células en presencia de ganciclovir. El ganciclovir
es un análogo de la guanina que puede ser incorporado en un ADN en
proceso de síntesis siempre que sea fosforilado previamente por la proteína
tk del virus del herpes, y después di y trifosforilado por enzimas celulares.
Una vez en forma de trifosfato, bloquea la replicación y produce la muerte
celular. El ganciclovir destruye todas las células en las que se expresa el gen
tkhs, es decir, todas las que han incorporado al azar toda la construcción. Las
células en las que se produce el intercambio del gen normal por el gen
clonado no incorporan el gen tkhs (ya que sólo recombina la región cuyos
extremos son exactamente homólogos) y son, por tanto, resistentes al
ganciclovir.
En resumen, se seleccionan las células que son doblemente resistentes a la
neomicina y al ganciclovir, es decir, las que contienen el gen neo y que no contienen
el gen tk. Son las células en las que se ha producido la recombinación homóloga: el
gen original ha sido reemplazado por el gen exógeno. Las células que no han
integrado el gen exógeno o que lo han integrado al azar en su genoma son
destruidas.
La recombinación homóloga puede emplearse para inactivar un gen. Es una estrategia de
estudio de la función precisa de un gen ya que se pueden conocer y evaluar las
consecuencias anatómicas o fisiológicas debidas a la ausencia de la proteína codificada por
este gen. Esta técnica es particularmente útil en mamíferos para estudiar la función de los
genes de los que no existen mutantes espontáneos. Se usa para obtener ratones knock-out,
son ratones modificados por ingeniería genética para que uno o más de sus genes estén
inactivados mediante una técnica llamada gene knockout. Inactivando el gen y estudiando
las diferencias que presenta el ratón afectado, los investigadores pueden inferir la probable
función de ese gen. El ratón es el organismo modelo más próximo a los seres humanos en el
que esta técnica se puede realizar con facilidad.
Existen algunas variaciones sobre el procedimiento para producir ratones knockout,
siendo el siguiente el ejemplo más habitual:
1. Se aísla el gen que se desea sustituir a partir de una genoteca de ratón.
Posteriormente se diseña una nueva secuencia de ADN muy parecida al gen
original, salvo que está cambiada lo suficiente para hacerla inoperante.
Normalmente también se incluye en la nueva secuencia genes marcadores (p.ej. gen
neoR y gen de la timidina quinasa del herpesvirus), que los ratones normales no

66 Alberto Fonte Polo
poseen y que les confieren resistencia a ciertos agentes tóxicos. Las oportunidades
de un suceso de recombinación con éxito son relativamente bajas, de modo que la
mayoría de las células alteradas tienen cambiado sólo uno de ambos cromosomas.
se dice que son heterocigotos.
2. A partir de un blastocisto de ratón (un embrión muy temprano que está formado por
una masa esférica de células indiferenciadas rodeada de células extraembrionarias)
se aíslan las células madre; después se cultivan in vitro. Como ejemplo, tomaremos
una célula madre de un ratón blanco.
3. Las células madre del paso 2 se combinan con la nueva secuencia del paso 1. Esto
se realiza mediante electroporación (empleando un pulso eléctrico para transferir
ADN a través de la membrana celular). Algunas de las células madre electroporadas
incorporarán la nueva secuencia en el lugar en el que el gen antiguo estaba en su
cromosoma, es decir, ocurre recombinación homóloga. La razón por la que tiene
lugar este proceso es que ambas secuencias, la vieja y la nueva, son muy similares.
Utilizando los genes marcadores del paso 1, se puede aislar rápidamente las células
que han incorporado la nueva secuencia, mediante el proceso de doble selección
positiva-negativa con neomicina y ganciclovir.
4. Las células madre del paso 3 se insertan en un blastocisto de ratón. En este caso
utilizamos blastocistos de ratón pardo. Estos blastocistos se implantan en el útero de
una madre adoptiva (denominada pseudopreñada, pues fue apareada previamente
con un macho estéril), para llevar a término la gestación. Los blastocistos contienen
dos tipos de células madre: las originales (ratón gris) y las modificadas (ratón
blanco). El ratón neonato será por tanto una quimera genética: parte del cuerpo
provendrá de las células madre originales y parte de las células modificadas. El
pelaje también mostrará zonas blancas sobre pardo.
5. Los ratones neonatos sólo serán útiles si la secuencia ha sido incorporada a la línea
germinal. Estos ratones se cruzan con otros del tipo blanco para obtener una
progenie completamente blanca. Estos ratones aún contienen una copia funcional
del gen y tienen que someterse a cruzamiento endógamo para producir un ratón que
no lleve ninguna copia funcional del gen original (es decir, que sea homocigótico
para ese alelo).

Fundamentos de Genética Aplicada 67
Inactivación de genes por ARN o ADN antisentido:
Moléculas que hibridan con ARN mensajeros específicos pueden inactivar genes
selectivamente. Los oligodesoxinucleótidos antisentido son los instrumentos más utilizados
para alterar la transcripción del ARNm. Éstas son secuencias nucleótidas cortas,
sintetizadas como complementos inversos exactos del ARNm diana, con el cual hibridan.
El dúplex resultante interferirá en la traducción ribosómica y tendrá como diana de
destrucción al ARNm.
Producción de ARN antisentido. Se inserta un gen, o una parte del mismo, en un vector mediante clonación
direccional en el lado 3' de un promotor y con orientación inversa a la normalmente presente en la célula de
origen. La transfección de este ADN recombinante en la célula paterna que lleva el gen normal, da lugar a la
transcripción de ARN a partir del ADN con polaridad inversa (ARN antisentido), junto con un tránscrito de
ARNm de la celular normal (ARN con sentido). Los dos ARN complementarios antiparalelos se pueden
hibridar dentro de la célula bloqueando la expresión (traducción) del tránscrito de ARNm normal.
5' 3'
3' 5'
Promotor T3
ARN polimerasa
del fago T3 5' 3'
ARNm funcional
3' 5'
ARNm antisentido
Transcripción del gen
de la cadena de ADN
complementaria
(cadena antisentido)
El ARN antisentido
hibrida con el ARN con
sentido bloqueando la
traducción del ARNm
Vector de expresión
recombinante
transfectado en la célula
Gen normal

68 Alberto Fonte Polo
La inhibición de la expresión genética puede obtenerse en el ámbito de la transcripción o en
el de la traducción. La inhibición de la expresión de los genes al nivel de la traducción suele
conllevar interferencia con el ARNm. El mensaje antisentido puede ser ADN o ARN, y
puede ser introducido en la célula diana o expresado in situ desde un vector vírico o un
plásmido transfectado. Mediante complementación inversa, el mensaje antisentido se
hibridará con el ARNm diana y el dúplex resultante convertirá el mensaje ARNm en
ilegible por el complejo ribosómico. Ciertas enfermedades podrían ser tratadas con estas
moléculas, por ejemplo, se ha conseguido en células tumorales revertir el fenotipo tumoral
a célula normal.
También, inyectando oligonucleótidos complementarios adyacentes al inicio de la
traducción (AUG) se consigue bloquear la expresión.

Fundamentos de Genética Aplicada 69
Aplicaciones en genética humana
Aplicaciones al diagnóstico clínico
Las técnicas de ingeniería genética se pueden aplicar en la medicina para determinar el
agente infeccioso, o para localizar el desorden genético, responsable de una enfermedad.
Existen varios tipos de metodologías básicas aplicadas al diagnóstico de enfermedades:
Basadas en técnicas de hibridación de ácidos nucleicos: en este caso, lo que se
requiere es una copia física del gen, que va a actuar como sonda. En el caso de una
infección, serán secuencias únicas del agente infeccioso y que no sean comunes;
mientras que en el caso de un desorden, es el gen causante del trastorno genético.
Basadas en la reacción en cadena de la polimerasa (PCR): no es preciso la
presencia de sondas, pero si es necesario conocer la secuencia nucleotídica de los
cebadores empleados.
Basadas en técnicas de secuenciación.
Detección de agentes infecciosos por hibridación:
Se emplea la técnica de Dot-Blot. Para este análisis se requieren sondas de regiones
genómicas de los microorganismos infecciosos, y ADN del cultivo de muestra del paciente
infectado.
El ADN del paciente se inmoviliza en forma de puntos (dot) en una membrana.
Luego, se añade la sonda marcada de cada patógeno. Se deja incubar y, tras el revelado, si
da positivo, es que la enfermedad del paciente es causada por el patógeno en cuestión.
También se puede inmovilizar el ADN del patógeno en la membrana e hibridarla
con el ADN muestra del paciente.
Diagnóstico de enfermedades genéticas por hibridación:
Si el gen clonado muestra polimorfismo entre los alelos que determinan fenotipo normal y
fenotipo enfermo, se puede poner de manifiesto la enfermedad por medio de los RFLPs
(polimorfismo para la longitud de fragmentos de restricción).
Por ejemplo, en el caso de la anemia falciforme se emplea el gen de la β–globina para el
análisis de esta enfermedad. En este gen existen dianas para las enzimas de restricción
MstII y DdeI. Mientras que, en el gen mutado (responsable de la patología) estas dianas se
han perdido.
Si se digiere el ADN de la muestra del paciente, con la enzima de restricción MstII,
se realiza un Southern Blot y se hibrida con la sonda del gen de la β–globina, se observan
dos bandas en los pacientes sanos, y una sola banda en los pacientes con anemia falciforme.
1111

70 Alberto Fonte Polo
Para diagnosticar la anemia falciforme también se puede utilizar un marcador ligado. En
este caso, se estudia el polimorfismo de longitud del fragmento de restricción HpaI
adyacente al gen de la β–globina.
Diagnóstico de enfermedades infecciosas por PCR:
Se requiere el diseño de oligonucleótidos que amplifiquen regiones específicas del
patógeno.
Se extrae ADN de sangre, vello coriónico o de una gota de saliva del paciente, y se
utiliza en una reacción de PCR con los oligonucleótidos específicos del patógeno (que
servirán de cebadores en la reacción de PCR).
La detección de una infección viral activa (por ejemplo, infección por HIV) requiere
análisis de RT-PCR.
Diagnóstico de enfermedades genéticas por PCR:
Por ejemplo, se estudiará el caso de la hemofilia A. Se trata de una enfermedad hereditaria,
caracterizada por una deficiencia en el factor VIII de coagulación, resultando sangrados
No
rmal
En
ferm
o Diana para DdeI Diana para MstII
----GTGCACCTGACTCCTGAGGAGAAG---- β-globina normal
Se pierden las dianas para las enzimas
----GTGCACCTGACTCCTGTGGAGAAG---- β-globina alterada

Fundamentos de Genética Aplicada 71
anormales de los pacientes. La enfermedad está causada por un rasgo recesivo ubicado en el
segmento diferencial del cromosoma X. Debido a que el gen defectuoso está localizado en
el cromosoma X, la enfermedad es mucho más común en los varones.
Localización y organización genómica del gen FVIII
El gen del factor VIII consta de 26 exones. En el intrón 22 hay dos genes (F8A y F8B), y el
primero tiene dos copias altamente homólogas en la región telomérica distal a 400 Kb del
gen FVIII.
Mecanismo de formación de la inversión del intrón 22 del gen FVIII
En la meiosis masculina en ausencia del cromosoma homólogo pueden ocurrir
recombinaciones desiguales entre el gen F8A intrónico y sus homólogos teloméricos, y se
produce una inversión, responsable del déficit de factor VIII. En las personas en las que ha
ocurrido la inversión se observa una banda más pequeña que en las personas sanas.
Estrategia de PCR de fragmentos largos para la detección de la inversión del intrón 22

72 Alberto Fonte Polo
Diagnóstico genético preimplantatorio (DGP):
El diagnóstico genético preimplantatorio, o preimplantacional, es el estudio del ADN de
embriones humanos para seleccionar los que cumplen determinadas características y/o
eliminar los que portan algún tipo de defecto congénito. Esta técnica nos permite examinar
genéticamente embriones obtenidos a través de fecundación in vitro, para establecer un
diagnóstico de salud genética en el embrión antes de ser transferido al útero materno.
Situaciones susceptibles de DGP:
En la selección de sexo para evitar enfermedades hereditarias ligadas a los
cromosomas sexuales (p. ej. hemofilia).
En casos de enfermedades monogénicas (p. ej. fibrosis quística, distrofia muscular
de Duchenne).
En casos de mujeres de edad avanzada con mayor riesgo de embarazos con
Síndrome de Down.
En casos de parejas con múltiples abortos como consecuencia de alteraciones
cromosómicas de sus embriones.
Cuando los padres presentan alteraciones cromosómicas en sus cariotipos.
La técnica consiste en extraer por aspiración una de las 8 células
del embrión de tres días de desarrollo, cuando estas son
pluripotenciales. Una vez realizada la biopsia, el embrión se
devuelve al incubador del laboratorio y se mantiene en el cultivo
in vitro con las condiciones adecuadas para que siga su normal
desarrollo hasta el momento de la transferencia al útero.
La localización de anormalidades cromosómicas en el
embrión se hace por FISH o por PCR (48 horas). La hibridación
fluorescente in situ (FISH) utiliza moléculas fluorescentes para
localizar genes o fragmentos de ADN.
Al quinto día los embriones diagnosticados como normales son transferidos al útero
de la paciente.
Terapia génica
La caracterización a nivel génico y proteico de un gran número de desórdenes hereditarios
ha abierto la posibilidad de que estos defectos puedan ser corregidos a nivel molecular.
La terapia génica consiste en la inserción de una copia funcional normal de un gen
defectivo, o ausente en el genoma de un individuo, en las células de los tejidos del
individuo con el objetivo de restaurar la función normal del tejido y así eliminar los
síntomas de la enfermedad.
Embrión con trisomía 21. El núcleo se
ha hibridado con sondas para los
cromosomas 13 (verde) y 21 (rojo), y
tiene claramente tres señales rojas.
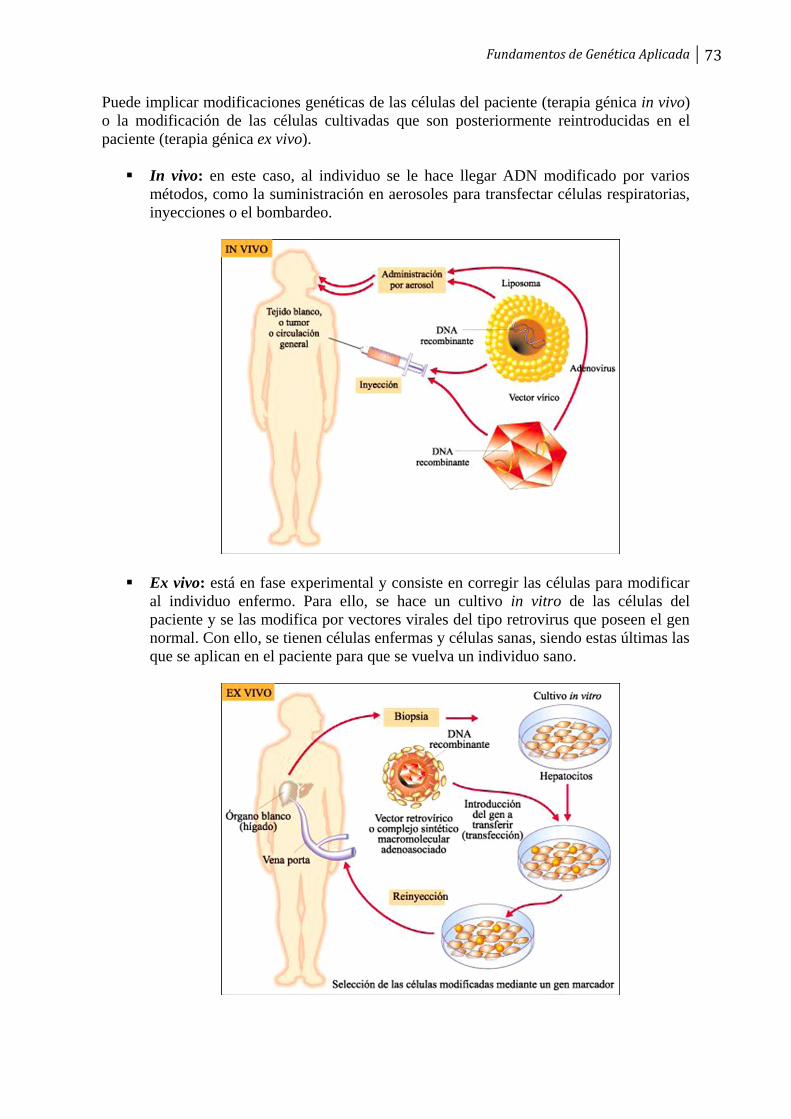
Fundamentos de Genética Aplicada 73
Puede implicar modificaciones genéticas de las células del paciente (terapia génica in vivo)
o la modificación de las células cultivadas que son posteriormente reintroducidas en el
paciente (terapia génica ex vivo).
In vivo: en este caso, al individuo se le hace llegar ADN modificado por varios
métodos, como la suministración en aerosoles para transfectar células respiratorias,
inyecciones o el bombardeo.
Ex vivo: está en fase experimental y consiste en corregir las células para modificar
al individuo enfermo. Para ello, se hace un cultivo in vitro de las células del
paciente y se las modifica por vectores virales del tipo retrovirus que poseen el gen
normal. Con ello, se tienen células enfermas y células sanas, siendo estas últimas las
que se aplican en el paciente para que se vuelva un individuo sano.
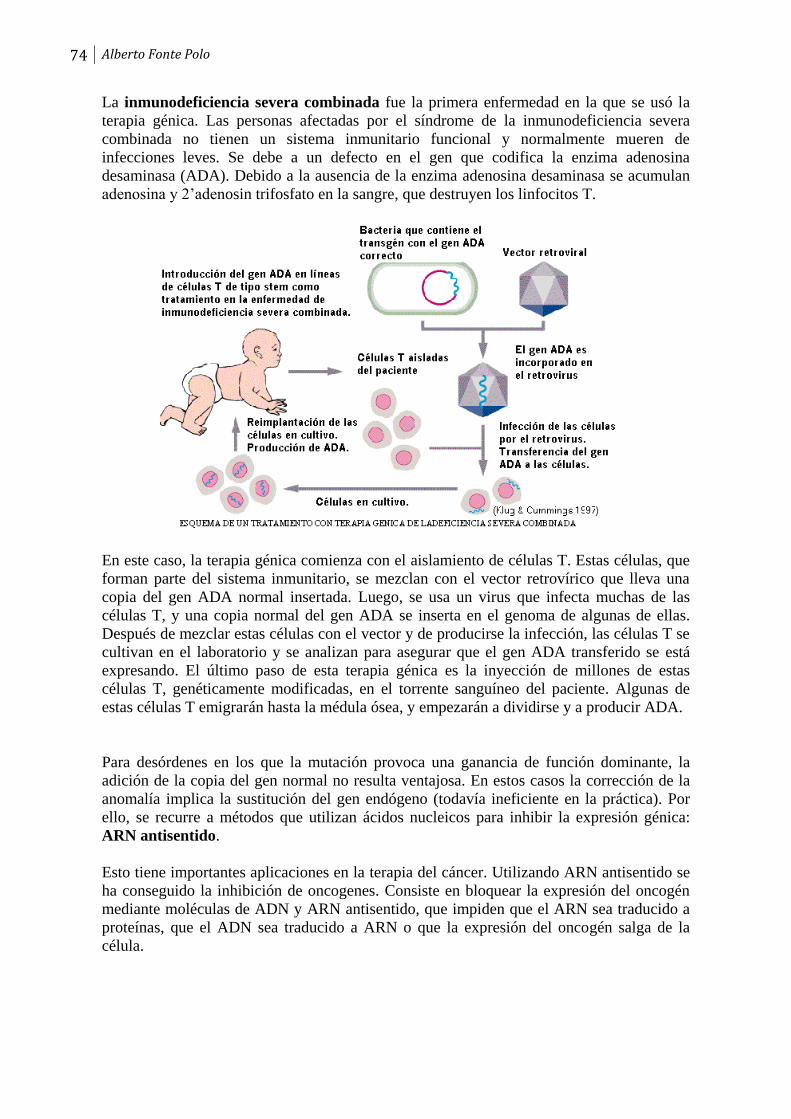
74 Alberto Fonte Polo
La inmunodeficiencia severa combinada fue la primera enfermedad en la que se usó la
terapia génica. Las personas afectadas por el síndrome de la inmunodeficiencia severa
combinada no tienen un sistema inmunitario funcional y normalmente mueren de
infecciones leves. Se debe a un defecto en el gen que codifica la enzima adenosina
desaminasa (ADA). Debido a la ausencia de la enzima adenosina desaminasa se acumulan
adenosina y 2’adenosin trifosfato en la sangre, que destruyen los linfocitos T.
En este caso, la terapia génica comienza con el aislamiento de células T. Estas células, que
forman parte del sistema inmunitario, se mezclan con el vector retrovírico que lleva una
copia del gen ADA normal insertada. Luego, se usa un virus que infecta muchas de las
células T, y una copia normal del gen ADA se inserta en el genoma de algunas de ellas.
Después de mezclar estas células con el vector y de producirse la infección, las células T se
cultivan en el laboratorio y se analizan para asegurar que el gen ADA transferido se está
expresando. El último paso de esta terapia génica es la inyección de millones de estas
células T, genéticamente modificadas, en el torrente sanguíneo del paciente. Algunas de
estas células T emigrarán hasta la médula ósea, y empezarán a dividirse y a producir ADA.
Para desórdenes en los que la mutación provoca una ganancia de función dominante, la
adición de la copia del gen normal no resulta ventajosa. En estos casos la corrección de la
anomalía implica la sustitución del gen endógeno (todavía ineficiente en la práctica). Por
ello, se recurre a métodos que utilizan ácidos nucleicos para inhibir la expresión génica:
ARN antisentido.
Esto tiene importantes aplicaciones en la terapia del cáncer. Utilizando ARN antisentido se
ha conseguido la inhibición de oncogenes. Consiste en bloquear la expresión del oncogén
mediante moléculas de ADN y ARN antisentido, que impiden que el ARN sea traducido a
proteínas, que el ADN sea traducido a ARN o que la expresión del oncogén salga de la
célula.
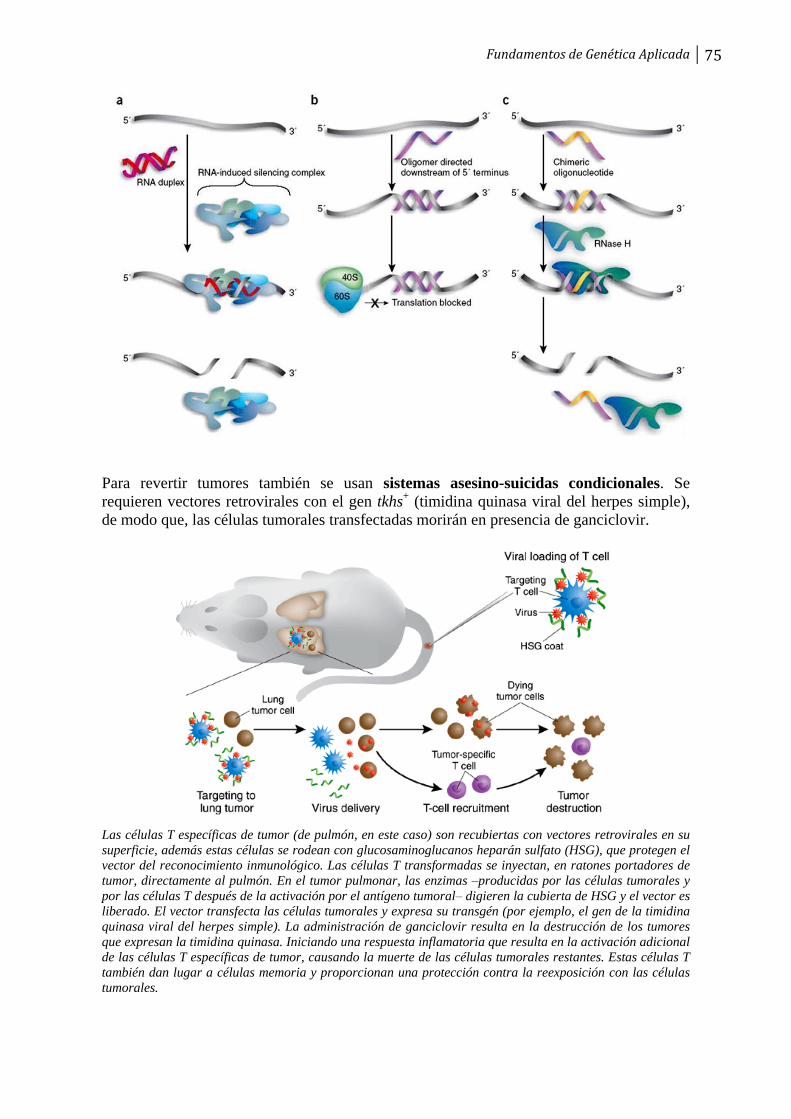
Fundamentos de Genética Aplicada 75
Para revertir tumores también se usan sistemas asesino-suicidas condicionales. Se
requieren vectores retrovirales con el gen tkhs+ (timidina quinasa viral del herpes simple),
de modo que, las células tumorales transfectadas morirán en presencia de ganciclovir.
Las células T específicas de tumor (de pulmón, en este caso) son recubiertas con vectores retrovirales en su
superficie, además estas células se rodean con glucosaminoglucanos heparán sulfato (HSG), que protegen el
vector del reconocimiento inmunológico. Las células T transformadas se inyectan, en ratones portadores de
tumor, directamente al pulmón. En el tumor pulmonar, las enzimas –producidas por las células tumorales y
por las células T después de la activación por el antígeno tumoral– digieren la cubierta de HSG y el vector es
liberado. El vector transfecta las células tumorales y expresa su transgén (por ejemplo, el gen de la timidina
quinasa viral del herpes simple). La administración de ganciclovir resulta en la destrucción de los tumores
que expresan la timidina quinasa. Iniciando una respuesta inflamatoria que resulta en la activación adicional
de las células T específicas de tumor, causando la muerte de las células tumorales restantes. Estas células T
también dan lugar a células memoria y proporcionan una protección contra la reexposición con las células
tumorales.

76 Alberto Fonte Polo
Medicina forense
Aunque los individuos de la misma especie comparten el mismo genoma básico, existen
diferencias entre genomas individuales basadas en unas secuencias muy polimórficas
(hipervariables) para las que existen muchos alelos distintos. Estas secuencias pueden ser
de dos tipos, minisatélite y microsatélite.
Las técnicas de DNA fingerprinting (huella genética) ponen de manifiesto estas
diferencias entre individuos.
Análisis de minisatélites:
Los minisatélites son secuencias repetidas en tándem (10-100 kb) que se encuentran en
distintas posiciones en el genoma (multilocus). El número de unidades repetidas por locus
es variable entre individuos.
El análisis consiste en hacer un Southern-blot y usar sondas específicas para detectar
los VNTR (repeticiones en tándem de número variable). En primer lugar el ADN que se va
a analizar debe cortarse en fragmentos de diferentes tamaños usando enzimas de restricción,
los fragmentos se separan por tamaño mediante electroforesis en gel. Y después se añade la
sonda específica marcada. Este proceso termina generando un patrón de bandas de ADN
característico de cada individuo, ya que los VNTRs son únicos para cada persona.
En otras ocasiones la sonda utilizada permite identificar una única región genómica
(unilocus) hipervariable.
Sonda multilocus Sonda unilocus

Fundamentos de Genética Aplicada 77
Análisis de microsatélites:
Los microsatélites son secuencias repetidas menores de 200 pb con unidades de repetición
muy cortas (1-4 pb) que se amplifican desde el ADN de los individuos por la técnica de
PCR.
Al contrario que en el caso anterior, los análisis de microsatélites son técnicas
unilocus, dado que se analiza, cada vez, un único locus. El principio de la variabilidad es el
mismo que el de minisatélites, pero las secuencias flanqueantes son iguales en toda la
población, por lo que se busca manifestar el polimorfismo por amplificación y determinar
su tamaño. Para este cometido, se diseñan oligonucleótidos con las secuencias flanqueantes
(que serán los cebadores de la PCR y estarán marcados con fluorocromos) y se realiza una
PCR. Tras esto, se introducen en un carril de un gel de electroforesis. Al final, se revela o
se tiñe el gel y se observan los resultados.
Las pruebas de paternidad genética se basan en comparar el ADN nuclear entre un niño y
su presunto padre, para aprobar o desaprobar la relación de parentesco. El ser humano al
tener reproducción sexual hereda un alelo de la madre y otro del padre, por lo tanto, un hijo
debe tener para cada locus un alelo que provenga del padre. Esta prueba se realiza
comparando entre 13 y 19 locus del genoma del hijo, del presunto padre y de la madre, en
regiones que son muy variables para cada individuo llamadas STR (short tandem repeat).
Proyectos genoma
Los proyectos genoma incluyen toda una serie de iniciativas internacionales apoyadas por
numerosos grupos de investigación que trabajan coordinadamente en la obtención de la
secuencia completa del genoma de una especie.
Diversos consorcios públicos han conseguido hasta la fecha la secuenciación de
numerosos genomas procariotas, mitocondriales y eucariotas.

78 Alberto Fonte Polo
Especie Tamaño del genoma (Mb) Número de genes
Saccharomyces cerevisiae 12 5.800
Drosophila melanogaster 180 13.700
Caenorhabditis elegans 97 19.000
Arabidopsis thaliana 125 25.500
Homo sapiens 3.000 27.000
Contenido en genes y tamaño del genoma de varios organismos
El Proyecto Genoma Humano (PGH) es un proyecto internacional de investigación
científica con el objetivo fundamental de determinar la secuencia de pares de bases que
componen el ADN e identificar y cartografiar los aproximadamente 25.000 genes del
genoma humano desde un punto de vista físico y funcional. La principal justificación del
PGH fue la promesa de avances importantes en medicina.
El Proyecto constituía el programa de investigación más ambicioso llevado a cabo
nunca en la biología: obtener el mapa de la secuencia genética completa del ser humano.
Tal empresa sería realizada por el llamado Consorcio Público Internacional, una entidad
formada por veinte laboratorios públicos de investigación de Estados Unidos, Reino Unido,
Francia, Alemania, Japón y China.
El Consorcio Público contaba inicialmente para desarrollar el Proyecto Genoma con
3.000 millones de dólares. El consorcio fue liderado inicialmente por el científico James
Watson (codescubridor de la doble hélice). El objetivo principal era determinar la secuencia
completa del ser humano leyendo los 3.000 millones de bases químicas de la cadena de
ADN. La metodología que se utilizaría, la secuenciación clon a clon, era una aproximación
laboriosa pero segura. La estrategia clon a clon de secuenciación consistía en fragmentar el
genoma en pedazos de unos 150.000 pares de bases. A partir de mapas genéticos
elaborados previamente se sitúan los fragmentos en las regiones del genoma. Los
fragmentos se vuelven a cortar, hasta 500 pb, y se superponen. Los fragmentos solapantes
que maximizan la longitud de superposición se secuencian. Por último se ensambla la
secuencia a través de las superposiciones sucesivas obtenidas en la etapa previa.
Ocho años más tarde del inicio del Proyecto, en 1998, apareció en escena una empresa
privada: Celera Genomics y a su cabeza Craig J. Venter. Venter lanzó el reto de obtener la
secuencia del genoma humano en un tiempo récord, antes que el Consorcio Público.
Utilizando para ello la estrategia de secuenciación del perdigonazo, o Shotgun, que está
basada en la fuerza bruta que supone disponer de una gran capacidad de secuenciación y de
cómputo informático. Se fragmenta al azar todo el genoma en pedazos que serán

Fundamentos de Genética Aplicada 79
secuenciados varias veces y posteriormente
ensamblados por potentísimos ordenadores
hasta obtener la secuencia borrador de todo el
genoma.
Al reto de Venter el Consorcio Público
reacciono doblando el presupuesto destinado a
los principales centros de secuenciación.
Paralelamente se iniciaron y terminaron
distintos proyectos de secuenciación de
organismos completos, los denominados
organismos modelos como el genoma de la
bacteria Escherichia coli, o determinados
eucariotas como la levadura Saccaromyces
cerevisiae, el gusano Caenorhabditis elegans, o
la mosca del vinagre Drosophila melanogaster
entre otros.
Debido a la amplia colaboración internacional, a los avances en el campo de la
genómica, así como los avances en la tecnología computacional, un borrador inicial del
genoma fue terminado en el año 2001, finalmente el genoma completo fue presentado en
abril del 2003, dos años antes de lo esperado.
Características principales del genoma humano:
Nuestro genoma contiene un número de genes bastante menor a lo esperado, entre
25.000 y 30.000 genes. Este es un número ligeramente superior al del gusano C.
elegans e incluso menor que el del genoma del arroz. Esta aparente paradoja indica
que son las interacciones entre los productos génicos, más que el número de genes
per se, lo que explicaría la complejidad morfológica y funcional.
El genoma humano tiene más de 3.000 millones de nucleótidos, pero sólo un 3%
codifica proteínas, el resto, la inmensa mayoría, parece no tener función.
El 50% de este ADN no codificante son secuencias repetitivas que derivan de
elementos transponibles.
Los genes no se distribuyen uniformemente por los cromosomas.
El número de intrones en los genes humanos varía desde 0 (en los genes de las
histonas) hasta 234 (el gen de la titina).
Cada persona comparte un 99,9% del mismo código genético con el resto de los
seres humanos.
La principal fuente de variabilidad en el genoma humano se corresponde a 3
millones de bases de la secuencia. Las variaciones entre dos personas en un sólo
nucleótido se conocen como SNPs (single nucleotide polimorphisms), en estas
variaciones se centra la mayor parte de los estudios.
Aplicaciones del conocimiento de la secuencia del genoma humano:
Técnicas de identificación forense.
Técnicas de diagnóstico de enfermedades: incluso mucho antes de que se
manifiesten. Ya hay unos 11.000 genes identificados relacionados con
enfermedades.
Diagnóstico Genético Preimplantacional.
Terapias génicas.
Ensamblaje de las secuencias por ordenador

80 Alberto Fonte Polo
Aplicaciones en biología animal y vegetal
Un organismo transgénico es aquel que ha sido modificado mediante la adición de genes
exógenos para conferirle nuevas propiedades. Se conoce como transgénesis al proceso de transferir genes en un organismo. La
transgénesis se usa actualmente para hacer plantas y animales transgénicos.
Obtención de plantas transgénicas
La mejora genéticas de las plantas ha sido una tarea lenta y difícil, pero la tecnología del
ADN recombinante promete cambios revolucionarios. Hoy en día es posible utilizar
técnicas genéticas in vitro para modificar un ADN vegetal y, a continuación, transformar
las células vegetales con ADN libre mediante: electroporación, por el método del
disparador de partículas, o utilizando vectores procedentes de Agrobacterium tumefaciens.
Es posible utilizar técnicas de cultivo de tejidos vegetales para seleccionar clones de
células vegetales que hayan sido genéticamente alteradas utilizando técnicas in vitro y, a
continuación, mediante tratamientos adecuados, inducir estos cultivos celulares a que
produzcan plantas complejas que puedan propagarse de forma vegetativa o por semillas.
1122

Fundamentos de Genética Aplicada 81
La bacteria fitopatógena gram-negativa de dicotiledóneas Agrobacterium tumefaciens
contienen un gran plásmido, plásmido Ti, que es responsable de su virulencia. El plásmido
contiene genes que movilizan el ADN para transferirlo a la planta. El segmento de ADN del
plásmido Ti que se transfiere a la planta recibe el nombre de T-DNA. Las secuencias de los
extremos del T-DNA son esenciales para la transferencia y el ADN que se va a transferir
debe estar entre estos extremos. Se sustituye la región del plásmido que produce el tumor, o
agalla de la corona, por el gen de interés.
Se ha construido un tipo de vector que se utiliza para transferir genes a plantas y se
denomina vector binario. Este vector contiene dos extremos del T-DNA a cada lado del
sitio que utiliza para la clonación, así como algún gen marcador de resistencia a antibióticos
que puede utilizarse en plantas. También contiene un origen de replicación que puede
replicarse tanto en Agrobacterium tumefaciens como en Escherichia coli, así como otro
marcador de resistencia a los antibióticos que se expresa en la bacteria. El ADN que debe
clonarse se inserta en el vector, que a continuación se transforma en Escherichia coli. Acto
seguido se transfiere a Agrobacterium tumefaciens.
Este vector de clonación no contiene los genes necesarios para transferir el T-DNA a una
planta, por lo que la bacteria Agrobacterium tumefaciens en la que se trasfiera debe
contener el otro miembro del sistema del vector binario. Este otro plásmido contiene la
región de virulencia (vir) de un plásmido Ti, pero está “desarmado”.
Aunque puede dirigir la transferencia de ADN en una planta, ya no tiene genes que
provoquen una enfermedad. Este plásmido desarmado, D-Ti, proporcionará todos los genes
necesarios para transferir el T-DNA desde el vector de clonación. El ADN clonado y el

82 Alberto Fonte Polo
marcador de resistencia a la kanamicina del vector pueden movilizarse mediante el
plásmido D-Ti y transferirse a una célula vegetal. Tras la recombinación con un
cromosoma del hospedador, el ADN foráneo puede expresarse y conferir así nuevas
propiedades a la planta. Muchos genes no se expresan de manera eficaz en las plantas, a
menos que se clonen en un vector de expresión que contenga un promotor vegetal.
El uso de Agrobacterium tumefaciens ha permitido la creación de varias plantas
transgénicas. Bien es verdad que se han obtenido más éxitos con plantas herbáceas
(dicotiledóneas) tales como el tomate, la patata, el tabaco, la soja, la alfalfa y el algodón,
pero Agrobacterium tumefaciens también se ha utilizado con dicotiledóneas leñosas como
pueden ser el nogal o el manzano. Los cultivos de plantas transgénicas de la familia de las
gramíneas (monocotiledóneas) han sido más difíciles de generar utilizando Agrobacterium
tumefaciens, pero parece que puede conseguirse buenos resultados con otros métodos de
introducción del ADN, como es inyección balística del ADN.
Las técnicas de biobalística emplean microesferas de oro o tungsteno, que se recubren del
ADN con el gen deseado, y se disparan a gran velocidad con una pistola, o cañón, de genes.
Las células en la línea directa del proyectil pueden morir, pero a su alrededor muchas
células captan el ADN sin daños.
Después, se induce la regeneración de la planta adulta a partir de los protoplastos o
de las células tratadas. Incluso se pueden transformar cloroplastos con este sistema. Sirve
para plantas que son más difíciles de cultivarse sus tejidos (cereales, leguminosas), aunque
tiene el inconveniente de que el ADN puede insertarse en copias, y puede ser inestable. El
bombardeo de microproyectiles presenta el mayor potencial para la transformación de
cereales. Por medio de esta técnica se han obtenido plantas transgénicas en
monocotiledóneas como maíz, arroz, trigo, avena y caña de azúcar.
Aplicaciones de la biotecnología vegetal:
Resistencia a herbicidas: se han desarrollado plantas transgénicas a partir de genes
de bacterias resistentes a herbicida. Así cuando se pulveriza un cultivo con
herbicidas sólo mueren las malas hierbas y no las plantas cultivadas.
Resistencia a plagas y a enfermedades: por ejemplo, se utiliza el gen de la proteína
tóxica de Bacillus thuringiensis. Esta bacteria produce una proteína cristalina,
llamada toxina Bt, que es tóxica para las larvas de las polillas y de las mariposas.
Mejoras de las propiedades nutritivas.
Resistencia a sequía y frío (estrés abiótico).
Otras aplicaciones: producción de variedades coloreadas, plantas transgénicas
productoras de vacunas, etc…
Planta
sensible
Planta
resistente A partir de las células transformadas
se obtienen plantas adultas
Las células que incorporan
el plásmido crecen en un
medio especial o selectivo
Partes de la planta se ponen en contacto
con las bacterias en medio de cultivo

Fundamentos de Genética Aplicada 83
Clonación animal
¿Qué entendemos por clonación animal? La obtención de uno o de varios individuos, bien
sea a partir de una célula (diferenciada o indiferenciada), o simplemente, a partir de un
núcleo. De modo que, los individuos clonados son idénticos o casi idénticos al original.
El principio de la clonación está en la obtención de organismos idénticos genéticamente, y
por tanto morfológica y fisiológicamente, como lo son dos gemelos univitelinos. Se pueden
utilizar dos métodos para conseguir clones de animales:
Disgregación de células embrionarias: se basa en el mismo principio por el que
nacen gemelos de forma natural. Se pueden separar las células de un embrión en
diferentes estados de desarrollo, desde el estado de 2 células hasta el estado de
mórula. Cada célula separada puede funcionar como un zigoto que puede
desarrollarse para dar un individuo completo.
Transferencia nuclear: se toman células embrionarias en fase de mórula o blástula,
obtenidas por disgregación, se cultivan in vitro, y después se transfieren a ovocitos a
los que se les ha quitado el núcleo.
Se provoca la fusión de las dos células animales de modo que el núcleo de la
célula embrionaria quede en el interior del ovocito, pudiendo éste empezar a
funcionar como un zigoto.
Cuanto más diferenciadas estén las células donantes de material genético, más difícil es
conseguir la reprogramación de dicho material genético para que pueda iniciar la
diferenciación de la célula receptora. Actualmente es posible obtener clones de células
totalmente diferenciadas de un animal adulto que actúan como donantes de su material
genético, como ocurrió en el caso de la famosa oveja Dolly.
La eficiencia de clonación a partir de un animal adulto es muy baja, lo que limita su
aplicación en la industria agropecuaria.

84 Alberto Fonte Polo
Aplicaciones de la clonación animal:
Mejoramiento genético.
Conservación de especies silvestres en peligro de extinción.
Producción de animales transgénicos:
o Productores de fármacos por sangre, leche u orina (p. ej. factor de
coagulación IX humano).
o Producción de órganos de cerdo para xenotransplantes.
o Producción de animales resistentes a enfermedades.
Transgénesis en animales
Generalmente, en animales, el ADN extraño, llamado transgén, se introduce en zigotos, y
los embriones que hayan integrado el ADN exógeno en su genoma, previamente a la
primera división, producirán un organismo transgénico; de modo que el transgén pasará a
las siguientes generaciones a través de la línea germinal (gametos).
La transgénesis puede efectuarse siguiendo dos estrategias distintas:
Transgénesis por microinyección de cigotos: en la primera fase, se aíslan un
número grande de óvulos fertilizados. Se consigue sometiendo a las hembras a un
tratamiento hormonal para provocar una superovulación. La fertilización puede
hacerse in vitro o in vivo. En la segunda fase, los zigotos obtenidos se manipulan
uno a uno y con una micropipeta a modo de aguja, y se introduce una solución que
contiene ADN. En la tercera fase, estos óvulos son reimplantados en hembras que
actuarán como nodrizas permitiendo la gestación hasta término. Por último, tras el
destete de los recién nacidos, éstos se chequean, para ver si ha ocurrido la
incorporación del transgén.
Transgénesis por manipulación de células embrionarias: una estrategia más
poderosa para la transgénesis implica la introducción de ADN extraño en células
embrionarias totipotentes (células ES) o células embrionarias madres (células EM).
Estas células se toman del interior de la blástula en desarrollo y se pasan a un medio
donde se tratan con distintos productos con lo que se conseguirá que las células no
se diferencien, y se mantiene su estado embrionario. El ADN extraño se introduce
en las células ES, posteriormente las células transfectadas son reintroducidas en una
blástula y ésta reimplantada en una hembra.
Con esta técnica los neonatos son quimeras, es decir, tienen células de origen
distinto, parte con el material genético original y parte transfectadas; pero mediante
el cruce de éstas con aquellas quimeras que hayan incorporado el transgén en su
línea germinal se consiguen animales transgénicos.
Top Related