







Idiomas
Páginas
Jurídico


CENTRO DE INVESTIGACIÓN EN QUÍMICA APLICADA
T E S I S
Síntesis de un ortoespirocarbonato aromático y su evaluación como agente
antiencogimiento para la fotopolimerización catiónica de monómeros
epóxicos
Presentada por:
JORGE LUIS ROBLES OLIVARES
PARA OBTENER EL GRADO DE:
MAESTRO EN TECNOLOGÍA DE POLÍMEROS
SALTILLO, COAHUILA SEPTIEMBRE 2011

i
RESUMEN
En el presente trabajo se reporta la preparación de un nuevo monómero del tipo
ortoespirocarbonato aromático (FSOC). Este monómero se obtiene al hacer reaccionar
fluoreno diol con tetraetilortocarbonato en relación estequiométrica 2:1. La reacción procedió
con un 93% de rendimiento. Una vez preparado y caracterizado se estudió el efecto de éstos
monómeros como agentes antiencogimiento en la fotopolimerización catiónica de los
monómeros comerciales BADGE y 3,4-EP.
Se corroboró la hipótesis de que el FSOC podría en un momento actuar como
fotosensibilizador absorbiendo energía de una determinada longitud de onda y después
transferir esta energía a un segundo compuesto DPPI, en base a los resultados obtenidos el
FSOC definitivamente presenta propiedades de fotosensibilización a concentraciones
relativamente grandes de DPPI.
Las reacciones de fotopolimerización procedieron rápidamente, alcanzando conversiones entre
50 y 60 % en los primeros 50 segundos y conversiones finales entre 70 y 80%. Dado que las
concentraciones de FSOC no fueron muy altas, la velocidad de fotopolimerización del
BADGE no se ve afectada significativamente por la presencia del FSOC.
El monómero FSOC resultó ser efectivo como agente antiencogimiento al incrementar su
concentración ya que el encogimiento para polímeros derivados de monómeros epóxicos se
encuentra reportado que es de -4 a -6 % y en las formulaciones preparadas con monómero
epóxico al aumentar la concentración de FSOC se observó no solo que se eliminó totalmente
el encogimiento sino que incluso se llegó a observar un ligero nivel de expansión de 0.19%.
El comportamiento antiencogimiento del FSOC se atribuye por un lado a la formación del
poliéter carbonato derivado de la doble apertura de anillo del FSOC, y por otro lado al
volumen estérico inducido por los grupos voluminosos fluorénicos localizados en el polímero
poliéter carbonato, lo cual impide la compactación adecuada de las cadenas poliméricas.

ii
ÍNDICE GENERAL
RESUMEN i
ÍNDICE GENERAL ii
ABREVIATURAS v
ÍNDICE DE FIGURAS vi
ÍNDICE DE TABLAS ix
CAPÍTULO 1. INTRODUCCIÓN 1
CAPÍTULO 2. ANTECEDENTES 3
2.1 ENCOGIMIENTO EN LOS POLÍMEROS 3
2.1.1 ENCOGIMIENTO POR TIPO DE POLIMERIZACIÓN 5
2.2 ESTRATEGIAS PARA REDUCIR EL ENCOGIMIENTO DURANTE LA POLIMERIZACIÓN 7
2.3 ORTOESPIROCARBONATOS (SOC) 10
2.4 POLIMERIZACIÓN CATIÓNICA POR APERTURA DE ANILLO 14
2.5 FOTOPOLIMERIZACIONES 18
2.5.1 FOTOPOLIMERIZACIONES CATIÓNICAS 20
2.5.2 FOTOINICIADORES CATIÓNICOS PARA FOTOPOLIMERIZACIONES UV 22
2.6 DETERMINACIÓN DE CINÉTICAS DE FOTOPOLIMERIZACIÓN EN TIEMPO REAL 27
2.6.1 ESPECTROSCOPÍA DE FT-IR EN TIEMPO REAL (RT-FTIR) 28
2.7 ANÁLISIS TÉRMICO DE POLÍMEROS 30
2.7.1 ANÁLISIS DINÁMICO MECÁNICO (DMA) 30
2.7.2 ANÁLISIS TERMOGRAVIMÉTRICO (TGA) 31
2.7.3 CALORIMETRÍA DIFERENCIAL DE BARRIDO (DSC) 32
2.8 JUSTIFICACIÓN DEL TRABAJO DE TESIS 33
CAPÍTULO 3. OBJETIVO E HIPÓTESIS: 34
3.1 OBJETIVO 34
3.1.1 OBJETIVOS PARTICULARES 34
3.2 HIPÓTESIS 34

iii
CAPÍTULO 4. PARTE EXPERIMENTAL CIQA 35
4.1 REACTIVOS 35
4.1.2 DISOLVENTES 36
4.1.3 PURIFICACIÓN DE LOS REACTIVOS Y LOS DISOLVENTES 36
4.2 METODOLOGÍA 36
4.3 SÍNTESIS DEL 9H-FLUORENO-9,9-DIMETANOL (FDIOH) 37
4.4 SÍNTESIS DE FSOC A PARTIR DEL FDIOH 38
4.5 CARACTERIZACIÓN DE LOS MONÓMEROS 39
4.5.1 CARACTERIZACIÓN POR ESPECTROSCOPÍA INFRARROJA (FT-IR) 39
4.5.2 CARACTERIZACIÓN POR RESONANCIA MAGNÉTICA NUCLEAR (RMN) 39
4.5.3 CROMATOGRAFÍA EN CAPA DELGADA (CCD) 39
4.6 DETERMINACIÓN DE LAS CINÉTICAS DE FOTOPOLIMERIZACIÓN MEDIANTE LA TÉCNICA DE
TIEMPO REAL (RT-FTIR) 40
4.7 DETERMINACIÓN DEL CAMBIO DE VOLUMEN MEDIANTE LA DETERMINACIÓN DE LA DENSIDAD
DE LAS FORMULACIONES DEL MONÓMERO ANTES Y DESPUÉS DE LA POLIMERIZACIÓN 41
4.8 ANÁLISIS DINÁMICO MECÁNICO (DMA) 44
4.9 CALORIMETRÍA DIFERENCIAL DE BARRIDO (DSC) 45
4.10 ANÁLISIS TERMOGRAVIMÉTRICO (TGA) 45
4.11 DETERMINACIÓN DEL PORCENTAJE DE GEL EN LA FORMULACIÓN DE FSOC Y BADGE 45
CAPÍTULO 5. PARTE EXPERIMENTAL POLITÉCNICO DE TORINO-ITALIA 46
5.1 METODOLOGÍA 46
5.2 DETERMINACIÓN DE LA CONCENTRACIÓN OPTIMA DE CANFORQUINONA Y RHODORSIL A USAR
PARA OBTENER EL MEJOR % DE CONVERSIÓN. 47
5.3 DETERMINACIÓN DE LAS CINÉTICAS DE POLIMERIZACIÓN DE LAS FORMULACIONES DEL
MONÓMERO MEDIANTE LA TÉCNICA DE TIEMPO REAL (RT-FTIR). 47
5.4 DETERMINACIÓN DEL CAMBIO DE VOLUMEN MEDIANTE LA MEDICIÓN DE LA DENSIDAD DE LAS
FORMULACIONES ANTES Y DESPUÉS DE LA POLIMERIZACIÓN. 49
5.5 DETERMINACIÓN DE LAS PROPIEDADES VISCOELÁSTICAS DE LAS PELÍCULAS OBTENIDAS
MEDIANTE LA TÉCNICA DE ANÁLISIS DINÁMICO MECÁNICO (DMA), ANÁLISIS TERMO
GRAVIMÉTRICO (TGA), CALORIMETRÍA DIFERENCIAL DE BARRIDO (DSC) Y %GEL. 51

iv
CAPÍTULO 6. RESULTADOS Y DISCUSIONES 52
6.1 SÍNTESIS DEL ORTOESPIROCARBONATO DEL FLUORENO (FSOC) 52
6.2 CARACTERIZACIÓN DEL ORTOESPIROCARBONATO DEL FLUORENO (FSOC) 54
6.3 ESPECTROMETRÍA UV 59
6.4 ESPECTROMETRÍA DE LUMINISCENCIA 62
6.5 FOTOPOLIMERIZACIÓN DEL FSOC CON LUZ UV Y DPPI COMO FOTOINICIADOR 65
6.6 DETERMINACIÓN DE LAS CINÉTICAS DE FOTOPOLIMERIZACIÓN CATIÓNICA DE LAS
FORMULACIONES DEL BADGE CON DIFERENTES CONCENTRACIONES DE FSOC. 69
6.7 ESTUDIOS POR DMA DE LAS PROPIEDADES VISCOELÁSTICAS DE LOS POLÍMEROS OBTENIDOS 72
6.8 DETERMINACIÓN DE LA TG POR DSC DE LAS FORMULACIONES BADGE-FSOC 75
6.9 DETERMINACIÓN DEL % DE ENCOGIMIENTO DE LAS FORMULACIONES FOTOCURABLES DE
BADGE CON FSOC 79
6.10 ESTUDIO DE ESTABILIDAD TÉRMICA POR TGA DE LOS POLÍMEROS PREPARADOS CON BADGE
Y FSOC 80
6.11 DETERMINACIÓN DEL % DE GEL DE LOS POLIÉTERES OBTENIDOS CON BADGE Y FSOC 81
6.12 ESTUDIO DE FOTOPOLIMERIZACIÓN DE MONÓMEROS EPÓXICOS FORMULADOS CON FSOC
UTILIZANDO LUZ VISIBLE 83
6.13 DETERMINACIÓN DE LAS CINÉTICAS DE FOTOPOLIMERIZACIÓN CATIÓNICA DE LAS
FORMULACIONES 3,4-EP CON DIFERENTES CONCENTRACIONES DE FSOC. 85
6.14 ESTUDIO DE ESTABILIDAD TÉRMICA POR TGA DE LOS POLÍMEROS PREPARADOS CON 3,4-EP
Y FSOC 88
6.15 DETERMINACIÓN DEL % DE ENCOGIMIENTO DE LAS FORMULACIONES FOTOCURABLES DE
3,4-EP CON FSOC 90
6.16 DETERMINACION DE LA TG POR DSC DE LAS FORMULACIONES 3,4-EP - FSOC 91
6.17 DETERMINACIÓN DEL % DE GEL DE LOS POLIÉTERES OBTENIDOS CON 3,4-EP Y FSOC 94
CAPÍTULO 7. CONCLUSIONES 96
CAPÍTULO 8. REFERENCIAS 98
!

v
ABREVIATURAS SOC Ortoespirocarbonato
FSOC Ortoespirocarbonato del Fluoreno
SOC DIOL 1,5,7,11-tetraoxaespiro [5.5]-3,9-undecanodiol
3,4-EP 3,4-Epoxiciclohexilmetil 3,4 Epoxiciclohexancarboxilato
BADGE Glicidil éter del Bisfenol A
DPPI (4-n-deciloxifenil) feniliodonio hexafluoroantimonato
FT-IR Espectroscopía infrarroja por transformada de Fourier
RT-FTIR Espectroscopía infrarroja por transformada de Fourier por tiempo real 1H RMN Resonancia Magnética Nuclear de Protón 13C RMN Resonancia Magnética Nuclear de Carbono
CCD Cromatografía de capa delgada
GE General Electric
Tg Temperatura de transición vítrea
! Cambio de Volumen
" Coeficiente de absortividad molar
DMA Análisis Dinámico Mecánico
DSC Calorimetría Diferencial de Barrido
TGA Análisis Termo Gravimétrico
FDiOH Dialcohol del Fluoreno
AM Mecanismo de monómero activado
ACE Mecanismo de cadena activa
UV Ultra violeta
P-FSOC Polímero del Ortoespirocarbonato del Fluoreno
LED Diodo Emisor de Luz
SOE Ortoespiroésteres
BOE Bicicloortoéster
PP Polipropileno
Fs Fotosensibilizador

vi
ÍNDICE DE FIGURAS
Figura 2.1 Cambios de enlace durante la fotopolimerización del 1,4,6- trioxaespiro [4,4] nonano
8
Figura 2.2 Diferentes monómeros expansivos y su apertura
9
Figura 2.3 Estructura base de los Ortoespirocarbonatos
10
Figura 2.4 Síntesis de los ortoespirocarbonatos realizada por Endo y colaboradores
10
Figura 2.5 Síntesis de los ortoespirocarbonatos
11
Figura 2.6 Ortoespirocarbonatos funcionalizados con grupos fotopolimerizables
11
Figura 2.7 SOC dihidroxílico
12
Figura 2.8 Estructura química de diferentes SOC´s reportados
12
Figura 2.9 SOC aromático preparado a partir de catecoles
13
Figura 2.10 SOC aromático reportado por Takata
13
Figura 2.11 Síntesis de SOC polimérico de tipo escalera
13
Figura 2.12 Síntesis de SOC´s asimétricos aromáticos
14
Figura 2.13 Esquema de polimerización de un monómero cíclico
14
Figura 2.14 Mecanismos de polimerización por apertura de anillo
16
Figura 2.15 Mecanismo de polimerización catiónica AM de resinas epoxi
16
Figura 2.16 Mecanismo de polimerización catiónica ACE de resinas epoxi
17
Figura 2.17 Fotoiniciadores catiónicos más utilizados en fotopolimerizaciones
22
Figura 2.18 Mecanismo de la fotólisis de las sales de diariliodonio
23
Figura 2.19 Iniciación indirecta utilizando fotosensibilizadores con espectros de absorción diferentes al fotoiniciador
24
Figura 2.20 Diferentes monómeros que pueden ser fotopolimerizados catiónicamente
25
Figura 2.21 Esquema del RT-FTIR utilizado
27
Figura 4.1 Reactivos de trabajo utilizados
34
Figura 5.1 Lámpara foto curable de luz azul Astralis 5 Ivoclar 45

vii
Figura 5.2 Moldes de preparación de probetas para determinar densidad
48
Figura 6.1 Ruta de síntesis propuesta inicialmente a partir de cetonas aromáticas
51
Figura 6.2 Ruta de síntesis propuesta a partir del Fluoreno
52
Figura 6.3 Espectro de 1H de FDiOH
53
Figura 6.4 Espectro de 13C de FDiOH
54
Figura 6.5 Espectro de RMN de 1H del compuesto FSOC
54
Figura 6.6 Espectro de RMN de 13C del FSOC
55
Figura 6.7 Comparación de espectro de FTIR del fluoreno, FDiOH y FSOC
56
Figura 6.8 Espectro de absorción UV del compuesto FSOC
57
Figura 6.9 Espectro de absorción UV del compuesto DPPI
58
Figura 6.10 Espectro comparativo de absorción entre el FSOC y DPPI
58
Figura 6.11 Espectro de emisión del FSOC
59
Figura 6.12 Espectro de emisión del DPPI
59
Figura 6.13 Espectros de emisión de mezclas de FSOC Y DPPI en solución de cloroformo
60
Figura 6.14 Comparación de los espectros de FTIR del FSOC y del polímero obtenido después de fotopolimerizar el FSOC
62
Figura 6.15 Espectro de RMN de 1H del polímero resultante (P-FSOC) de la fotopolimerización del FSOC
63
Figura 6.16 Especie formada al polimerizar el monómero FSOC
63
Figura 6.17 Mecanismo de polimerización del monómero FSOC
64
Figura 6.18 Curvas comparativas de conversión contra tiempo para las formulaciones fotocurables usando BADGE como monómero base a diferentes concentraciones molares de FSOC
65
Figura 6.19 Copolímero de FSOC y BADGE
67
Figura 6.20 Efecto de la concentración de fotoiniciador sobre la velocidad de fotopolimerización de la formulación fotocurable de BADGE con 10 % de FSOC
68

viii
Figura 6.21 Comparación del almacenamiento de módulo de los polímeros obtenidos con las formulaciones de BADGE a diferentes concentraciones de FSOC
69
Figura 6.22 Curva de tan delta para los polímeros obtenidos 70
Figura 6.23
Termogramas comparativos de DSC de las muestras poliméricas derivadas de las formulaciones de BADGE con diferentes concentraciones de FSOC
71
Figura 6.24 Termograma obtenidos por TGA para las muestras de polímeros derivados de las formulaciones de BADGE con diferentes concentraciones de FSOC
75
Figura 6.25 Espectro de Absorción de la Canforquinona
77
Figura 6.26 Mecanismo de fotosensibilización de sales de diariliodonio
78
Figura 6.27 Comparación de curvas de conversión contra tiempo para diferentes formulaciones de 3-4-EP con FSOC irradiando con una lámpara de dentista con LED de 415 nm y 600 mW/cm2
79
Figura 6.28 Espectro comparativo de la fotopolimerización de FSOC con luz UV (curva roja) y con luz visible fotosensibilizada con Canforquinona (curva azul)
81
Figura 6.29 Comparación de las curvas de pérdida en peso contra temperatura de los polímeros derivados de las formulaciones a diferentes concentraciones de FSOC
82
Figura 6.30 Termogramas obtenidos al correr los DSC de las muestras poliméricas derivadas de las formulaciones de 3,4-EP con diferentes concentraciones de FSOC
84

ix
ÍNDICE DE TABLAS
Tabla 6.1 Valores obtenidos de " para el FSOC
57
Tabla 6.2 Valores obtenidos de " para el DPPI
58
Tabla 6.3 Temperaturas de transición vítrea (Tg) obtenidas por DMA para los polímeros formulados con FSOC
70
Tabla 6.4 Valores de Tg para polímeros derivados de las formulaciones fotocurables con y sin FSOC
71
Tabla 6.5 Valores de % de encogimiento para las diferentes formulaciones BADGE-FSOC
73
Tabla 6.6 Valores de % de gel obtenidos con BADGE y FSOC
76
Tabla 6.7 Valores de % de encogimiento usando el monómero 3,4-EP
83
Tabla 6.8 Valor de Tg para los polímeros derivados de las formulaciones fotocurables de 3,4-EP con y sin FSOC
85
Tabla 6.9 Valores de % de gel obtenidos con 3,4-EP y FSOC
86

1. INTRODUCCIÓN
Hace cien años al mencionar el término plástico, se podía entender como algo relativo a las
artes plásticas en la reproducción de formas, llámese pintura, escultura o modelado. En la
actualidad esta palabra se utiliza con mayor frecuencia y tiene un significado que no solo
implica al arte, sino también a la tecnología y a la ciencia.
Hoy en día se puede afirmar que no hay un solo lugar, donde no exista un artículo de plástico,
a cualquier lugar que dirijamos la mirada encontraremos plásticos: gracias a la gran
versatilidad de propiedades que presentan estos materiales cuya estructura interna es la que
determina sus propiedades fundamentales las cuales son la causa de las diferencias con otros
materiales.
En base a la aplicación o finalidad que vaya a tener cada plástico, estos requieren de ciertas
propiedades para lograr tal fin. Estos requerimientos incluyen una combinación de
propiedades físicas, mecánicas y eléctricas tales, como fuerza de tensión, fuerza de flexión,
resistencia al impacto, resistencia a los solventes, constante dieléctrica, factor de poder, dureza
y temperatura de transición vítrea, solo por mencionar algunos. Estas propiedades se ven
afectadas por las condiciones de polimerización como pueden ser pureza de monómeros,
temperatura, tiempo, tipo y cantidad de iniciador, estequiometría de los comonómeros, peso
molecular del polímeros y grado de entrecruzamiento.
Otro factor que con frecuencia no se considera, es el cambio de volumen que se origina
durante la polimerización. Un cambio de volumen negativo o dicho en otras palabras,
encogimiento, acompaña en un determinado nivel todos los tipos de polimerización. En la
mayoría de los casos este encogimiento se tolera y no se toma ninguna previsión o acción para
eliminar o disminuir este efecto. En el pasado, la principal razón para tolerar este
encogimiento era que no existía un método para eliminarlo completamente. Además, aún no
se habían desarrollado monómeros que al polimerizar indujeran un cambio positivo de
volumen (expansión). Si fuera posible encontrar un método en el cual se logrará polimerizar

2
monómeros con el nivel de cambio de volumen requerido para cada aplicación, entonces se
mejorarían notablemente las propiedades del polímero y esto resultaría en la fabricación de
mejores productos. Por ejemplo, en ciertas aplicaciones, tales como materiales compuestos,
adhesivos, recubrimientos y materiales para aislamiento eléctrico, sería muy deseable tener
monómeros que al polimerizar exhibieran cero encogimiento, para producir materiales libres
de tensiones internas. Para otras aplicaciones como materiales poliméricos producidos en
moldes, rellenos dentales y en materiales poliméricos para fibra óptica, sería ventajoso, contar
con monómeros que indujeran una ligera expansión al polimerizar con el fin de llenar
completamente el molde o la cavidad dental. Por otro lado, en ciertas aplicaciones en la cual la
pieza de plástico se produce en el molde en una sola pieza, sería deseable contar con
monómeros que produjeran solo un ligero encogimiento durante la polimerización, con el fin
de remover más fácilmente el molde de la pieza.
A partir de los años setenta del siglo pasado el investigador William J Bailey 1,2 descubrió que
ciertos compuestos bicíclicos al polimerizar exhibían un cambio positivo de volumen y a partir
de este hecho se han realizado múltiples investigaciones tendientes a obtener materiales con
cero encogimiento o ligera expansión.
En este trabajo se sintetizó un nuevo tipo de ortoespirocarbonato aromático derivado del
Fluoreno, mediante una metodología sintética para obtener un dialcohol proveniente del
fluoreno. El dialcohol se transesterificó con tetraetilortocarbonato para obtener el respectivo
ortoespirocarbonato del Fluoreno (FSOC).
Este compuesto FSOC fue evaluado como agente antiencogimiento en la fotopolimerización
catiónica del monómero glicidil éter del bisfenol A (BADGE por sus siglas en inglés
Bisphenol A Diglycidyl Éter). Se determinó el cambio de volumen y se encontró que el
compuesto preparado no solo inhibe el encogimiento sino que produce un ligero nivel de
expansión.

3
2. ANTECEDENTES
2.1 Encogimiento en los polímeros
El encogimiento es un fenómeno inherente a todos los procesos de polimerización. Se define
como la reducción de volumen ocasionada por un incremento en la densidad. Esto es debido a
la formación de enlaces covalentes durante el proceso de polimerización.
Una de las principales causas de este encogimiento es que en el monómero líquido, las
moléculas se localizan a distancia de van der Waals unas de otras, mientras que en el
polímero, las unidades correspondientes al monómero se encuentran a una distancia de enlace
covalente.3 El paso de enlaces de Van der Waals a enlaces covalentes, que tienen una distancia
de enlace más corta, provoca que las unidades monoméricas se muevan más cerca las unas de
las otras, dentro del polímero, empaquetándose más juntas y provocando un incremento de la
densidad en el polímero y, por consiguiente, una contracción. Otros factores menos
importantes que afectan el cambio de volumen son el cambio de entropía al pasar del estado
líquido al sólido, el volumen libre en polímeros amorfos, y que tan bien empacados están tanto
el monómero como el polímero.4 Por ejemplo, en algunos casos el polímero se empaca u
ordena mejor que el monómero, como sería el caso de un monómero líquido que se transforma
a un polímero semicristalino, lo que produce un valor de encogimiento más alto que lo normal.
Por el contrario, cuando un monómero cristalino se convierte a un polímero amorfo, esto
traería como consecuencia una cantidad considerable de volumen libre, resultando en
encogimiento menor que el normal.
La contracción de volumen causa a menudo una acumulación de tensiones internas en la resina
así como cambios volumétricos y dimensionales. La eliminación o control de esta contracción
durante la polimerización es de gran importancia en el diseño de materiales que requieren de
dimensiones precisas o para la preparación de materiales poliméricos compuestos. El
encogimiento puede ser un factor adverso en ciertas aplicaciones como el desarrollo de
materiales compuestos, materiales para aislamiento eléctrico, fabricación de fibra óptica y la
preparación de materiales dentales poliméricos.

4
También puede originarse encogimiento del polímero durante el período de aplicación, si este
se lleva a cabo a temperaturas altas, como sería el caso de los barnices de cable de cobre de
un motor eléctrico que opera a 125 °C. Generalmente las reacciones de polimerización no se
llevan a cabo al 100% por lo que existen pequeñas cantidades de monómero sin reaccionar y
especies de bajo peso molecular en el producto final. Durante el período de aplicación de una
pieza como la que se mencionó anteriormente cuya temperatura de operación es de 125 °C, el
monómero y las especies de bajo peso molecular se encuentran alrededor de la matriz
polimérica y los primeros podrían reaccionar con algún punto terminal reactivo del polímero
para inducir un crecimiento posterior en las cadenas poliméricas y producir de esta manera
encogimiento adicional.3
Las consecuencias del encogimiento producido durante la polimerización pueden observarse
de varias formas. Por ejemplo, en materiales para aplicaciones eléctricas, el encogimiento
puede producir tensiones compresivas internas que son capaces de fracturar o comprimir
algunos componentes delicados o frágiles dentro del dispositivo. En algunos componentes
poliméricos que se usan como aislantes, las tensiones internas pueden producir
microcavidades o microfracturas las cuales reducen la capacidad aislante del material.
En aplicaciones que requieren de moldeo, el encogimiento resulta en llenado incompleto del
molde que resulta en fallas en la pieza y no repetibilidad. En recubrimientos poliméricos, el
encogimiento puede dar lugar a baja adhesión del recubrimiento sobre el sustrato o incluso
separación del recubrimiento. Si la adhesión del recubrimiento sobre el sustrato no es buena,
esto puede dar lugar a permeación de la humedad o del oxígeno, causando corrosión o
formación de otros compuestos sobre la superficie del sustrato.
Muchos materiales compuestos que involucran fibras de alta fuerza tensil en una matriz
polimérica, fallan debido ya sea a la pobre adhesión entre la matriz y la fibra, o también puede
deberse a defectos o microfracturas en la matriz. En ambos casos, el problema está relacionado
con el hecho de que cuando éstos materiales polimerizan se produce un nivel alto de
encogimiento. En materiales producidos en polimerización por masa, parte de estas tensiones
generadas pueden ser aliviadas por un encogimiento en las dimensiones del artículo. Sin

5
embargo en un material compuesto, el material reforzante que tiene un módulo alto, no
permitirá el encogimiento en la dimensión total del artículo y como resultado de esto, se
generan una enorme tensión en el material. Esta tensión puede liberarse súbitamente,
originando una falla en el adhesivo, en la cual la matriz polimérica se separa del material
reforzante, o por una falla cohesiva en la cual se forma un defecto o una microfractura.
El valor total del encogimiento durante la polimerización según Carsten,3 es determinado por
los siguientes factores:
• Grado de Polimerización (número de pasos de adición)
• Tamaño de los monómeros (moléculas grandes muestran más encogimiento
que monómeros pequeños).
• Estado del monómero y polímero resultante, ejemplo líquido isotrópico,
cristalino o sólido amorfo.
• Tipo de polimerización (condensación, poliadición, apertura de anillo, doble
apertura de anillo)
2.1.1 Encogimiento por tipo de polimerización
En general, se utilizan tres diferentes tipos de procesos de polimerización para convertir
monómeros a polímeros: polimerización por crecimiento de cadena, policondensación y
polimerización por apertura de anillo,5,6 En éstos tres tipos de polimerización el encogimiento
ocurre en diferentes niveles, dependiendo de los cambios de enlace involucrados, el tamaño
de monómero, estructura y composición elemental.1
De los diferentes tipos de polimerización, el que presenta una menor contracción es la
polimerización por apertura de anillo, y la más desfavorable es la policondensación, ya que el
grado de encogimiento depende del tamaño de la molécula liberada durante la
polimerización.1

6
En una reacción de polimerización por condensación, el encogimiento es más pronunciado
debido a que una molécula pequeña se elimina durante la formación de un nuevo enlace.
Generalmente se observan niveles altos de encogimiento en las polimerizaciones por
condensación. Entre más grande sea la molécula liberada en la polimerización, mas grande es
el nivel de encogimiento (22-66% de encogimiento).1
En las reacciones de polimerización por adición, aun y cuando no hay liberación de una
molécula, aun así hay un encogimiento apreciable en muchos casos (6-66%).7 Esto se debe a
que los átomos en el polímero están más cerca uno del otro, que lo que estaban en el
monómero.
En polimerizaciones por apertura de anillo, el encogimiento es menor respecto a los dos casos
discutidos anteriormente (2-20%).8,9 Esto se debe a que no se desprende ninguna molécula
durante la polimerización y a que por cada enlace que sufre un cambio yendo de la distancia
de Van der Waals a la distancia de enlace covalente, otro enlace va de la distancia de enlace
covalente a la distancia de Van der Waals.
En el caso particular de las resinas termofijas, esta contracción puede dar lugar a tensiones
internas del material, una reducción en la adherencia del sustrato y formación de microgrietas
y microagujeros, que pueden reducir su vida útil.9
En el caso de las fotopolimerizaciones catiónicas de monómeros epóxicos, el encogimiento
alcanzado es del orden del 2-6%10 lo cual es relativamente bajo comparado con el 10-12 %
para las fotopolimerizaciones de monómeros acrílicos,11 pero aun así, y con este bajo nivel de
encogimiento se generan grandes tensiones así como variaciones en las dimensiones finales de
los polímeros obtenidos. Esto resulta en fallas en las formas y dimensiones de los artículos
finales, así como una disminución en la fuerza mecánica o propiedades de adhesividad.

7
2.2 Estrategias para reducir el encogimiento durante la polimerización
Se han empleado tres estrategias para reducir el encogimiento en volumen:
a) uso de agentes reforzantes,12
b) uso de prepolímeros13
c) uso de agentes antiencogimiento.
Las tres presentan ventajas y desventajas, por ejemplo; una de las formas más requeridas para
reducir el encogimiento es usando agentes reforzantes como fibras y rellenos. En este caso,
aunque existe la tendencia del polímero a encoger, los reforzantes disminuyen esta tendencia
hasta cierto grado. Sin embargo, el esfuerzo inducido por el polímero al encogerse, produce
altas tensiones que pueden resultar en fisuras o fallas en el polímero. Sin embargo este sería el
método más económico ya que los agentes de refuerzo son baratos.
Al usar prepolímeros, el encogimiento es mucho menor, ya que éste ya ocurrió a cierto nivel
durante la preparación del prepolímero. Además, facilita el procesado de los artículos. La
ventaja es que los prepolímeros no son tan caros pero el nivel de reducción del encogimiento
no es óptimo.
Por último, se ha demostrado que el uso de agentes antiencogimiento es la mejor opción
tecnológica ya que no solo se reduce el encogimiento sino que en ciertos casos y dependiendo
del tipo de agente, se puede lograr hasta expansión en el volumen del polímero. Sin embargo,
la principal desventaja de estos compuestos es que son relativamente caros.
Otra de las maneras que se han encontrado para producir materiales libres de encogimiento y
con bajos niveles de tensión, ha sido la incorporación de monómeros bicíclicos o
multicíclicos altamente tensionados como los bicicloortoésteres14-16 y los
ortoespirocarbonatos,17-20 en formulaciones curables catiónicamente. Especialmente los
ortoespirocarbonatos son los monómeros más eficientes en reducir el encogimiento y en
algunos casos las polimerizaciones de estos ocurren con expansión de volumen. Además, los

8
ortoespirocarbonatos pueden copolimerizar con el monómero base introduciendo flexibilidad a
la cadena polimérica lo que disminuye la tensión interna en la resina.21-24
Las razones para el bajo nivel de encogimiento que imparten estos compuestos durante la
polimerización pueden explicarse al comparar el monómero original con el polímero final que
es un poliéster con enlace éter en la cadena. Si tomamos el caso de los ortoespiroésteres como
ejemplo, podemos observar que existen dos procesos que podrían conducir al encogimiento
del polímero: un enlace pasa de una distancia de Van der Waals a la distancia de un enlace
covalente y otro enlace pasa de la distancia de enlace covalente a distancia de doble enlace.
Esta contracción se ve balanceada por los dos enlaces que pasan de la distancia de enlace
covalente a la distancia de Van der Waals en el polímero final,25 lo anterior esta representado
en la siguiente figura:
Figura 2.1. Cambios de enlace durante la fotopolimerización del 1,4,6- trioxaespiro [4,4] nonano
Para que se de una polimerización con un cambio positivo en volumen o cero encogimiento
podemos hablar de ciertos requerimientos que deben cumplir los monómeros tales como:
• Los anillos han de tener como mínimo un átomo en común.
• Cada anillo ha de contener como mínimo un heteroátomo.
• Los anillos no se han de abrir simétricamente.
El resultado de polimerizar este tipo de monómeros es la formación de poliéter-éster como en
el caso de los ortoespiroésteres (SOE) y los bicicloortoéster (BOE), y de un poliéter-

9
carbonato, como en el caso de los ortoespirocarbonatos (SOC), como se puede observar en la
figura 2:
Figura 2.2. Diferentes monómeros expansivos y su apertura dando lugar a la expansión.
Los altos niveles de expansión que inducen estos compuestos bicíclicos se deben a la
liberación de estructuras muy compactas y tensionadas del monómero inicial al polimerizar
por doble apertura de anillo.
Los monómeros ortoespiroéster y ortoespirocarbonato se consideran difuncionales por lo tanto
al polimerizar por doble apertura de anillo dan lugar a cadenas lineales. Para obtener redes
tridimensionales es necesaria una funcionalidad mayor, que puede conseguirse usando
monómeros que contengan al menos dos de estas estructuras o mediante copolimerización con
un monómero con la funcionalidad adecuada, como es el caso de las resinas epoxi.
Actualmente se han reportado un gran número de artículos y patentes en los que se describe
nuevos tipos de monómeros bicíclicos tales como ortoespirocarbonatos (SOC) y
ortoespiroésteres (SOE) así como también procesos de evaluación de la actividad
antiencogimiento. Sin embargo muchos de estos compuestos son cristalinos bajo condiciones
!
"!!
Bailey y colaboradores [1], fueron los primeros en reportar en 1975, que compuestos
bicíclicos como los ortoespiroesteres y los espiroortocarbonatos producían un cambio
positivo de volumen al polimerizar. De entonces a la fecha se han reportado un gran
número de artículos y patentes en los que se describe nuevos tipos de ortoesteres y
ortoespirocarbonatos, así como también procesos de evaluación de la actividad
antiencogimiento [3].
Los monómeros expandibles han de cumplir los siguientes requerimientos:
· Los anillos han de tener como mínimo un átomo en común.
· Cada anillo ha de contener como mínimo un heteroátomo.
· Los anillos no se han de abrir simétricamente.
Entre los monómeros que cumplen estos requerimientos se encuentran los
espiroortoésteres (SOEs), espiroortocarbonatos (SOCs) y bicicloortoésteres (BOEs).
Siendo los espiroortocarbonatos uno de los monómeros más eficientes con los cuales
las polimerizaciones ocurren con expansión de volumen. Además, los
espiroortocarbonatos pueden copolimerizar con el monómero base introduciendo
flexibilidad a la cadena polimérica lo que disminuye la tensión interna en la resina.
La polimerización de monómeros con estas estructuras resulta en la formación de
polioeter-ester en el primer y tercer caso, y poliéter-carbonato en el segundo, como se
puede observar en la figura 2:
!
"!!
Figura 2. Esquema de los diferentes monómeros expansivos y de su apertura dando lugar a la
expansión.
Los altos niveles de expansión que inducen estos compuestos bicíclicos se deben a la
liberación de estructuras muy compactas y tensionadas del monómero inicial al
polimerizar por doble apertura de anillo.
Los monómeros ortoester y ortoespirocarbonato se consideran difuncionales por lo
tanto al polimerizar por doble apertura de anillo dan lugar a cadenas lineales. Para
obtener redes tridimensionales es necesaria una funcionalidad mayor, que puede
conseguirse usando monómeros que contengan al menos dos de estas estructuras o
mediante copolimerización con un monómero con la funcionalidad adecuada, como es
el caso de las resinas epoxi.
Desde los primeros reportes sobre polimerizaciones por doble apertura de anillo en
1972, se han desarrollado muchos monómeros bicíclicos tales como Espiroortoesteres
(SOE) y Espiroortocarbonatos (SOC). Sin embargo muchos de estos compuestos son
cristalinos bajo condiciones ambientales o polimerizan solo bajo doble apertura de
anillo parcial, lo que limita seriamente su aplicabilidad práctica. Por ello tales
monómeros son comúnmente polimerizados en fundido a elevadas temperaturas, donde
su densidad es más baja que en el estado cristalino, lo cual no es de uso práctico.
Por otra parte, los fragmentos de bajo peso molecular formados durante el fotocurado,
pueden actuar como plastificantes reduciendo de esta manera la contracción del
volumen. La transición de fase explica la aparente expansión en volumen. [2]
!
"!!
Bailey y colaboradores [1], fueron los primeros en reportar en 1975, que compuestos
bicíclicos como los ortoespiroesteres y los espiroortocarbonatos producían un cambio
positivo de volumen al polimerizar. De entonces a la fecha se han reportado un gran
número de artículos y patentes en los que se describe nuevos tipos de ortoesteres y
ortoespirocarbonatos, así como también procesos de evaluación de la actividad
antiencogimiento [3].
Los monómeros expandibles han de cumplir los siguientes requerimientos:
· Los anillos han de tener como mínimo un átomo en común.
· Cada anillo ha de contener como mínimo un heteroátomo.
· Los anillos no se han de abrir simétricamente.
Entre los monómeros que cumplen estos requerimientos se encuentran los
espiroortoésteres (SOEs), espiroortocarbonatos (SOCs) y bicicloortoésteres (BOEs).
Siendo los espiroortocarbonatos uno de los monómeros más eficientes con los cuales
las polimerizaciones ocurren con expansión de volumen. Además, los
espiroortocarbonatos pueden copolimerizar con el monómero base introduciendo
flexibilidad a la cadena polimérica lo que disminuye la tensión interna en la resina.
La polimerización de monómeros con estas estructuras resulta en la formación de
polioeter-ester en el primer y tercer caso, y poliéter-carbonato en el segundo, como se
puede observar en la figura 2:

10
ambientales o polimerizan solo bajo doble apertura de anillo parcial, lo que limita seriamente
su aplicabilidad práctica.26 Por ello tales monómeros son comúnmente polimerizados en
fundido a elevadas temperaturas, donde su densidad es más baja que en el estado cristalino, lo
cual no es de uso práctico.
2.3 Ortoespirocarbonatos (SOC)
Los Ortoespirocarbonatos (SOC’s por sus siglas en ingles: Spiro Ortho Carbonates) son
monómeros que muestran expansión de volumen al polimerizar por el mecanismo de apertura
de anillo. Los SOC’s generalmente polimerizan con ácidos de Lewis como BF3-OEt2. 27
Están compuestos por dos anillos con un átomo central de carbono conectado a cuatro átomos
de oxígeno. Estos también llamados acetales cíclicos dobles son estables bajo condiciones
básicas pero fácilmente experimentan polimerización por apertura de anillo catalizadas por
catalizadores catiónicos.
Figura 2.3. Estructura base de los Ortoespirocarbonatos
Estos compuestos se sintetizan por dos rutas diferentes. En la primera de ellas se hace
reaccionar por transesterificación un diol con otro ortocarbonato más sencillo,28-31 tal como se
muestra en la Figura 2.4.
Figura 2.4. Síntesis de los ortoespirocarbonatos realizada por Endo y colaboradores.
(CH2)n
O O
O O
(CH2)n

11
En la Figura 2.5 se muestra el segundo de los métodos que implica la reacción de un
compuesto cíclico de organoestaño con un diol. El compuesto cíclico se hace reaccionar con
un compuesto de tipo tiocarbonato. La reacción de este tiocarbonato con el mismo u otro
compuesto de estaño resulta en un ortoespirocarbonato simétrico o asimétrico.32-34
Figura 2.5. Síntesis de los ortoespirocarbonatos
El grupo de fotopolimerizaciones del CIQA ha venido impulsando fuertemente esta área, en
colaboración con el grupo de investigación del Dr. Marco Sangermano del Politécnico de
Torino. Se han desarrollado varios tipos de ortoespirocarbonato funcionalizados con grupos
epoxiciclohexano (SOC CEP)35 y oxetano (SOC OX).36 Al tener grupos fotopolimerizables en
su estructura se logra que copolimerizen con el monómero, obteniéndose de esta manera
mejores resultados que con ortoespirocarbonatos simples.
SOC CEP SOC OX
Figura 2.6. Ortoespirocarbonatos funcionalizados con grupos fotopolimerizables
Así mismo este mismo grupo ha reportado la síntesis de un compuesto de tipo SOC
dihidroxílico (SOC DIOL).37, 38 Se ha encontrado que este compuesto SOC reduce
notablemente el encogimiento de monómeros epóxicos.

12
SOC DIOL
Figura 2.7. SOC dihidroxílico
Por otro lado, Bailey y colaboradores reportaron la síntesis de diferentes SOC´s aromáticos.39
En el esquema 2.8 se muestran las estructuras químicas de los compuestos preparados. En
todos los casos los compuestos fueron preparados a partir del catecol y otros dioles y tetraoles
aromáticos.
Figura 2.8. Estructura química de diferentes SOC´s reportados.
Otra referencia encontrada en la búsqueda bibliográfica relacionada con SOC´s aromáticos
corresponde a una patente japonesa en la que utilizan ésteres del ácido 2,2-dihalo-1,3-
benzodioxocarboxilico con catecoles alcoxicarbonílicos40 para preparar compuestos con la
siguiente fórmula general:
!
! !
!
!
!
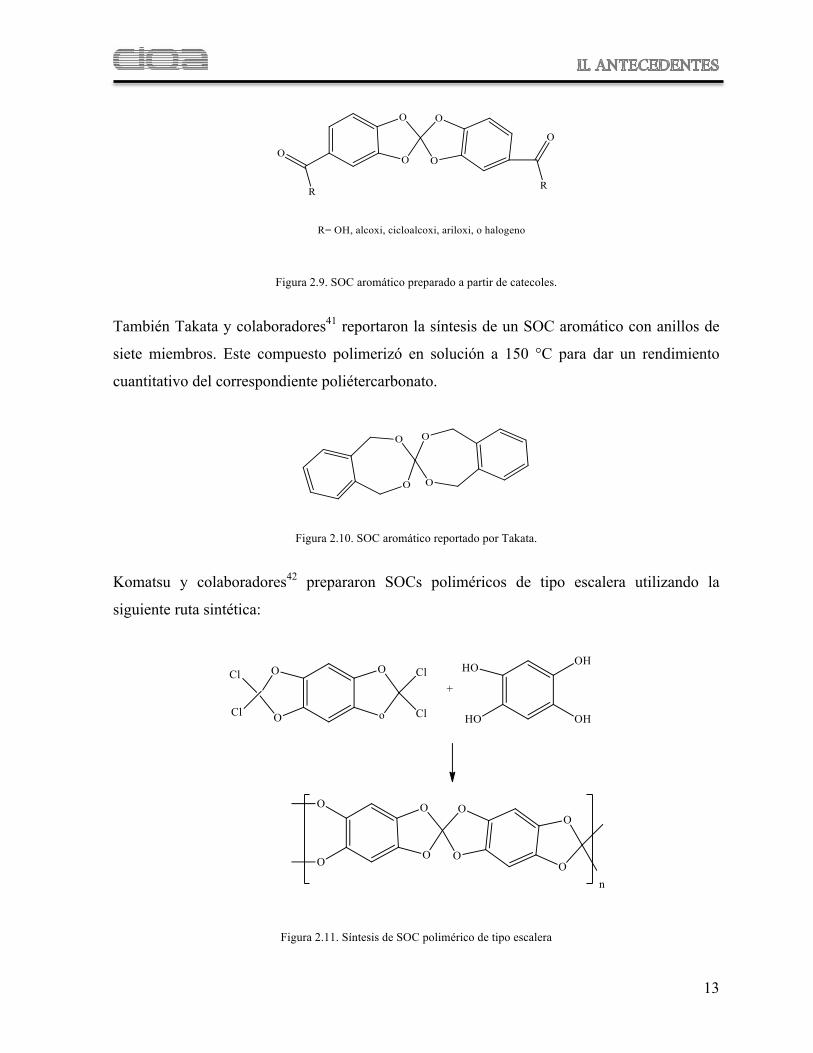
13
Figura 2.9. SOC aromático preparado a partir de catecoles.
También Takata y colaboradores41 reportaron la síntesis de un SOC aromático con anillos de
siete miembros. Este compuesto polimerizó en solución a 150 °C para dar un rendimiento
cuantitativo del correspondiente poliétercarbonato.
Figura 2.10. SOC aromático reportado por Takata.
Komatsu y colaboradores42 prepararon SOCs poliméricos de tipo escalera utilizando la
siguiente ruta sintética:
Figura 2.11. Síntesis de SOC polimérico de tipo escalera
!

14
Se encontró que una ventaja importante de todos los compuestos aromáticos de tipo SOC es su
mayor estabilidad contra la humedad y medios ácidos, ya que mientras un SOC alifático se
descomponía prácticamente en un día, los compuestos aromáticos presentaban una estabilidad
más prolongada hacia la humedad, soportando más de un mes sin almacenar en desecador.43
Así mismo, se reportó también la síntesis de SOCs asimétricos aromáticos44 utilizando la
siguiente ruta sintética:
Figura 2.12. Síntesis de SOC´s asimétricos aromáticos.
2.4 Polimerización Catiónica por Apertura de Anillo
Es un tipo de polimerización en el cual un monómero cíclico es convertido en un polímero que
no contiene anillos. Los anillos del monómero se abren y se extienden a lo largo de la cadena
polimérica, así:
Figura 2.13 Esquema de polimerización de un monómero cíclico.
Los monómeros que pueden polimerizar por este método son los monómeros cíclicos que se
pueden abrir debido a la acción de un reactivo o un catalizador ácido. Dentro de este grupo se
pueden encontrar las lactonas y los éteres cíclicos.

15
La polimerización por apertura de anillo es una forma de polimerización por crecimiento de
cadena en el cual el grupo terminal de la cadena actúa como un centro reactivo, al cual el
posterior ataque de una segunda molécula de monómero cíclico propicia la formación de una
cadena polimérica a través de propagación iónica.45 Algunos ejemplos de polímeros obtenidos
por este mecanismo son las poliamidas derivadas de las lactamas, como la caprolactama, los
poliésteres que provienen de las lactonas como la caprolactona y los poliéteres que se derivan
de éteres cíclicos como pueden ser los éteres glicídicos o los monómeros epóxicos.
En general, es posible que las reacciones de polimerización por apertura de anillo procedan
por medio de tres diferentes tipos de mecanismo (ver Figura 2.14). En el primer tipo el
monómero nucleofílico (por ejemplo un éter cíclico) reacciona con el iniciador electrofílico
para abrir el anillo y en el proceso se forma un nuevo electrófilo, el cual reacciona nuevamente
con una segunda molécula de monómero para formar a su vez una nueva especie electrófila, y
así sucesivamente hasta que se consuma todo el monómero. En ausencia de especies
nucleófilas que terminen la reacción, el grupo terminal queda activo para reiniciar la
polimerización al agregar más monómero o al agregar un segundo tipo de monómero para
obtener un copolímero.
El segundo tipo de mecanismo es similar, sin embargo en este caso queda un grupo terminal
que activa la cadena, el cual no se ha abierto aún después del ataque del nucleófilo cíclico
sobre el electrófilo. El ataque subsecuente de otra molécula de monómero al grupo terminal,
abre el ciclo y deja nuevamente un grupo activado sin abrir que será posteriormente atacado
por el monómero.
En el tercer tipo de mecanismo la iniciación se lleva a cabo sobre el monómero, dando lugar a
un monómero activado. El ataque de una molécula neutra resulta en el crecimiento de la
cadena. Pero con un grupo terminal neutro, debido a que hay una transferencia de protón
desde la especie cargada a una molécula neutra de monómero. Las especies cargadas
positivamente activan otras moléculas de monómero y de esta manera procede la
polimerización. A este mecanismo se le conoce comúnmente como mecanismo de monómero
activado.46

16
La selección del tipo de iniciador, sustrato, solvente, y condiciones de reacción pueden afectar
el tipo de mecanismo que opere en determinado caso.
Figura 2.14 Mecanismos de polimerización por apertura de anillo.
Kubisa y Penczek,46 han propuesto que la polimerización catiónica por apertura de anillo de
éteres cíclicos puede proceder por dos mecanismos, conocidos como AM (monómero activo)
y ACE (cadena activa). En la Figura 2.15 se presenta el mecanismo del caso particular de la
polimerización catiónica por monómero activo para el caso de los grupos epoxi.
Figura 2.15. Mecanismo de polimerización catiónica AM de resinas epoxi

17
Como se puede ver en la figura anterior el mecanismo AM consta de diferentes pasos. En la
iniciación (a), el monómero activado reacciona con un alcohol dando lugar a un catión oxonio
secundario con un hidrógeno activo y un grupo alcohol. En la propagación (b), este catión
reacciona con un monómero cediendo un protón y dando lugar a otro monómero activo. La
reacción puede continuar por reacción de este monómero activado con el alcohol anterior,
dando lugar al crecimiento de la cadena polimérica, o bien, con otro alcohol, presente en el
medio de reacción, para dar lugar de nuevo una iniciación.
El mecanismo ACE tiene lugar en ausencia de grupos hidroxilo. Como se puede ver en la
siguiente figura, se produce un ataque del grupo epoxi al monómero o a la cadena activa,
alargando en una unidad la cadena polimérica.
Figura 2.16. Mecanismo de polimerización catiónica ACE de resinas epoxi
Estos dos mecanismos no son excluyentes y compiten en un sistema dado. El balance entre
uno y otro vendrá dado, entre otros factores, por la relación en la cantidad de grupos epoxi y la
cantidad de grupos hidroxilo. Los grupos hidroxilo tienen un carácter nucleófilo más elevado
que los grupos epoxi, y por tanto, en presencia de grupos hidroxilo, el mecanismo
predominante será el AM.

18
2.5 Fotopolimerizaciones
Las fotopolimerizaciones son reacciones químicas de polimerización en las cuales los centros
activos, ya sean radicales libres o iones, se producen por una reacción fotoquímica. Una vez
que se generan estos centros activos reaccionan con los monómeros generando cadenas en
crecimiento hasta formar un polímero.
Ventajas de las reacciones de fotopolimerización sobre las polimerizaciones térmicas: 47
• Alta velocidad de reacción que generalmente se encuentra dentro del intervalo de
microsegundos a minutos.
• Utilizan solo una fracción de la energía que se utiliza en la polimerización térmica
• No utilizan solventes, por que el proceso es más barato y ecológico que en el proceso
térmico
• Los equipos utilizados ocupan menor espacio ya que son más compactos que los
hornos utilizados en la polimerización convencional.
Los sistemas fotocurables requieren de tres distintos componentes para que ocurra la
polimerización:
• Monómeros y /o prepolímeros
• Fotoiniciadores y posiblemente fotosensibilizadores y co-iniciadores
• Aditivos como pigmentos o estabilizadores de luz UV o fotoantioxidantes
El fotoiniciador es el componente más importante en una formulación fotopolimerizable. Un
fotoiniciador es cualquier compuesto que absorbe energía de un fotón y posteriormente genera
especies reactivas como radicales o iones iniciando de esta manera la polimerización. La
absorción directa de luz por el fotoiniciador, es un proceso fotofísico que provoca que el
compuesto pase de un estado basal a uno excitado o reactivo. El estado reactivo excitado tiene
más energía que el estado basal y provee la energía necesaria para que la reacción fotoquímica

19
ocurra. Dado que la energía en exceso que tiene el fotoiniciador es proporcionada por la luz
absorbida, la longitud de onda de absorción determina la máxima energía disponible. Por
ejemplo, la energía de la luz UV es mayor de 70 kcal / mol la cual en muchos casos es
suficiente para romper enlaces carbono-carbono (83 Kcal / mol). La luz visible provee menos
de 70 Kcal/mol de energía por lo que es insuficiente para romper los enlaces carbono-
carbono.48 Los fotoiniciadores generalmente contienen enlaces que son lábiles, es decir que se
rompen al absorber luz UV generando radicales o iones.
La principal ventaja del uso de fotoiniciadores radica en la posibilidad de definir exactamente
los puntos de inicio y terminación del proceso de polimerización mediante la duración del
periodo de irradiación. Además, la velocidad de fotólisis del fotoiniciador es casi
independiente de la temperatura de reacción, pero depende fuertemente de la intensidad de la
luz UV.
Para que un fotoiniciador desempeñe su función, este tiene que absorber energía y a través de
una interacción intermolecular o intramolecular producir un reactivo intermedio, el cual debe
de inducir la polimerización de un monómero o mezcla de monómeros. La especie reactiva
generada inicia la reacción con una molécula de monómero que a su vez reaccionará con otra
y así sucesivamente hasta producir una red entrecruzada de polímero.
En función del mecanismo de reacción de la iniciación de la fotopolimerización éstas pueden
ser divididas dos grupos:
• Fotopolimerizaciones catiónicas en las cuales se usan monómeros epóxicos, éteres
vinílicos y algunos heterociclos como derivados de tetrahidrofurano (THF) y oxetanos.
• Fotopolimerizaciones radicálicas que usan monómeros vinílicos o acrílicos.
Tanto las fotopolimerizaciones catiónicas como las de radicales libres presentan ventajas y
limitaciones. Por ejemplo, las polimerizaciones catiónicas se pueden considerar vivientes y no
se ven inhibidas por el oxígeno como las de tipo radical. Además los monómeros utilizados en
estas polimerizaciones no son tan tóxicos, volátiles o irritantes como los monómeros

20
polimerizables por radicales libres. Este tipo de fotopolimerizaciones se inicia mediante la
generación in situ o mediante la adición de un ácido de Lewis o un ácido fuerte de Bronsted a
monómeros polimerizables catiónicamente como lo son los epóxidos, vinil éteres y
compuestos heterocíclicos.
Por otro lado, las polimerizaciones por radicales libres no se ven inhibidas por agua o
humedad y son mucho más rápidas que las polimerizaciones catiónicas fotoiniciadas. Para
iniciar este tipo de fotopolimerizaciones se usan compuestos químicos generalmente del tipo
cetona aromática que absorben luz UV y generan radicales libres al fotolizarse. Estos radicales
libres primarios, al estar en contactos con monómeros con dobles enlaces generan una
reacción en cadena de polimerización.
Los monómeros acrílicos pueden ser fácilmente modificables en el grupo éster, obteniendo de
esta manera materiales con una amplia variedad de propiedades.
Se han desarrollado composiciones “híbridas” fotocurables que combinan tanto componentes
catiónicos así como de radicales libres, ya sea como mezclas de monómeros, por ejemplo, un
acrilato mezclado con un epóxido, o un monómero que contenga en su estructura ambas
funcionalidades como el acrilato y vinil éter.49 La mezcla de ambos sistemas generalmente
resulta en materiales con mejores propiedades que la de los componentes por separado. Por
ejemplo, se ha determinado que las formulaciones epóxicas no se ven afectadas por la
inhibición por oxígeno y se reduce el nivel de encogimiento que caracteriza a los sistemas
acrílicos. Además las excelentes propiedades físicas y mecánicas de los polímeros epóxicos,
refuerzan las obtenidas de los sistemas acrílicos.20
2.5.1 Fotopolimerizaciones catiónicas
Las fotopolimerizaciones catiónicas se inician mediante la generación in situ de un ácido de
Lewis o un ácido fuerte de Bronsted a monómeros polimerizables catiónicamente como los
epóxidos, vinil éteres y compuestos heterocíclicos.50

21
Las fotopolimerizaciones catiónicas se diferencian de las fotopolimerizaciones radicálicas de
la siguientes manera:
• La especie inicial, la cual es un ácido de Bronsted o de Lewis, comúnmente es un
compuesto estable, que genera especies cargadas positivamente. En ausencia de bases
o nucleófilos, tienen un tiempo de vida ilimitado, a diferencia de los fotoiniciadores
radicálicos los cuales pueden sufrir reacciones de terminación por colisión de radicales
libres.
• Es necesaria una activación térmica además de la activación fotoquímica del
fotoiniciador para permitir el curado de la película a excepción de los monómeros más
reactivos.51 La fotopolimerización UV vía radicálica, una vez iniciada, no requerirá
energía térmica para alcanzar el curado de la película.
• La fotopolimerización catiónica, una vez iniciada, continúa por un largo período de
tiempo en la ausencia de luz UV. Este comportamiento fue demostrado por Rodríguez
y Neumann52 al realizar la fotopolimerización del THF utilizando una sal de diaril
sulfonio como fotoiniciador.
• Dado que no se involucran radicales libres en la reacción de polimerización, la
fotopolimerización catiónica no se inhibe por el oxígeno.
• La basicidad del substrato puede inhibir el curado.
• Se puede presentar ácido residual en las películas curadas.
La polimerización deberá empezar inmediatamente después de la formación del ácido o solo
después de que la formulación ha sido calentada dependiendo del tipo de monómero utilizado.
En la práctica, la formulación líquida contiene los monómeros multifuncionales, un
fotoiniciador y otros tipos de componentes, como pigmentos o aditivos, recubre el substrato el
cual puede ser vidrio, plástico, papel, piel, madera o metal, entonces, el recubrimiento se
irradia con luz UV a una longitud de onda apropiada.

22
2.5.2 Fotoiniciadores catiónicos para fotopolimerizaciones UV
Los fotoiniciadores que se utilizan en polimerizaciones catiónicas fotoinducidas son
moléculas que al ser irradiadas por una fuente de luz UV generan, directa o indirectamente, un
ácido de Lewis o de Bronsted el cual debe ser capaz de iniciar la polimerización a través de un
mecanismo catiónico sin importar que la reacción sea térmica.
Una característica clave de la polimerización catiónica es el uso de contraiones de muy baja
nucleofilidad lo que permite que el catión propagante no sufra reacciones de terminación con
la especie aniónica. Por lo tanto, la mayoría de los fotoiniciadores catiónicos son sales
orgánicas que tienen contraiones como hexafluoroantimonato, hexafluoroarsenato,
tetrafluoroborato, los cuales son aniones complejos no-nucleofílicos.53
El descubrimiento de fotoiniciadores que generan eficientemente ácidos de Lewis o Bronsted a
partir de la irradiación UV o luz visible ha hecho posible el desarrollo de varias nuevas
tecnologías disponibles comercialmente.54, 55
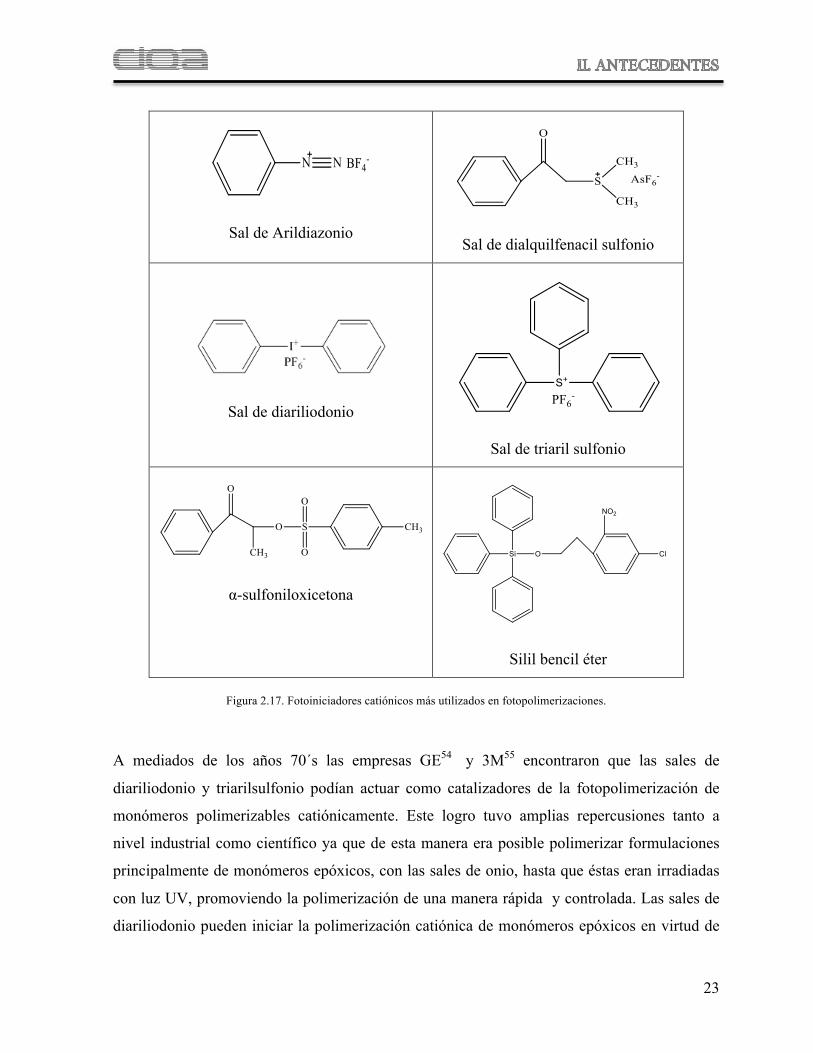
En la Figura 2.17 se muestra la mayoría de estos fotoiniciadores catiónicos, los cuales
considera Crivello56 cubren el 99% de los fotoiniciadores catiónicos utilizados actualmente.
23
Sal de Arildiazonio
Sal de dialquilfenacil sulfonio
Sal de diariliodonio
Sal de triaril sulfonio
#-sulfoniloxicetona
Silil bencil éter
Figura 2.17. Fotoiniciadores catiónicos más utilizados en fotopolimerizaciones.
A mediados de los años 70´s las empresas GE54 y 3M55 encontraron que las sales de
diariliodonio y triarilsulfonio podían actuar como catalizadores de la fotopolimerización de
monómeros polimerizables catiónicamente. Este logro tuvo amplias repercusiones tanto a
nivel industrial como científico ya que de esta manera era posible polimerizar formulaciones
principalmente de monómeros epóxicos, con las sales de onio, hasta que éstas eran irradiadas
con luz UV, promoviendo la polimerización de una manera rápida y controlada. Las sales de
diariliodonio pueden iniciar la polimerización catiónica de monómeros epóxicos en virtud de

24
su habilidad de generar súper ácidos de tipo Bronsted al ser fotolizadas. Al realizarse estudios
de la fotólisis de las sales de diariliodonio se postuló el siguiente mecanismo:57
Figura 2.18. Mecanismo de la fotólisis de las sales de diariliodonio
Al absorber la luz de longitudes de onda entre 190 y 400 nm la sal de diariliodonio pasa a un
estado triplete a partir del cual pueden suceder dos tipos de eventos: desactivación del estado
excitado por transferencia de energía a un segundo compuesto y/o reacción química. En este
último evento, la sal de diariliodonio puede sufrir la ruptura tanto homolítica como heterolítica
del enlace C-I, generando un radical-catión en el primer caso y un catión arenio en el segundo.
Las especies catiónicas reaccionan inmediatamente con el solvente o el monómero para
generar un ácido de Bronsted. Una vez que se genera el ácido, este es capaz de protonar una
molécula de monómero en una reacción “obscura”. El subsiguiente ataque de nuevas
moléculas de monómero resulta en la formación de polímero.
La absorción de energía por el fotoiniciador, en el intervalo del espectro electromagnético
puede ser insuficiente para iniciar la polimerización completa. Esta región del espectro
electromagnético está definida por la fuente de luz disponible y por la absorción de los
distintos componentes en la formulación. Las lámparas de mercurio, comúnmente usadas
como fuentes de luz, proveen emisiones substanciales de 313 nm a 366 nm. Así, el uso de
Ar2I+MtXn- h! Ar2I
+MtXn-! Ar "#MtXn- # Ar"
Ar+ MtXn- # ArI
ArI#MtXn-Ar+Ar#MtXn-#"Ar HMtXn
HMtXn # O O+ H
O+ H # On O+ O Hn
solventemonomero
o

25
estas lámparas requiere que el fotoiniciador absorba adecuadamente a estas longitudes de
onda. Componentes, como pigmentos, pueden absorber a longitudes de onda similares a la del
fotoiniciador, promoviendo una competencia por la luz incidente, resultando en una
disminución de la eficiencia de iniciación. Este problema puede ser evitado mediante el uso de
fotosensibilizadores con espectros de absorción diferentes al del fotoiniciador.
Figura 2.19. Iniciación indirecta utilizando fotosensibilizadores (Sen) con espectros de absorción diferentes al fotoiniciador.
El fotosensibilizador transferirá la energía absorbida ya sea por resonancia o por la formación
de un compuesto complejo excitado llamado exciplex el cual transferirá un electrón del
fotosensibilizador excitado al fotoiniciador, posteriormente se llevará a cabo la reducción de la
sal de diariliodonio, como se muestra en la Figura 2.19. La descomposición rápida e
irreversible del radical libre de diariliodonio impide la retroreacción de transferencia del
electrón haciendo que este proceso sea más o menos eficiente.
En términos del tipo de monómeros que pueden ser polimerizados, la fotopolimerización
catiónica es la más versátil de las polimerizaciones por adición. La cantidad de monómeros
26
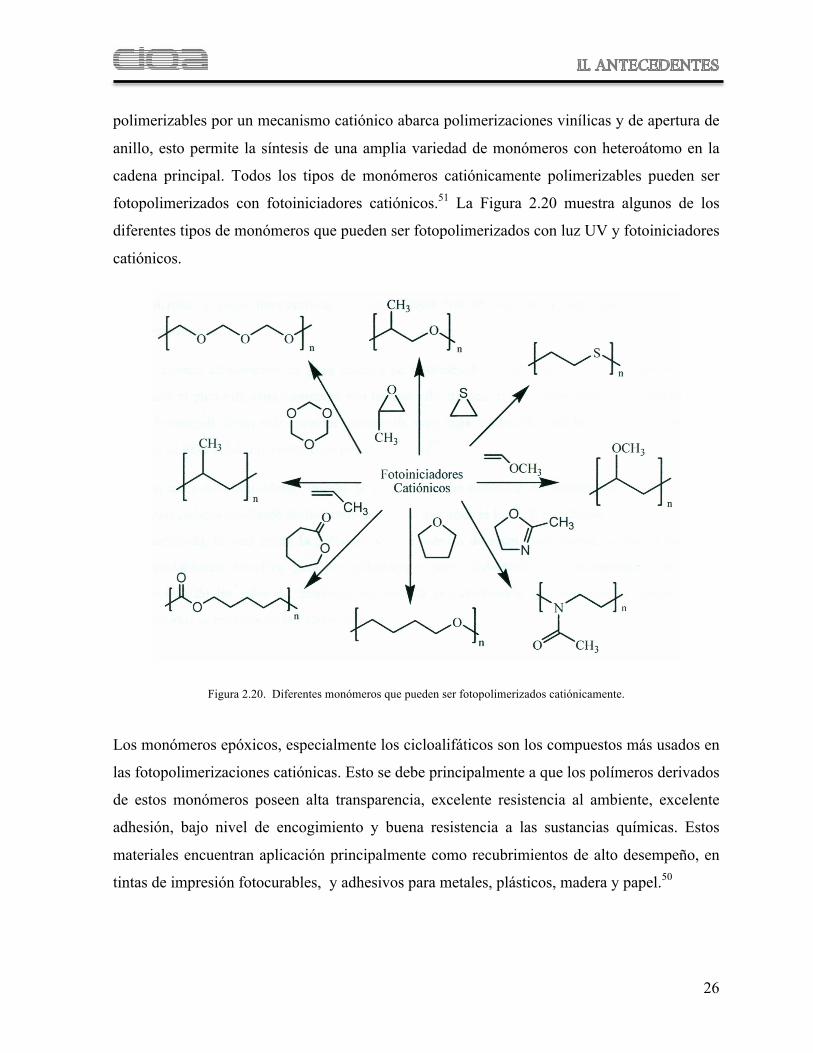
polimerizables por un mecanismo catiónico abarca polimerizaciones vinílicas y de apertura de
anillo, esto permite la síntesis de una amplia variedad de monómeros con heteroátomo en la
cadena principal. Todos los tipos de monómeros catiónicamente polimerizables pueden ser
fotopolimerizados con fotoiniciadores catiónicos.51 La Figura 2.20 muestra algunos de los
diferentes tipos de monómeros que pueden ser fotopolimerizados con luz UV y fotoiniciadores
catiónicos.
Figura 2.20. Diferentes monómeros que pueden ser fotopolimerizados catiónicamente.
Los monómeros epóxicos, especialmente los cicloalifáticos son los compuestos más usados en
las fotopolimerizaciones catiónicas. Esto se debe principalmente a que los polímeros derivados
de estos monómeros poseen alta transparencia, excelente resistencia al ambiente, excelente
adhesión, bajo nivel de encogimiento y buena resistencia a las sustancias químicas. Estos
materiales encuentran aplicación principalmente como recubrimientos de alto desempeño, en
tintas de impresión fotocurables, y adhesivos para metales, plásticos, madera y papel.50
CAPÍTULO II ANTECEDENTES
~o/'...o/'...ol
Ñ•
~oi"PCH3
~S{n
V'OCH3• NFotoiniciadores
Catiónicos
~N~n
OACH3
o LrCH3N
~oin
Figura 2.4. Diferentes monómeros que pueden ser fotopolimerizados catiónicamente.
2.3.1 Fotoiniciadores Catiónicos
A mediados de los años 70's las empresas GEI4 y 3MI5 encontraron que las sales de
diariliodonio y triarilsulfonio podían actuar como catalizadores de la fotopolimerización de
monómeros polimerizables catiónicamente. Este logro tuvo amplias repercusiones tanto a
nivel industrial como científico ya que de esta manera era posible polimerizar
formulaciones de monómeros epóxicos principalmente, con las sales de onio, hasta que
éstas eran irradiadas con luz UV, promoviendo la polimerización de una manera rápida y
controlada.
El grupo de Crivello en GE fue uno de los pioneros en el desarrollo de sales de onio y
prepararon una amplia variedad de éstas entre las que se incluyen sales de diariliodonio,
triarilsulfonio, triarilselenonio y dialquilfenacil sulfonio 16, 17-19.
La principal ventaja de los fotoiniciadores catiónicos es que presentan altas velocidades de
reacción y requieren de baja energía para pasar a estados excitados. Además, pueden
8

27
2.6 Determinación de cinéticas de Fotopolimerización en tiempo real
Una de las principales ventajas del curado por radiación es la rapidez con la que se da este
evento, el cual se desarrolla normalmente en fracciones de segundo para formar una red
polimérica altamente entrecruzada. Para los científicos que trabajan en esta área ha sido muy
difícil seguir adecuada y exactamente las cinéticas de estas reacciones ultrarrápidas cuando se
llevan a cabo con las lámparas de luz UV de alta intensidad que se usan normalmente en
sistemas prácticos.
Para un mejor entendimiento y control del proceso de curado es importante conocer
parámetros como la velocidad de polimerización (Rp), el rendimiento cuántico de la
polimerización (Øp), la longitud de la cadena cinética, la fotosensibilidad, así como las
constantes de propagación y terminación. Estas evaluaciones son esenciales para llevar a cabo
comparaciones de la reactividad de diferentes sistemas fotocurables y para estudiar el
desempeño de curado de nuevos fotoiniciadores y monómeros, así como el efecto de
diferentes factores físicos como la temperatura, intensidad de la luz y longitud de onda de la
luz.
Tomado en cuenta lo antes mencionado, se han desarrollado varias técnicas de análisis
resueltas en el tiempo que comúnmente se han denominado en tiempo real, para analizar el
comportamiento de los sistemas fotocurables. En estas técnicas se lleva a cabo el análisis de
cierta propiedad del sistema o cambios químicos en el mismo, al mismo tiempo que se irradia
la muestra con luz UV o visible. Es indispensable que el equipo de análisis tenga la habilidad
de llevar cabo barridos sucesivos en intervalos cortos de tiempo, con el fin de seguir el cambio
en propiedades físicas o químicas llevadas a cabo en el sistema a fotopolimerizar, como
resultado de la irradiación. A continuación se describe la técnica de espectroscopía de FTIR en
tiempo real (RT-FTIR) que se utilizó en este estudio.

28
2.6.1 Espectroscopía de FT-IR en tiempo real (RT-FTIR)
La técnica de RT-FTIR58,59 puede implementarse en equipos de IR que tengan la capacidad de
realizar barridos sucesivos a intervalos cortos de segundos o aun milisegundos. La muestra se
coloca entre dos pastillas de KBr o en su defecto en un sándwich de película de polipropileno
de preferencia con tratamiento corona. La salida de una fuente de luz UV acoplada a una fibra
óptica se posiciona a 45 ° con respecto al lugar en el que se encuentra el portamuestras. La
lámpara se enciende al mismo tiempo que se inicia el barrido de la muestra, así que los
cambios que se registran en grupos funcionales conforme se lleva a cabo la polimerización,
son registrados y además son proporcionales a la concentración por lo cual se puede
determinar la cinética de polimerización.
Figura 2.21. Esquema del RT-FTIR utilizado
El seguimiento continuo de la variación en la intensidad de señal IR bajo la radiación UV
permite la obtención de las curvas conversión vs tiempo para polimerizaciones que ocurren en
menos de 1 seg. Se pueden obtener curvas cinéticas obtenidas en tiempo real para
fotopolimerizaciones que ocurren en escalas de tiempo cortas (50% de conversión en menos
de 0.2 seg), y que transforma un monómero líquido a un material polimérico sólido. La
rapidez de la respuesta del detector IR permite un análisis cinético cuantitativo de
polimerizaciones donde la velocidad máxima es alcanzada en 30 microsegundos de
exposición.
Una ventaja importante de la espectroscopía RT-FTIR está en su alta sensibilidad; cambios
tan pequeños del orden del 1 % en la concentración de monómeros pueden ser detectados
instantáneamente en una capa de un micrómetro de espesor. Otra característica destacada es

29
que la película de polímero curada puede retenerse para una nueva investigación de sus
propiedades, tales como dureza, resistencia a la abrasión, flexibilidad, brillo, resistencia al
calor y/o solventes.
La velocidad de polimerización (Rp) puede ser determinada a cualquier tiempo del
experimento a partir de la pendiente de la curva cinética:
Donde:
[M]0 es la concentración de monómero inicial;
De los perfiles obtenidos por esta técnica de RT-FTIR se puede obtener la siguiente
información:
a) El periodo de inducción, el cual muestra como efectivamente el oxígeno o inhibidores
interfieren con el proceso de polimerización.
b) La velocidad de polimerización (Rp), la cual puede ser determinada en algún momento de la
reacción a partir de la pendiente del perfil RT-FTIR.
c) El cálculo del rendimiento cuántico (Øp) que corresponde al número de funciones
polimerizables por fotón absorbido y es calculado a partir de la relación de Rp a la intensidad
de luz absorbida.
d) La fotosensibilidad, la cual es a menudo definida como la cantidad de energía requerida
para polimerizar la mitad de las funcionalidades reactivas y es determinada fácilmente del
50% del tiempo de conversión
e) Contenido de insaturaciones residuales (RU) del polímero curado, que puede evaluarse
precisamente de la absorción de IR del producto final.
Rp= [M]0x2-x1
t2-t1

30
2.7 Análisis Térmico de Polímeros
Cuando se requiere un conocimiento más profundo del comportamiento de los plásticos por
efecto de la aplicación de calor se utilizan métodos térmicos donde se registran los cambios
físicos y químicos que sufren los plásticos al variar la temperatura de los mismos con respecto
al tiempo. Los cambios que se pueden estudiar son cambio de peso o dimensiones y la energía
asociada a una transición. En este estudio se utilizaron la técnica de análisis dinámico
mecánico, el análisis termogravimétrico y la calorimetria diferencial de barrido.
2.7.1 Análisis Dinámico Mecánico (DMA)
El análisis dinámico mecánico (DMA) es una técnica analítica que se utiliza para medir las
propiedades mecánicas de los materiales poliméricos y de algunos metales, en un régimen
dinámico, en función del tiempo, la temperatura y la frecuencia. Este análisis utiliza el
principio de estímulo-respuesta, para esto se aplica una fuerza oscilante a la muestra y se mide
el desplazamiento resultante. Por medio de la medición del lapso entre la fuerza aplicada y el
desplazamiento es posible determinar las propiedades de deformación del material. Debido a
esto se determina la rigidez de la muestra y se calcula su módulo elástico.60
Este análisis puede realizarse en el estado elástico de un sólido o bien en un fluído viscoso;
por ejemplo se estudia el comportamiento del componente elástico y del inelástico en pruebas
de tensión.
Mediante DMA es posible determinar el reblandecimiento de materiales para predecir su
utilización en procesos de extrusión, en los que la temperatura de transición vítrea (Tg) es
fundamental. El DMA se emplea en estudios de procesos de relajación y en reología para
caracterizar el comportamiento de materiales viscoelásticos como los polímeros, asi como la
respuesta de estos ante ciertos impulsos, esfuerzo/deformación en función del tiempo y la
frecuencia.

31
En un análisis de DMA se obtienen en la misma corrida, la curva de almacenamiento de
módulo (Storage Modulus que se representa por E´ ó también por G´), la curva para la pérdida
de módulo (loss modulus que se representa por E´´ o por G´´ ) y la curva tan delta la cual
proviene de la relación que resulta de dividir la pérdida de módulo entre el almacenamiento de
módulo (E´´/E´).
El almacenamiento de módulo E´, mide la energía almacenada y representa la porción elástica
del polímero, mientras que la pérdida de módulo mide la energía disipada como calor y
representa la porción viscosa del polímero. El tan delta representa el ángulo de la fase. En la
práctica se ha asumido que el punto máximo de la curva Tan delta representa la Tg del
polímero.
Con este tipo de estudios se comprende la mecánica de los materiales poliméricos en general
como hules fibras, textiles, empaques plásticos y espumas para optimizar y mejorar sus
propiedades.
2.7.2 Análisis Termogravimétrico (TGA)
El análisis termogravimétrico consiste en determinar la cantidad y el porcentaje de cambio en
el peso que sufre una muestra, como una función de la temperatura o el tiempo, bajo una
atmósfera controlada. El equipo consiste de una balanza muy sensible en la cual uno de los
extremos se introduce en el horno mientras que el otro es el sistema de referencia. La
termobalanza registra continuamente y en tiempo real el peso de la muestra.
La mayoría de las pruebas se hacen con nitrógeno como gas de arrastre y como gas inerte, en
estos casos se estudia estabilidad térmica, degradación, carbonización, deshidratación y
determinación de compuestos volátiles de la muestra ya sea algún disolvente, agua, gas
atrapado o un compuesto que se evapora conforme avanza la prueba.
Cuando se quieren estudiar cambios en los materiales, asociados a la presencia de oxígeno u
otro gas reactivo, se cambia el gas de arrastre por este y se procede a la prueba. Así se estudian

32
reacciones en medios oxidantes, estabilidad a la oxidación, degradación oxidativa y según el
material, reacciones de oxidación.
También se pueden realizar análisis en medio gaseoso de tipo isotérmico, es decir, se fija la
temperatura y se registra el cambio en peso de la muestra en función del tiempo. La muestra se
puede mantener sin cambios o bien, después de cierto tiempo, cambiar el peso de la muestra.
Este tipo de pruebas contribuyen a establecer la estabilidad de los materiales a esa temperatura
y respecto al medio que rodea o rodeará la muestra.
En una curva termogravimetrica se puede determinar cualitativa y cuantitativamente las etapas
del proceso de pérdida de peso, que se puede traducir en desorción de agua o desprendimiento
de material volátil; pero en una curva termogravimetrica derivada se puede realizar, además, el
análisis de la cinética de una reacción, es decir, de cómo se lleva a cabo un cambio químico en
la resina.
2.7.3 Calorimetría Diferencial de Barrido (DSC)
En este método se registra la diferencia de temperatura entre el espécimen de prueba y la
resina de referencia como función del tiempo o de la temperatura seleccionada. Los dos
materiales están sujetos a las mismas condiciones de temperatura, lo cual produce un ambiente
controlado de variación de energía.
La muestra problema de una resina y una muestra de referencia se colocan en celdas
independientes dentro de la cavidad de un bloque de transferencia de calor. A cada muestra se
le acopla un termopar para registrar los cambios de temperatura y a partir de estos valores, se
determinan las diferencias entre ellas; estos datos se grafican con respecto al tiempo o a la
temperatura previamente seleccionada en el equipo.
La muestra de referencia no debe sufrir fenómenos de transición conforme se lleva a cabo la
experimentación porque tiene como propósito comparar las mediciones de temperatura de la

33
muestra problema, además de minimizar las pérdidas térmicas debidas a la naturaleza del
equipo.
Las diferencias de temperatura dependen de las propiedades térmicas del equipo, celdas y de
la conductividad térmica de la muestra problema que varía de acuerdo a la Entalpía asociada a
sus transiciones.
El registro de los resultados de las diferencias de temperatura se realiza en termogramas, que
son, en esencia, ejes cardinales cuya ordenada indica la temperatura y la abscisa, el tiempo, la
temperatura del sistema (Ts) o la temperatura del espécimen de prueba. La ordenada se divide
en valores positivos y valores negativos, que se localizan al igual que en un eje de
coordenadas común, pero con intersección de la abscisa en la línea de valores negativos de la
ordenada. El tratamiento gráfico anterior se realiza para identificar si el fenómeno de
transición es exotérmico o endotérmico.
2.8 Justificación del trabajo de tesis
Tomando en consideración la eficiencia demostrada de los ortoespirocarbonatos para reducir o
eliminar el encogimiento, se ha propuesto que introduciendo grupos aromáticos al
ortoespirocarbonato, además de llevarse a cabo la polimerización por apertura de anillo, estos
compuestos al polimerizar con otro monómero epóxico podrían tener un menor nivel de
encogimiento, incluso hasta lograr un aumento en el volumen.
Es por eso que en esta investigación se propuso la preparación de un nuevo
ortoespirocarbonato a partir de un compuesto aromático. Este ortoespirocarbonato puede
prepararse mediante la reacción de transesterificación del tetraetilortocarbonato con el
Fluoreno diol.
El alcance de este trabajo de investigación abarca únicamente la síntesis y evaluación del
desempeño como agente antiencogimiento del ortoespirocarbonato aromático en la
fotopolimerización catiónica de monómeros epóxicos.

34
3. OBJETIVO E HIPÓTESIS:
3.1 Objetivo
Sintetizar un nuevo tipo de ortoespirocarbonato aromático que pueda tener actividad como
agente antiencogimiento en la fotopolimerización catiónica de monómeros epóxicos.
3.1.1 Objetivos Particulares
• Sintetizar el monómero Ortoespirocarbonato del Fluoreno (FSOC)
• Caracterizar el monómero, mediante las técnicas de espectroscopía de FT-IR, RMN de 1H y 13C, UV-Vis y Luminiscencia.
• Evaluar el monómero como agente antiencogimiento en la fotopolimerización
catiónica del monómero epóxico BADGE y 3,4-EP.
3.2 Hipótesis
Se ha registrado en la literatura que los ortoespirocarbonatos aromáticos son más estables a la
humedad que los ortoespirocarbonatos alifáticos, lo cual les confiere una ventaja al utilizarse
como agentes antiencogimiento. Se propone la hipótesis que el ortoespirocarbonato aromático
derivado del fluoreno puede disminuir o eliminar completamente el encogimiento de resinas
epóxicas. Además, se conoce que el fluoreno exhibe propiedades como fotosensibilizador, por
lo que otra hipótesis de este trabajo es que el FSOC podría fotosensibilizar la fotólisis de la sal
de yodonio (DPPI) usada como fotoiniciador ya sea con luz UV o visible.
35
4. PARTE EXPERIMENTAL CIQA
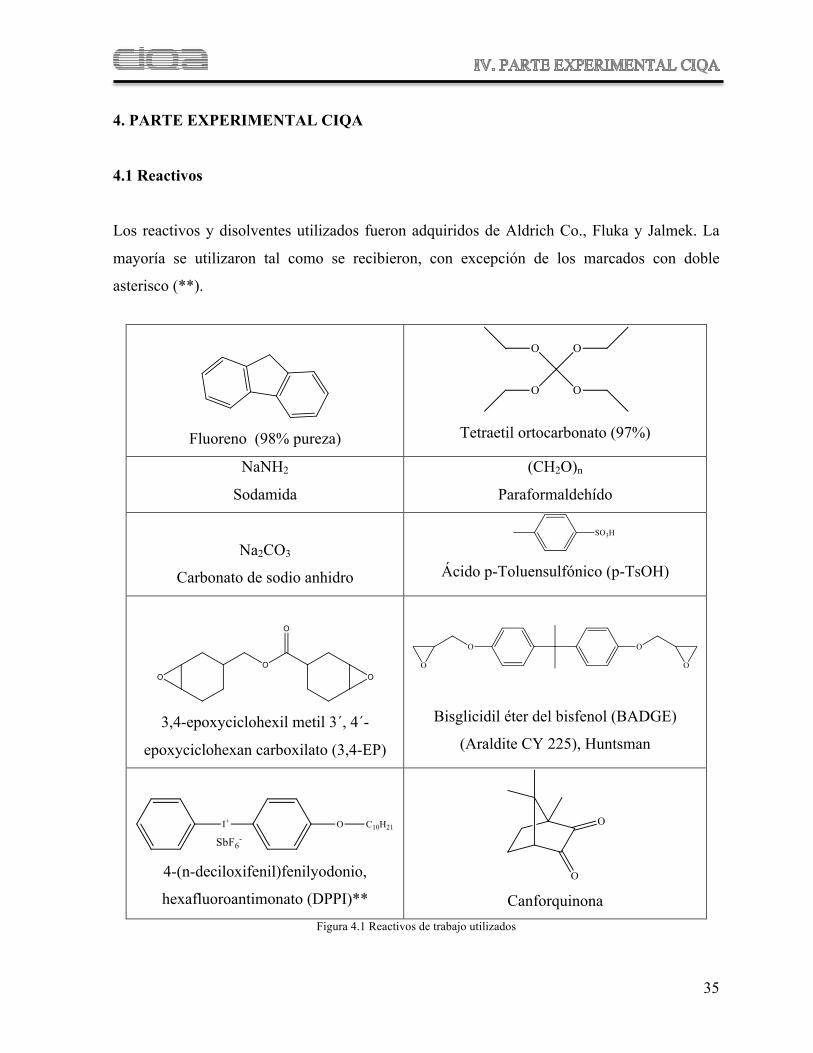
4.1 Reactivos
Los reactivos y disolventes utilizados fueron adquiridos de Aldrich Co., Fluka y Jalmek. La
mayoría se utilizaron tal como se recibieron, con excepción de los marcados con doble
asterisco (**).
Fluoreno (98% pureza)
Tetraetil ortocarbonato (97%)
NaNH2
Sodamida
(CH2O)n
Paraformaldehído
Na2CO3
Carbonato de sodio anhidro
Ácido p-Toluensulfónico (p-TsOH)
3,4-epoxyciclohexil metil 3´, 4´-
epoxyciclohexan carboxilato (3,4-EP)
Bisglicidil éter del bisfenol (BADGE)
(Araldite CY 225), Huntsman
4-(n-deciloxifenil)fenilyodonio,
hexafluoroantimonato (DPPI)**
Canforquinona Figura 4.1 Reactivos de trabajo utilizados
!
!
!
! !
!
!

36
4.1.2 Disolventes
**Piridina anhidra, pureza 99.8%, CAS 110-86-1, Aldrich
**Éter etílico, pureza 98%, CAS 60-29-7, Jalmek
Metanol, pureza 98%, CAS 67-56-1, Jalmek
Hexano
Cloroformo
4.1.3 Purificación de los reactivos y los disolventes
El (4-n-deciloxifenil) feniliodonio hexafluoroantimonato (DPPI) fue sintetizado por el método
reportado por Crivello.61
El éter etílico se seco con un complejo de sodio/benzofenona por 3 horas, previo a su uso.
La piridina se secó con sistema de destilación con AlLiH4 a 113° C.
4.2 Metodología
La metodología que se siguió para llevar a cabo este trabajo de investigación fue la siguiente:
1.- Síntesis de un compuesto dihidroxílico aromático como el 9H-Fluoreno-9,9-dimetanol
(FDiOH) a partir del Fluoreno en presencia de sodamida y paraformaldehído.
2.- Caracterización del compuesto FDiOH por espectroscopía de FT-IR, RMN de 1H y 13C.
3.- Síntesis del ortoespirocarbonato FSOC a partir del FDiOH.
Una vez formado el diol se transesterifica con el tetraetilortocarbonato para obtener el
monómero ortoespirocarbonato del Fluoreno (FSOC).

37
4.- Caracterización del monómero FSOC por espectroscopía de FT-IR, RMN de 1H y 13C.
5.- Determinación de las cinéticas de fotopolimerización mediante espectroscopía de FT-IR en
tiempo real (RT-FTIR), variando la concentración del FSOC desde 0 hasta 10 % molar usando
tanto luz UV como luz visible proveniente de una lámpara de LED.
6.- Evaluación del monómero ortoespirocarbonato como agente antiencogimiento variando la
concentración del FSOC desde 0, 2.5, 5, 7.5 y 10% molar utilizando el monómero Bisglicidil
éter del Bisfenol A (BADGE) como monómero base.
7.- Determinación de las propiedades viscoelásticas de las probetas obtenidas por
fotopolimerización de las formulaciones de BADGE con diferentes concentraciones de FSOC,
utilizando la técnica de Análisis dinámico mecánico (DMA).
8.- Determinación de las propiedades térmicas del polímero utilizando la técnica de análisis
diferencial de barrido (DSC).
9.- Determinación de la estabilidad térmica de los polímeros obtenidos mediante la técnica de
análisis termogravimétrico (TGA).
10.- Determinación del porcentaje de gel de las diferentes muestras fotopolimerizadas para
corroborar el grado de entrecruzamiento obtenido en el polímero.
4.3 Síntesis del 9H-Fluoreno-9,9-dimetanol (FDiOH)
El sistema de reacción estuvo conformado por un matraz bola de 3 bocas esmeriladas 14/20 de
50 ml, provisto de un refrigerante, termómetro y agitación magnética. Por la parte superior del
refrigerante se acopló un adaptador para la entrada de gas nitrógeno. Todo este sistema fue
instalado sobre una parrilla de agitación y sujetado por pinzas de tres dedos.

38
Para eliminar la humedad se flameó el sistema con un mechero Bunsen, haciendo vacío al
mismo tiempo, para eliminar la humedad y la presencia de oxígeno. Después de flamear se
dejó enfriar el sistema y se cambió un tapón de vidrio por un septum para poder agregar los
reactivos previamente pesados o medidos. Se adicionaron 9 ml de piridina seca y 2 g (12.03
mmol) de fluoreno. Una vez que el fluoreno se disolvió totalmente en la piridina se agregaron
0.9385 g (24.06mmol) de sodamida; la mezcla se mantuvo en agitación para permitir que se
disolvieran los reactivos y por último se agregaron 0.7225 g (24.06 mmol) de
paraformaldehído. La mezcla de reacción se dejó reaccionar por 24 horas manteniendo todo
este tiempo la agitación constante en el sistema y la atmósfera de nitrógeno. Pasado este
tiempo se procedió a neutralizar la piridina en la reacción, agregando poco a poco HCl al 10%
hasta disminuir el pH de la reacción de 14 a 7. Una vez logrado lo anterior, el producto de la
reacción se agregó a un matraz kitazato con 100 ml de agua, con la finalidad de disolver las
sales formadas. El sólido formado se filtró y se descartó el filtrado. El sólido obtenido se secó
en una estufa de vacío a 50°C por 2 horas. El producto crudo fue purificado por cromatografía
en columna utilizando metanol como eluente y posteriormente la columna se barrió con
cloroformo. Se obtuvieron 1.59 g de un sólido con punto de fusión de 142-145 °C, en un
rendimiento de 80%.
4.4 Síntesis de FSOC a partir del FDiOH
En un sistema provisto de un matraz bola de 3 bocas esmeriladas de 50 ml, refrigerante,
agitación magnética y termómetro, el cual se flameó en presencia de mallas moleculares, y
una vez enfriado se colocó en un baño de hielo, se agregaron 40 ml de éter seco, además de
4g (17.69 mmol) de FDiOH y 4.87 x 10-2 g (0.2834mmol) de p-TsOH. La mezcla se mantuvo
en agitación por 5 min y luego se adicionaron mediante una jeringa gota a gota, 1.47 g
(7.66mmol) de tetraetil ortocarbonato. Después, se retiró el baño de hielo/agua y se mantuvo
nuevamente la agitación por 10 min. Finalizado ese tiempo, se agregaron 8.93 x10 -2 g
(0.8428mmol) de Na2CO3. Por último se dejó reaccionar por aprox. 9 hrs a temperatura
ambiente. Una vez finalizada la reacción se filtró en un matraz de una boca de 100 ml con
entrada 24/40. El producto de la filtración se llevó a otro matraz del mismo tipo pero esta vez
se le dieron lavados con cloroformo junto con las mallas y se volvió a filtrar con papel filtro

39
cuantitativo, obteniéndose un líquido incoloro el cual se llevó al rotavapor para eliminar el
solvente y finalmente obtener 3.75 g de un polvo blanco con punto de fusión de 238-240 °C
que representa un 93.75% de rendimiento.
4.5 Caracterización de los monómeros
Los monómeros obtenidos de la síntesis fueron caracterizados por FTIR, RMN y CCD.
4.5.1 Caracterización por Espectroscopía Infrarroja (FT-IR)
Aproximadamente 5 mg de la muestra se mezclaron en un mortero con 30 mg de KBr, se
homogenizaron y se preparó una pastilla delgada con la prensa especial del equipo de FTIR.
Después del prensado la pastilla quedó fija en el soporte y ésta se montó en el portamuestras
del equipo. El espectrómetro de FTIR se ajustó para correr primero un background de aire y
después se corrieron las muestras realizando 25 barridos para cada muestra. Una vez
finalizado el proceso de barrido, se manipula el espectro obtenido para determinar las bandas
de absorción características de los compuestos preparados.
4.5.2 Caracterización por Resonancia Magnética Nuclear (RMN)
Se pesaron adecuadamente 20 mg de los compuestos de interés y se disolvieron en 5 ml de
deuterocloroformo. Los análisis fueron realizados en un espectrómetro modelo eclipse de 300
MHz de Jeol.
4.5.3 Cromatografía en capa delgada (CCD)
Esta técnica se utilizó para monitorear el avance de la síntesis. La técnica se describe a
continuación. Por medio de un capilar de vidrio se toma una pequeña muestra ya sea de la
mezcla de reacción o de los productos disueltos en solvente. Se coloca una microgota en la
parte inferior de la placa cromatográfica de silica-gel a una distancia de 5 mm del borde. Se
permite que se seque el solvente de la muestra y la placa se coloca dentro de una cuba que

40
contiene una mezcla de solventes llamada eluente. El eluente utilizado varió dependiendo del
producto, pero en la mayoría de los casos fue Hexano:Acetato de Etilo (6:4). Al introducir la
muestra en la cuba que contiene 5 -10 ml de eluente, por capilaridad sube el eluente en la
placa cromatográfica arrastrando la muestra mediante un proceso de adsorción. El grado de
adsorción depende de la polaridad de la muestra. Antes de llegar el eluente al borde superior
de la placa cromatográfica se saca de la cuba se permite secar por unos minutos y después se
revela dentro de un frasco que contiene unos cuanto gránulos de iodo.
4.6 Determinación de las cinéticas de fotopolimerización mediante la técnica de tiempo
real (RT-FTIR)
Las cinéticas de fotopolimerización se realizaron utilizando el compuesto BADGE como
monómero base. A este monómero se agregaron diferentes cantidades de FSOC y el
fotoiniciador DPPI se agregó a cada formulación en una concentración del 1 % molar
Las formulaciones elaboradas con el monómero BADGE y el monómero FSOC fueron
97.5/2.5, 95/5, 92.5/7.5 y 90/10 de relación en peso respectivamente, usando 1, 2 y 3% en peso
del fotoiniciador DPPI. Se prepararon 0.5g de cada formulación a evaluar así como un blanco
de cada monómero.
Las formulaciones fueron preparadas pesando en un vial la cantidad determinada de
monómero BADGE y la cantidad determinada de monómero utilizado como agente
antiencogimiento FSOC. Se agregaron unas pocas gotas de cloroformo para ayudar a disolver
perfectamente la mezcla de monómeros. El fotoiniciador DPPI se agregó y se mezcló
perfectamente con ayuda de una espátula. Después de tener las formulaciones listas se preparó
el equipo de espectroscopía infrarroja (FT-IR) de la siguiente manera:
Al equipo se le acopló una lámpara de luz UV marca UVEXS modelo SCU 110 a través de un
soporte universal para que el haz de la luz proveniente de la lámpara llegara a la muestra en un
ángulo de 45 °. La configuración del sistema se muestra en la Figura 2.21.

41
Hecho esto se determinó el background utilizando para esto un sándwich formado con dos
películas de PP con tratamiento corona, en lugar de aire, ya que en estas películas de PP se
corrieron las muestras y fue necesario restar las bandas de las películas de PP. Después se
manipuló el programa Series del software OMNIC para poder utilizar la técnica de tiempo real
en el espectrómetro de IR.
Se encendió la lámpara de luz UV y se ajustó la intensidad de esta a 10.1 mW/cm2 con un
radiómetro marca UV ProcessSupply Inc. La dosis fue de 606 mJ a la misma intensidad en un
intervalo de 60 segundos.
Una vez logrado lo anterior se depositaron 2 a 3 gotas de la formulación evaluada sobre la
película de PP y se colocó encima otra película con el fin de obtener un sándwich, el cual se
sujetó al portamuestras metálico del equipo de FT-IR mediante un imán. El portamuestras ya
con el sándwich se colocó en la cámara del equipo y se inició el barrido con el rayo IR del FT-
IR al mismo tiempo que se encendió la lámpara de luz UV. Se realizaron barridos a la muestra
cada segundo por espacio de 180 segundos.
Por medio del software OMNIC se obtuvo un perfil de disminución de la absorbancia
correspondiente a los picos de interés, el cual al graficarlo en Excel produce la curva cinética
de fotopolimerización del monómero. En nuestro caso se siguió el pico de 920 cm-1
correspondiente al grupo epóxido del BADGE, y el de 790 cm-1 para el 3,4-EP
4.7 Determinación del cambio de volumen mediante la determinación de la densidad de
las formulaciones del monómero antes y después de la polimerización
Las mediciones del cambio de volumen fueron determinadas midiendo la densidad de las
formulaciones antes y después de la polimerización. Las formulaciones fueron elaboradas con
el monómero BADGE el cual se evaluó con el monómero FSOC y el fotoiniciador DPPI en
cuatro formulaciones diferentes. Se preparó primero una probeta testigo únicamente con el
monómero BADGE y DPPI al 1 % molar. Las formulaciones elaboradas con el monómero

42
BADGE y el FSOC fueron de 97.5/2.5, 95/5 , 92.5/7.5 y 90/10 de relación molar
respectivamente.
Las mediciones de la densidad de las formulaciones (!L) se llevaron a cabo pesando una
cantidad específica (1 mL) de la formulación líquida. Después de esto se elaboraron las
probetas de las formulaciones las cuales se prepararon con dos placas de cuarzo, y un molde
de PP para dar el espesor y las dimensiones correctas.
El molde vacío se colocó sobre la placa de cuarzo a la cual se le aplicó previamente una capa
de silicón para evitar que se adhiera la muestra al cuarzo, al fotopolimerizar. Después de
depositar la formulación dentro del molde, éste se cubrió con otra placa de cuarzo encima,
ajustándose con unas pinzas. Una vez que se tuvo lista la formulacion dentro del molde y
sobre las placas de cuarzo, estas se colocaron sobre un reflector con el fin de que la muestra se
curara tanto por la parte superior como por la parte inferior como resultado de la
polimerización inducida por la luz reflejada en el reflector.
Figura 4.2. Sistema de fotopolimerización para curado de muestras
Se procedió a polimerizar las muestras en un horno equipado con una lámpara de luz UV
Marca Fusion UV de alta intensidad (300 W). Las placas colocadas sobre el reflector se
introdujeron en el horno de luz UV por espacio de 15 minutos. Después de este tiempo las
muestras se introdujeron en una estufa por 60 minutos a 120°C para completar el curado de la

43
resina epóxica con el agente antiencogimiento. El tamaño de las probetas preparadas fue de 10
x 40 mm con un espesor de 2mm.
La densidad del polímero curado se mide con un equipo de medición de densidad, aplicando el
principio de Arquímedes. Se determina la gravedad específica de un sólido al introducirlo en
un líquido de densidad conocida y se expone a la fuerza de empuje del sólido. El valor de este
empuje es el mismo que el peso del líquido desplazado por el volumen del sólido. El volumen
específico de las formulaciones antes y después de la polimerización se define como la inversa
de la densidad y se determina mediante la siguiente fórmula:
!" ! !!
Donde:
Vp = Volumen específico
! = Densidad
La densidad de las placas se determinó en una balanza Sartorius YDK 01 con el kit de
determinación de densidad, la cual permite obtener el peso del sólido en el aire así como
también en un líquido.
La densidad de las placas fueron calculadas con la siguiente fórmula:
!s ! !!!rel ! !!!liq
!!rel = !
! ! ! ! !
Donde:
!S = densidad absoluta del sólido (placa)
!rel = densidad relativa

44
!liq = densidad del líquido utilizado
!= peso de la muestra al aire
!= peso del alambre parcialmente inmerso
!= peso del alambre parcialmente inmerso + muestra totalmente inmersa
La densidad del líquido a usar debe ser a una temperatura conocida y para este caso se uso
agua a 23°C cuya ! = 0.9976 g/cc
El volumen específico de las formulaciones antes y después de la polimerización se define
como la inversa de la densidad y el cambio de volumen se obtiene de la siguiente fórmula:
! ! !!" ! !!!! !!!!""
Donde:
! = cambio de volumen
VS= volumen específico del polímero curado (placa)
VL= volumen específico de la formulación líquida
4.8 Análisis Dinámico Mecánico (DMA)
Se utilizó un Equipo TA Instrument Q800 provisto de sistema de enfriamiento con Nitrógeno
líquido mordaza tipo Cantiliver simple, empleando una frecuencia de 1Hz y una amplitud de
15 µm. El análisis se realizó con velocidad de calentamiento de 5°C/ min en un rango de 30°C
a 200°C. En este análisis se utilizaron las mismas probetas preparadas para el análisis de
cambio de volumen.

45
4.9 Calorimetría Diferencial de Barrido (DSC)
Se utilizó un equipo TA instrument modelo DSC2920, para el análisis se tomó 3-13 mg de las
probetas preparadas anteriormente que fueron colocados en una cápsula de aluminio; el
análisis se realizó utilizando una rampa de 10 °C/min bajo una atmósfera de nitrógeno con un
flujo de 50 ml/min en un rango de 30 a 200 °C.
4.10 Análisis Termogravimétrico (TGA)
Se utilizó un equipo TA instrument Q500. Se utilizaron de 3-7 mg de las probetas preparadas
anteriormente. La muestra se colocó en una canastilla de platino, iniciando el análisis a
temperatura ambiente hasta llegar a 600°C en atmósfera de nitrógeno y 600- 800 °C bajo
atmósfera de oxígeno con una velocidad de 10 °C/min.
4.11 Determinación del porcentaje de gel en la formulación de FSOC y BADGE
El contenido de gel se determinó en las placas curadas siguiendo el método ASTM D2765-84,
midiendo el peso perdido de las placas después de 24h de extracción con cloroformo a
temperatura ambiente. Producto de esta extracción se obtiene la fracción insoluble que
corresponde al gel producido por el entrecruzamiento de las especies poliméricas. Después de
la extracción las especies insolubles son removidas y secadas, volviéndose a pesar. La
cantidad de material extraído es calculado de la siguiente manera:
!!"# ! !!!
!!!!!!""
Donde:
Wf = Peso final, peso perdido durante la extracción
Wo = Peso inicial, peso original de la especie polimérica

46
5. PARTE EXPERIMENTAL POLITÉCNICO DE TORINO-ITALIA
5.1 Metodología
Se evaluó el FSOC como agente antiencogimiento en la fotopolimerización catiónica del
monómero 3,4-Epoxiciclohexilmetil (3,4-EP) utilizando Canforquinona y sal de diaril iodonio
(Rhodorsil 2074) como fotoiniciador usando una lámpara foto curable de luz azul (Astralis 5
Ivoclar Vivadent) de intensidad 600 mW/cm2 para inducir la Fotopolimerización.
Figura 5.1. Lámpara foto curable de luz azul Astralis 5 Ivoclar
Esta evaluación consistió en:
• Determinación de la concentración optima de Canforquinona y Rhodorsil a usar para
obtener el mejor % de conversión.
• Determinación de las cinéticas de polimerización de las formulaciones del monómero
mediante la técnica de tiempo real FT-IR (RT-FTIR).
• Determinación del cambio de volumen mediante la medición de la densidad de las
formulaciones antes y después de la polimerización.
• Determinación de las propiedades viscoelásticas de las películas obtenidas mediante la
técnica de análisis dinámico mecánico (DMA).
• Determinación del contenido de gel en las placas curadas.

47
5.2 Determinación de la concentración optima de Canforquinona y Rhodorsil a usar
para obtener el mejor % de conversión.
Para conocer la concentración optima de Canforquinona y Rhodorsil necesaria para obtener
mejores tiempos de curado y altas conversiones se prepararon 3 diferentes formulaciones con
el monómero 3,4-EP las cuales contenían el 1, 2 y 3% molar de Canforquinona y Rhodorsil
respectivamente. Cada una de estas formulaciones se irradió con una lámpara de luz azul por
espacios de tiempo de 15 segundos durante 120 segundos, obteniendo espectros de FTIR
correspondientes a cada formulación a diferentes tiempos de irradiación.
De estos espectros se obtuvieron los datos necesarios para conocer la concentración óptima a
usar de Canforquinona y Rhodorsil en la fotopolimerización del monómero 3,4-EP con el
agente antiencogimiento FSOC.
5.3 Determinación de las cinéticas de polimerización de las formulaciones del monómero
mediante la técnica de tiempo real (RT-FTIR).
Las cinéticas de fotopolimerización se realizaron con el monómero 3,4-EP, este se evaluó con
el monómero FSOC, Canforquinona y el fotoiniciador Rhodorsil, para la fotopolimerización
las muestras se irradiaron con una lámpara fotocurable de luz azul a 0, 15, 30, 45, 60 y 120
segundos.
Las formulaciones elaboradas con el monómero 3,4-EP y el monómero FSOC fueron 97.5/2.5,
95/5, 92.5/7.5 y 90/10 de relación en peso respectivamente, usando 2% en peso de Rhodorsil
y Canforquinona. Se preparó 1g de cada formulación a evaluar así como un blanco de cada
monómero.
Las formulaciones fueron preparadas pesando en un vial la cantidad determinada de
monómero 3,4-EP, Canforquinona, Rhodorsil y la cantidad determinada de monómero
utilizado como agente antiencogimiento FSOC.

48
Después de tener las formulaciones listas se preparó el equipo de espectroscopía infrarroja
(FT-IR) y se alistó una lámpara foto curable de luz azul.
Hecho esto se determinó el background utilizando para esto una placa de silicio (Microscope
Slide PEARL) ya que en esta placa se corrieron las muestras; una vez que se obtuvo el
espectro de background se almaceno en un dispositivo USB para usarlo siempre antes de cada
corrida.
Para correr el espectro de la primer formulación se colocó sobre la placa de silicio una gota de
esta, la cual fue esparcida sobre la placa por medio de una varilla para dejarla con un espesor
uniforme de 25 micras.
Esta placa se colocó en el equipo de FTIR y se corrió el primer espectro que fue el
correspondiente al tiempo de irradiación de 0 segundos de luz azul.
Una vez realizado lo anterior se sacó la placa y se irradió con la luz azul durante 15 segundos
para llevar de nuevo al FTIR y obtener el segundo espectro.
Posteriormente se hizo lo mismo con la misma muestra, se sacó y se irradió por 15 segundos y
se obtuvo el espectro correspondiente a 30 segundos de irradiación con luz azul.
Así sucesivamente se irradio la muestra por un tiempo de 15 segundos y se obtuvieron los
espectros a 45 y 60 segundos de irradiación con luz azul para que finalmente la última
irradiación que se le dio a la muestra durara 60 segundos que fue el espectro correspondiente
al tiempo de 120 segundos de irradiación con luz azul.
La misma metodología fue aplicada para las formulaciones subsecuentes. Al final de estas
pruebas se obtuvieron 6 espectros por cada formulación que correspondían al monitoreo de la
muestra cuando esta estuvo expuesta desde 0 hasta 120 segundos a la luz azul, con los datos
obtenidos de los espectros se calcularon los % de conversión para poder realizar una
comparación de datos.

49
Se realizaron barridos a la muestra cada segundo por un espacio de 120 segundos. Por medio
del software OMNIC se obtuvieron espectros de cada barrido que fueron guardados en un
dispositivo de almacenamiento USB para su posterior análisis.
5.4 Determinación del cambio de volumen mediante la medición de la densidad de las
formulaciones antes y después de la polimerización.
Las mediciones del cambio de volumen fueron determinadas midiendo la densidad de las
formulaciones antes y después de la polimerización. Las formulaciones fueron elaboradas con
el monómero 3,4-EP el cual se evaluó con el monómero FSOC y el fotoiniciador Rhodorsil en
cuatro formulaciones diferentes. Se preparó primero una probeta testigo únicamente con el
monómero 3,4-EP, Canforquinona y Rhodorsil al 2% molar. Las formulaciones elaboradas
con el monómero CE y el FSOC fueron de 97.5/2.5, 95/5 , 92.5/7.5 y 90/10 de relación molar
respectivamente.
Las mediciones de la densidad de las formulaciones (!L) se llevaron a cabo pesando una
cantidad específica (5 mL) de la formulación líquida. Después de esto se elaboraron las
probetas de las formulaciones,las cuales se prepararon en moldes de parafilm de 2 x 0.5 cm
colocados sobre un portaobjetos como se muestra en la figura 5.2.
Figura 5.2. Moldes para preparar probetas para determinar densidad
Antes de depositar las muestras sobre los moldes de parafilm en el portaobjetos se roció tanto
el molde como otro portaobjetos que funcionaría como tapa con un Spray desmoldante
(Resina 30, Gold Line).

50
Una vez realizado lo anterior se depositó la muestra sobre los huecos en el portaobjetos hasta
rellenarlos completamente, se tapó con el otro portaobjetos y se presionó de modo que casi
quedaran sellados.
Después se procedió a polimerizar las formulaciones irradiando con una lámpara fotocurable
de luz azul cada una de las muestras en el molde por un espacio de 2 minutos por cada lado.
Las probetas obtenidas se separaron de los moldes y se dejaron en una estufa a 80° por un
tiempo de 2 horas para completar el curado de la resina epóxica con el agente
antiencogimiento.
La densidad del polímero curado se mide con un equipo de medición de densidad, aplicando el
principio de Arquímedes. Se determina la gravedad específica de un sólido al introducirlo en
un líquido de densidad conocida y se expone a la fuerza de empuje del sólido. El valor de este
empuje es el mismo que el peso del líquido desplazado por el volumen del sólido. Las medidas
de densidad se llevan a cabo en un equipo especial con balanza hidrostática que permite pesar
el sólido tanto en el aire así como en el líquido. La gravedad específica del sólido se
determina conociendo la densidad del líquido que se desplazó mediante la siguiente fórmula:
! ! ! ! ! !!!!! ! !!!!!
Donde:
! = densidad del sólido
!(l) = densidad del líquido utilizado
W(a) = peso del sólido en el aire
W(l) = peso del sólido en el líquido
La densidad de las placas se determinó en una balanza Sartorius YDK 01 con el kit de
determinación de densidad, la cual permite obtener el peso del sólido en el aire así como
también en un líquido.

51
El volumen específico de las formulaciones antes y después de la polimerización se define
como la inversa de la densidad y el cambio de volumen se obtiene de la siguiente fórmula:
! ! !!" ! !!!! !!!!""
Donde:
! = cambio de volumen
VS= volumen específico del polímero curado (placa)
VL= volumen específico de la formulación líquida
5.5 Determinación de las propiedades viscoelásticas de las películas obtenidas mediante
la técnica de análisis dinámico mecánico (DMA), análisis termo gravimétrico (TGA),
calorimetría diferencial de barrido (DSC) y %Gel.
Para realizar este tipo de pruebas se procedió de manera similar a los análisis hechos en
México y reportados previamente en la sección 4.8 a 4.11.

52
6. RESULTADOS Y DISCUSIONES
6.1 Síntesis del Ortoespirocarbonato del Fluoreno (FSOC)
En este trabajo de tesis inicialmente se planteó desarrollar una metodología sintética basada en
el uso de cetonas aromáticas como materias primas para preparar los ortoespirocarbonatos
aromáticos. Dadas las propiedades del esqueleto de fluoreno como fotosensibilizador 62,63 se
decidió usar la fuorenona como materia prima. La metodología inicialmente planteada,
involucraba cinco etapas: la primera consistía en hacer reaccionar la fluorenona, con el
cloroetil acetato en presencia de una base fuerte como la sodamida, (Reacción de Darzens)
para obtener el éster glicídico de fluoreno (1,2), el cual después de hidrólisis y
descarboxilación, resulta en el aumento de un carbono en el compuesto aromático,
obteniéndose el aldehído correspondiente (3).
Figura 6.1. Ruta de síntesis propuesta inicialmente a partir de cetonas aromáticas
! O
Cl CH2 CO2 CH2CH3 NaNH2
CO2
OCH2CH3 COOH
O
H3O+
COOHO CO2
C
O
H
C
O
H CH2O NaOHOH
OH
OH
OH
CH3CH2OC
OCH2CH3
CH3CH2O OCH2CH3O
O
O
O
+ +
1) NaOH/MeOH
2)
-
1)
2)
3)
4) + +
5) + p-TsOH

53
Este aldehído se haría reaccionar con formaldehido en medio básico (Reacción de Cannízaro)
para obtener el diol aromático (4). Una vez preparado este diol se podría transesterificar con el
tetraetilcarbonato para obtener el SOC aromático (5).
Se procedió a validar esta ruta sintética usando primeramente la fluorenona como materia
prima. Se inició con la experimentación de la primera de las etapas de la síntesis. Se realizaron
diversos experimentos en los que se variaron las condiciones de reacción, como temperatura,
tipo de solvente y relación estequiométrica de reactivos, sin embargo se obtuvieron resultados
negativos ya que en todos los casos se obtuvo un producto sólido precipitado, el cual al ser
analizado presentó el mismo punto de fusión que la fluorenona así como un espectro de IR y
RMN muy similar al de la materia prima, por lo que se llegó a la conclusión de que por alguna
razón, la reacción química no se estaba llevando a cabo satisfactoriamente.
Una posible explicación de estos resultados es que los electrones del carbonilo de la cetona
aromática forman un solo sistema pi conjugado el cual es rico en electrones, por lo tanto al
tratar de llevar a cabo un ataque nucleofílico con una especie también rica en electrones como
el carbanión generado en el cloroacetato de etilo, probablemente haya repulsión entre las dos
especies y eso imposibilite el ataque del carbanión al carbonilo aromático.
Dado que nos estaba consumiendo demasiado tiempo el seguir intentando esta ruta, se decidió
seguir una ruta alternativa de síntesis para preparar dioles aromáticos.75 Se realizó una nueva
búsqueda bibliográfica para revisar las posibles rutas sintéticas y se diseñó la siguiente:
Figura 6.2. Ruta de síntesis propuesta a partir del Fluoreno
!

54
Esta ruta presentaba la ventaja de que se podía obtener el diol en un solo paso. Se realizaron
experimentos y se encontró que mediante esta ruta se obtuvo el diol deseado en un 80% de
rendimiento. La reacción procede mediante un mecanismo de adición nucleofílica del anión
generado en el fluoreno, sobre el formaldehido, el cual se agrega a la forma de
paraformaldehído.
6.2 Caracterización del Ortoespirocarbonato del Fluoreno (FSOC)
Es difícil saber si la reacción se lleva en etapas sucesivas, o simultáneamente, aunque se ha
reportado en Wikipedia64 que al hacer reaccionar el fluoreno con potasio metálico utilizando
dioxano a reflujo se forma relativamente fácil el dianión del fluoreno. Por lo cual podríamos
deducir que el dianión es relativamente estable y en éste caso se podrían adicionar las dos
moléculas de formaldehído para formar el dialcóxido y mediante acidulación formar el diol.
Además si la reacción se llevara en etapas se observaría la presencia clara de los protones
bencílicos a 3.88 ppm del monoalcohol, lo cual se observa apenas en trazas en el espectro de
protón de RMN para el FDiOH que se muestra en la Figura 6.3. Donde se observan las señales
correspondientes a los protones aromáticos entre 7 y 8 ppm que integran para 4 mientras que
en 4.05 ppm se encuentra un doblete que integra para dos y a campo alto se tiene un triplete
que integra para 1 en 2.07 ppm.
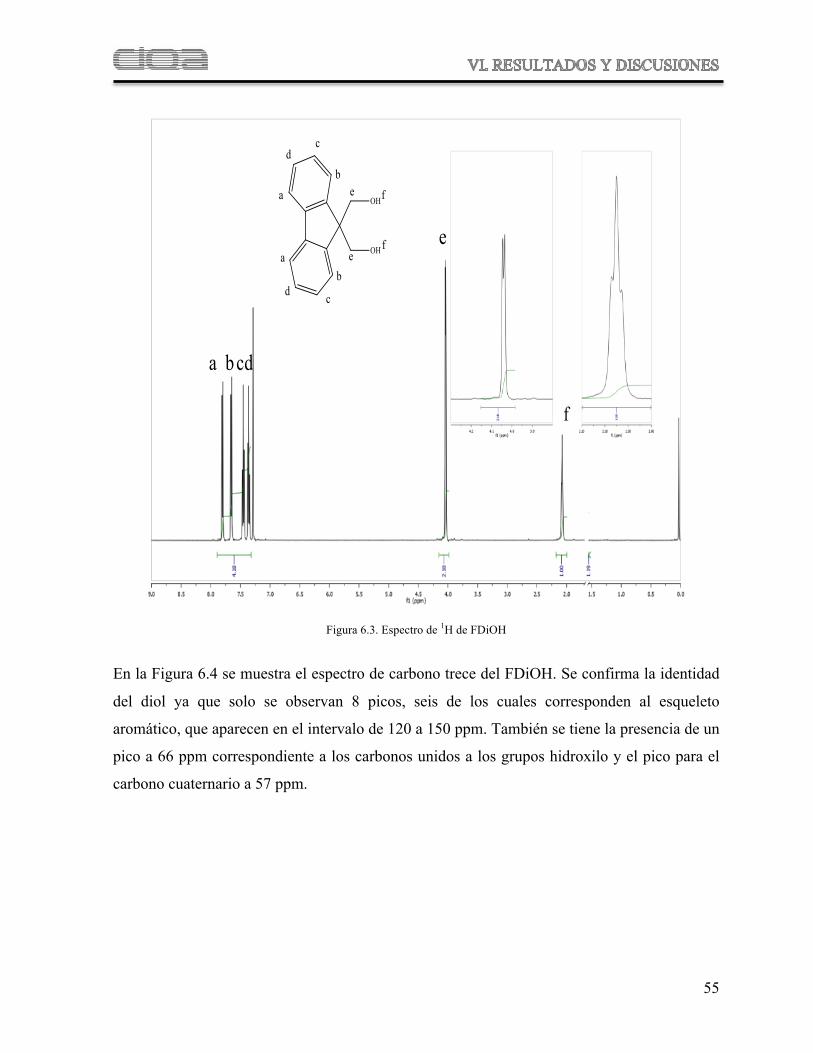
55
Figura 6.3. Espectro de 1H de FDiOH
En la Figura 6.4 se muestra el espectro de carbono trece del FDiOH. Se confirma la identidad
del diol ya que solo se observan 8 picos, seis de los cuales corresponden al esqueleto
aromático, que aparecen en el intervalo de 120 a 150 ppm. También se tiene la presencia de un
pico a 66 ppm correspondiente a los carbonos unidos a los grupos hidroxilo y el pico para el
carbono cuaternario a 57 ppm.
a bcd
e
f
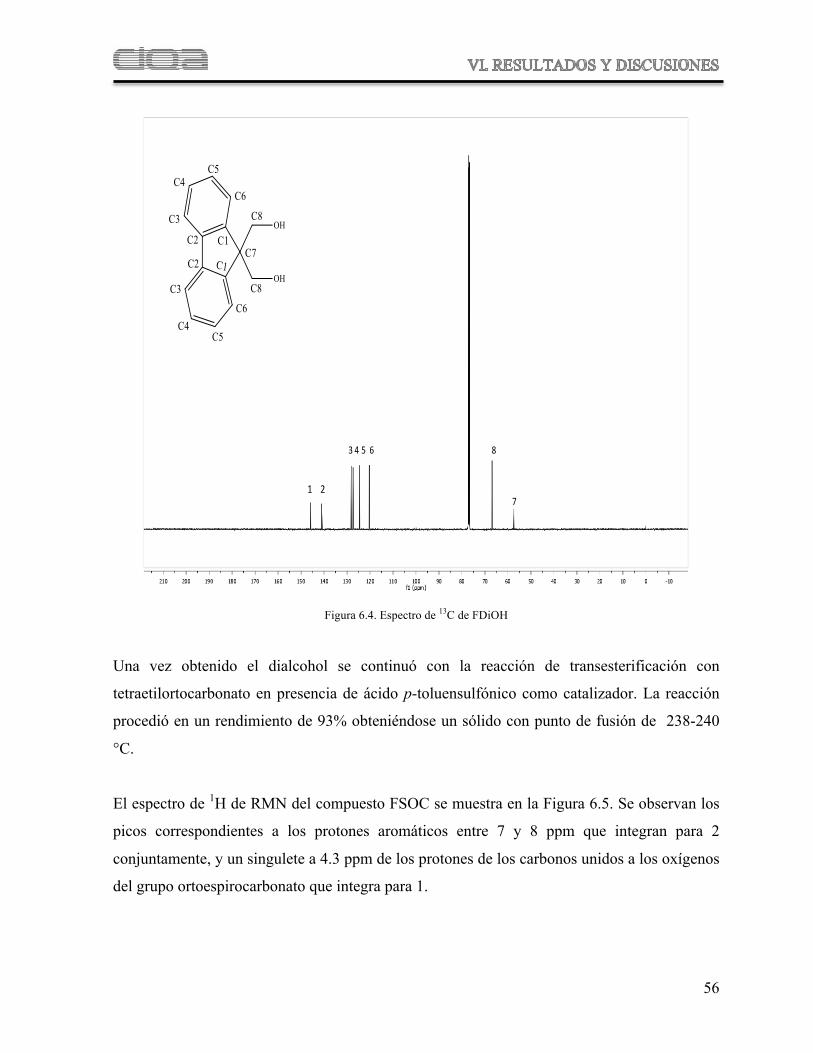
56
Figura 6.4. Espectro de 13C de FDiOH
Una vez obtenido el dialcohol se continuó con la reacción de transesterificación con
tetraetilortocarbonato en presencia de ácido p-toluensulfónico como catalizador. La reacción
procedió en un rendimiento de 93% obteniéndose un sólido con punto de fusión de 238-240
°C.
El espectro de 1H de RMN del compuesto FSOC se muestra en la Figura 6.5. Se observan los
picos correspondientes a los protones aromáticos entre 7 y 8 ppm que integran para 2
conjuntamente, y un singulete a 4.3 ppm de los protones de los carbonos unidos a los oxígenos
del grupo ortoespirocarbonato que integra para 1.
!""""#
$"%"&""' (
)
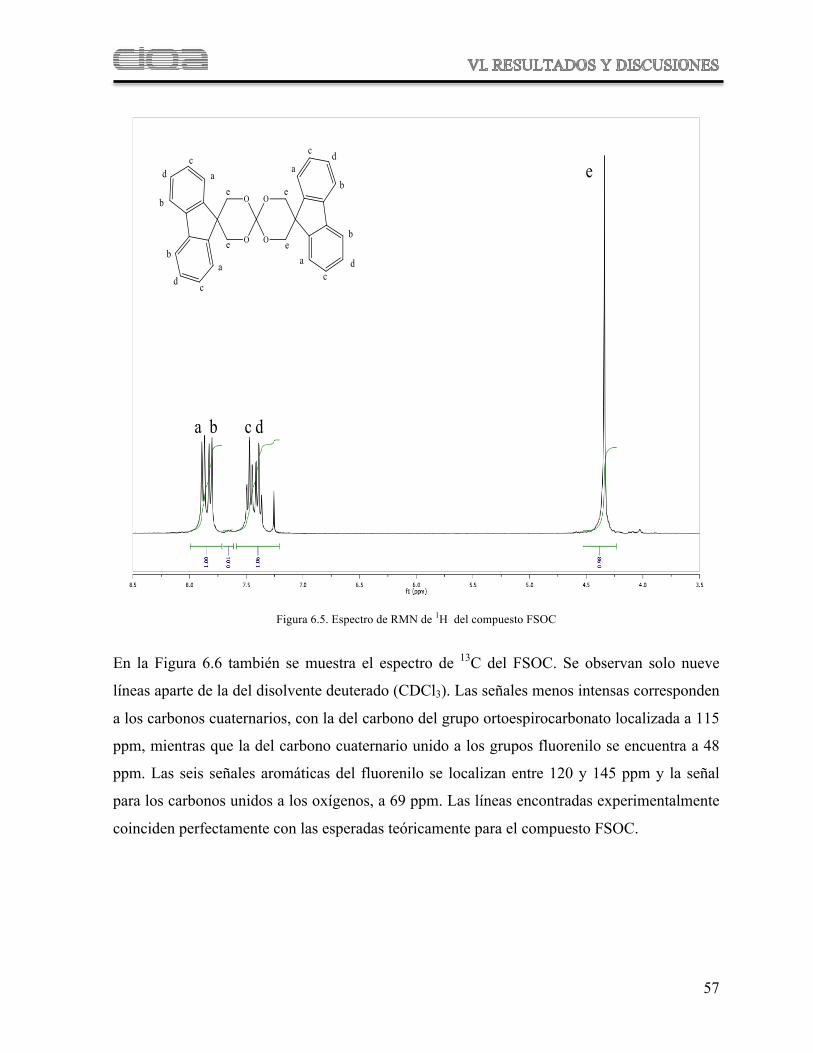
57
Figura 6.5. Espectro de RMN de 1H del compuesto FSOC
En la Figura 6.6 también se muestra el espectro de 13C del FSOC. Se observan solo nueve
líneas aparte de la del disolvente deuterado (CDCl3). Las señales menos intensas corresponden
a los carbonos cuaternarios, con la del carbono del grupo ortoespirocarbonato localizada a 115
ppm, mientras que la del carbono cuaternario unido a los grupos fluorenilo se encuentra a 48
ppm. Las seis señales aromáticas del fluorenilo se localizan entre 120 y 145 ppm y la señal
para los carbonos unidos a los oxígenos, a 69 ppm. Las líneas encontradas experimentalmente
coinciden perfectamente con las esperadas teóricamente para el compuesto FSOC.
e
a b c d
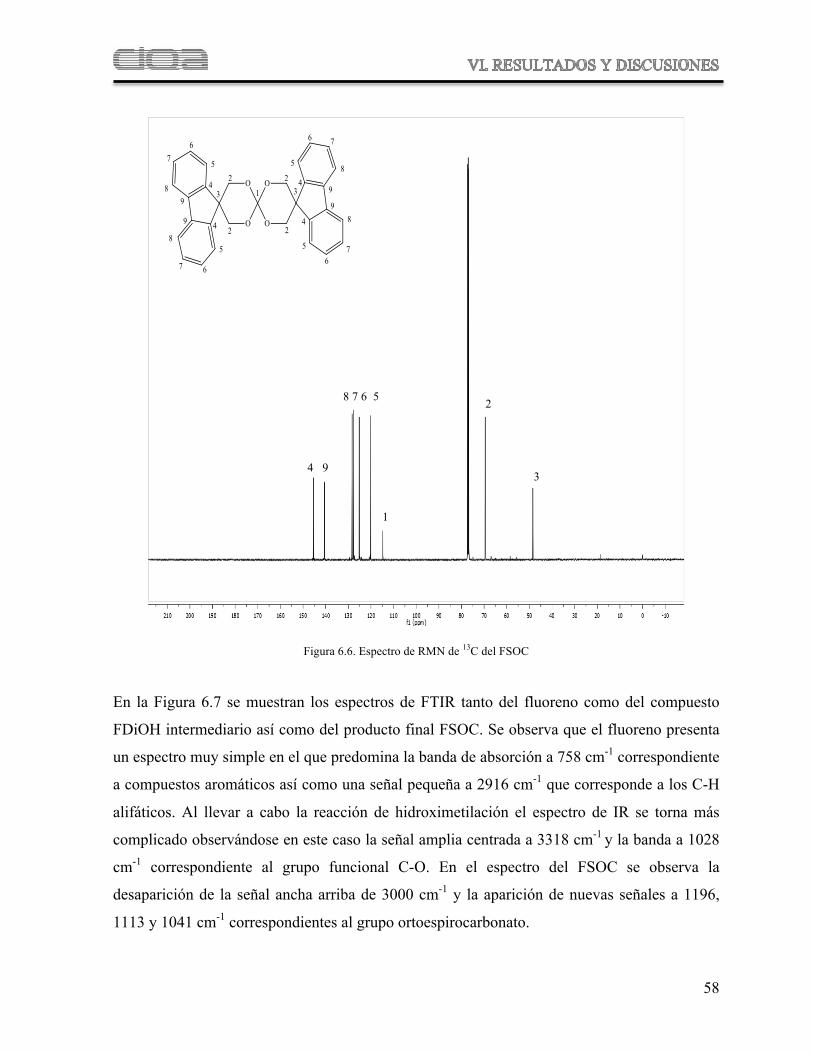
58
Figura 6.6. Espectro de RMN de 13C del FSOC
En la Figura 6.7 se muestran los espectros de FTIR tanto del fluoreno como del compuesto
FDiOH intermediario así como del producto final FSOC. Se observa que el fluoreno presenta
un espectro muy simple en el que predomina la banda de absorción a 758 cm-1 correspondiente
a compuestos aromáticos así como una señal pequeña a 2916 cm-1 que corresponde a los C-H
alifáticos. Al llevar a cabo la reacción de hidroximetilación el espectro de IR se torna más
complicado observándose en este caso la señal amplia centrada a 3318 cm-1 y la banda a 1028
cm-1 correspondiente al grupo funcional C-O. En el espectro del FSOC se observa la
desaparición de la señal ancha arriba de 3000 cm-1 y la aparición de nuevas señales a 1196,
1113 y 1041 cm-1 correspondientes al grupo ortoespirocarbonato.
1
2
3 4 9
8 7 6 5
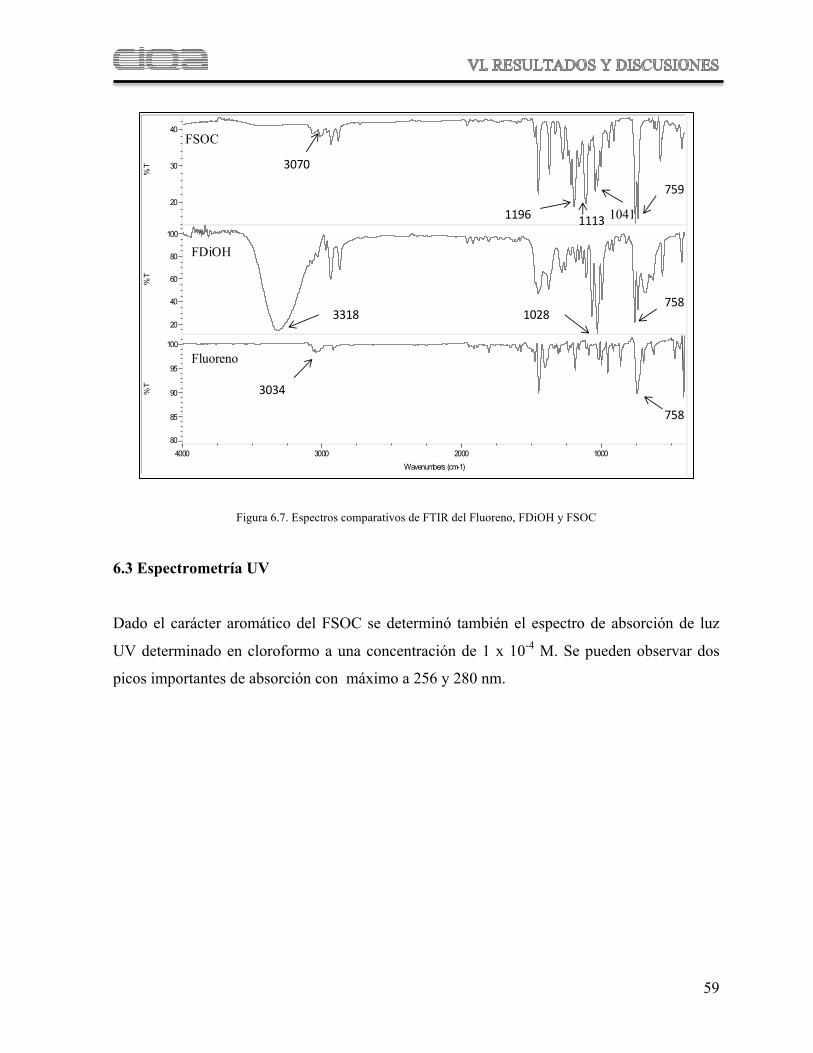
59
Figura 6.7. Espectros comparativos de FTIR del Fluoreno, FDiOH y FSOC
6.3 Espectrometría UV
Dado el carácter aromático del FSOC se determinó también el espectro de absorción de luz
UV determinado en cloroformo a una concentración de 1 x 10-4 M. Se pueden observar dos
picos importantes de absorción con máximo a 256 y 280 nm.
20
30
40
%T
20
40
60
80
100
%T
80
85
90
95
100
%T
1000 2000 3000 4000 Wavenumbers (cm-1)
FSOC
FDiOH
Fluoreno
!!"# "$%#
""&' 1041
()#
()&
()#
!$!*
!$($
"""!
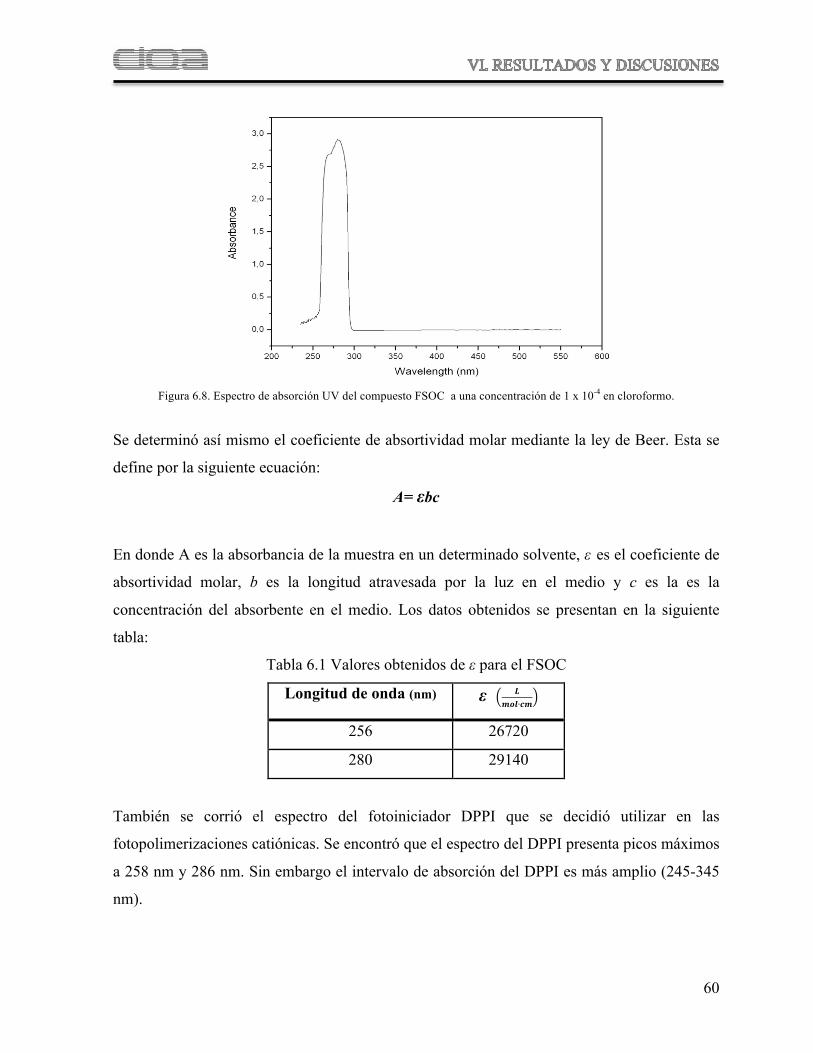
60
Figura 6.8. Espectro de absorción UV del compuesto FSOC a una concentración de 1 x 10-4 en cloroformo.
Se determinó así mismo el coeficiente de absortividad molar mediante la ley de Beer. Esta se
define por la siguiente ecuación:
A= !bc
En donde A es la absorbancia de la muestra en un determinado solvente, ! es el coeficiente de
absortividad molar, b es la longitud atravesada por la luz en el medio y c es la es la
concentración del absorbente en el medio. Los datos obtenidos se presentan en la siguiente
tabla:
Tabla 6.1 Valores obtenidos de ! para el FSOC
Longitud de onda (nm) ! !!"#!!"
256 26720
280 29140
También se corrió el espectro del fotoiniciador DPPI que se decidió utilizar en las
fotopolimerizaciones catiónicas. Se encontró que el espectro del DPPI presenta picos máximos
a 258 nm y 286 nm. Sin embargo el intervalo de absorción del DPPI es más amplio (245-345
nm).
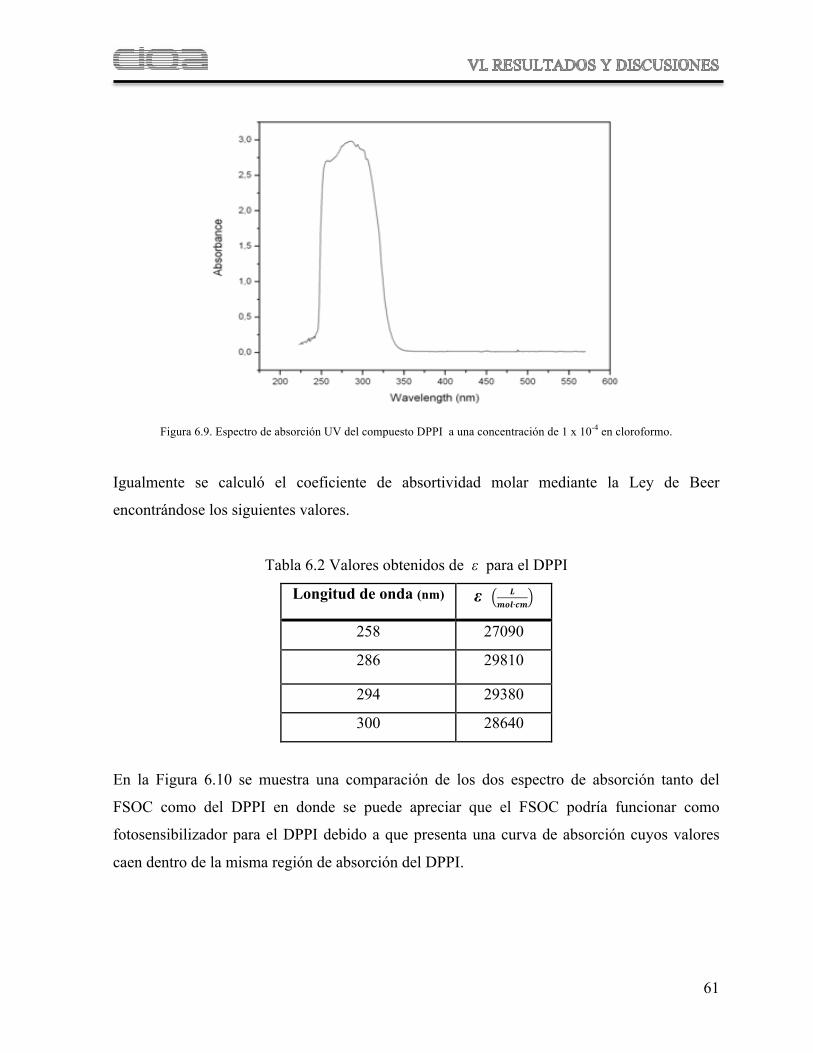
61
Figura 6.9. Espectro de absorción UV del compuesto DPPI a una concentración de 1 x 10-4 en cloroformo.
Igualmente se calculó el coeficiente de absortividad molar mediante la Ley de Beer
encontrándose los siguientes valores.
Tabla 6.2 Valores obtenidos de ! para el DPPI
Longitud de onda (nm) ! !!"#!!"
258 27090
286 29810
294 29380
300 28640
En la Figura 6.10 se muestra una comparación de los dos espectro de absorción tanto del
FSOC como del DPPI en donde se puede apreciar que el FSOC podría funcionar como
fotosensibilizador para el DPPI debido a que presenta una curva de absorción cuyos valores
caen dentro de la misma región de absorción del DPPI.
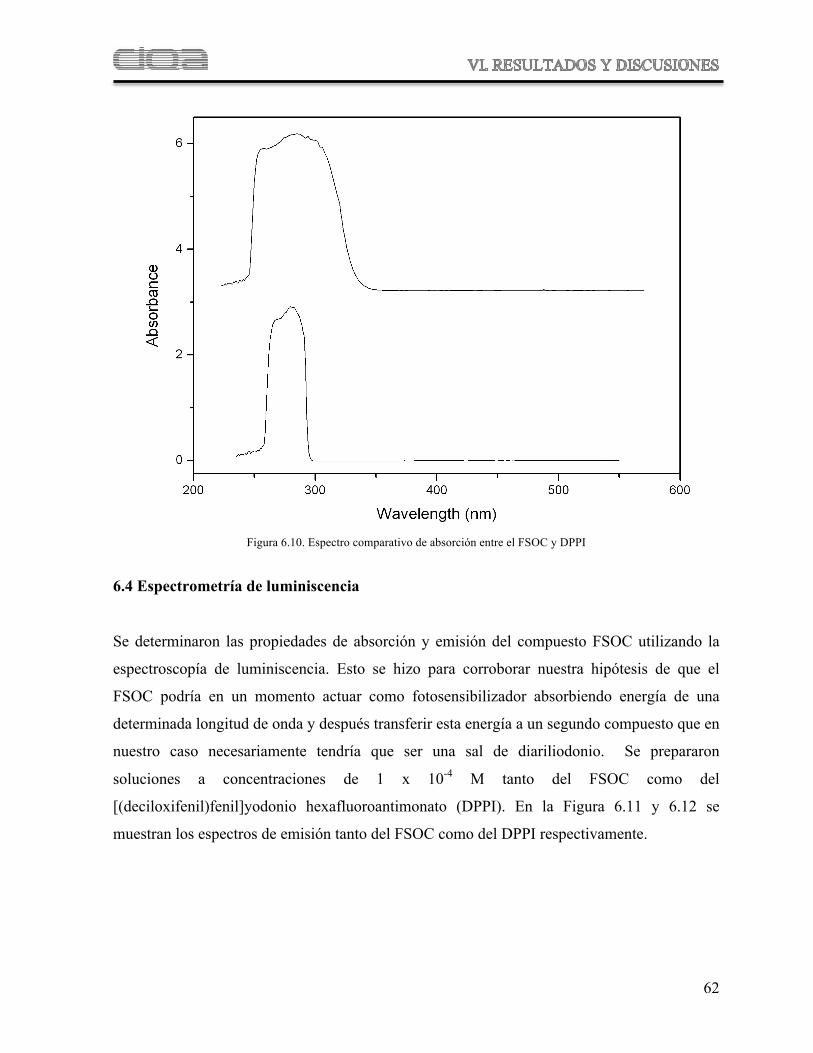
62
Figura 6.10. Espectro comparativo de absorción entre el FSOC y DPPI
6.4 Espectrometría de luminiscencia
Se determinaron las propiedades de absorción y emisión del compuesto FSOC utilizando la
espectroscopía de luminiscencia. Esto se hizo para corroborar nuestra hipótesis de que el
FSOC podría en un momento actuar como fotosensibilizador absorbiendo energía de una
determinada longitud de onda y después transferir esta energía a un segundo compuesto que en
nuestro caso necesariamente tendría que ser una sal de diariliodonio. Se prepararon
soluciones a concentraciones de 1 x 10-4 M tanto del FSOC como del
[(deciloxifenil)fenil]yodonio hexafluoroantimonato (DPPI). En la Figura 6.11 y 6.12 se
muestran los espectros de emisión tanto del FSOC como del DPPI respectivamente.

63
Figura 6.11. Espectro de emisión del FSOC
Figura 6.12. Espectro de emisión del DPPI
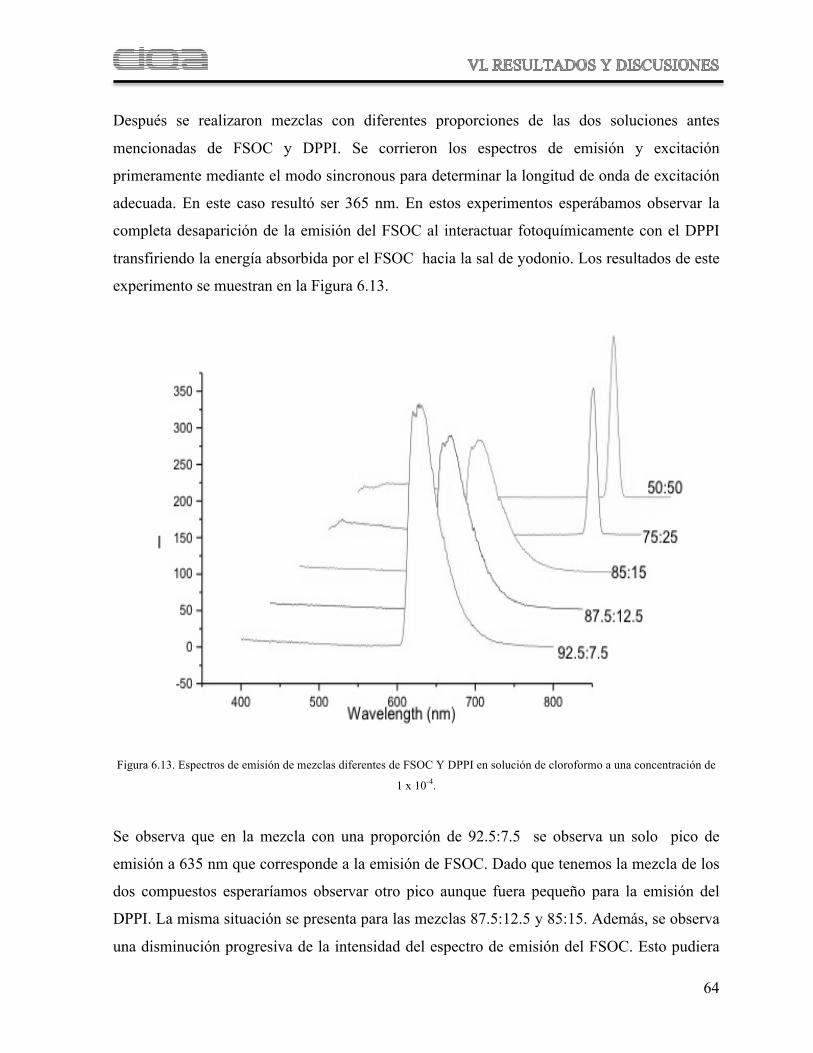
64
Después se realizaron mezclas con diferentes proporciones de las dos soluciones antes
mencionadas de FSOC y DPPI. Se corrieron los espectros de emisión y excitación
primeramente mediante el modo sincronous para determinar la longitud de onda de excitación
adecuada. En este caso resultó ser 365 nm. En estos experimentos esperábamos observar la
completa desaparición de la emisión del FSOC al interactuar fotoquímicamente con el DPPI
transfiriendo la energía absorbida por el FSOC hacia la sal de yodonio. Los resultados de este
experimento se muestran en la Figura 6.13.
Figura 6.13. Espectros de emisión de mezclas diferentes de FSOC Y DPPI en solución de cloroformo a una concentración de
1 x 10-4.
Se observa que en la mezcla con una proporción de 92.5:7.5 se observa un solo pico de
emisión a 635 nm que corresponde a la emisión de FSOC. Dado que tenemos la mezcla de los
dos compuestos esperaríamos observar otro pico aunque fuera pequeño para la emisión del
DPPI. La misma situación se presenta para las mezclas 87.5:12.5 y 85:15. Además, se observa
una disminución progresiva de la intensidad del espectro de emisión del FSOC. Esto pudiera

65
explicarse en base a que sí está ocurriendo el fenómeno de transferencia de energía del FSOC
hacia el DPPI, ya que conforme se aumenta la concentración del DPPI se disminuye la
emisión del FSOC y llega un momento en que éste desaparece por completo, observándose
únicamente el espectro de emisión del DPPI.
De esta manera se puede deducir de estos experimentos que el FSOC definitivamente presenta
propiedades de fotosensibilización pero a concentraciones relativamente grandes de DPPI, lo
cual lo hace impráctico en la experiencia real ya que los niveles comunes de fotoiniciador
catiónico no rebasan el 2.5% en peso.
6.5 Fotopolimerización del FSOC con luz UV y DPPI como fotoiniciador
Una vez obtenido, purificado y caracterizado el FSOC se procedió a fotopolimerizar el
compuesto FSOC bajo las condiciones en las que se llevarían a cabo las cinéticas de
fotopolimerización por espectroscopía de RT-FTIR para asegurarnos que el FSOC
polimerizaba adecuadamente. De esta manera, dado que nuestro compuesto es sólido se tuvo
que disolver en un solvente inerte como es en este caso el cloroformo. Se disolvieron 200 mg
de FSOC en 5 ml de cloroformo y se agregaron 5 mg de DPPI. La muestra se irradió por 30
minutos con la lámpara de luz UV SCU con conducción de luz UV por fibra óptica y con una
intensidad de aproximadamente 10-20 mW/cm2. Al cabo de este tiempo la solución irradiada
se tornó café. El solvente se evaporó y se determinó tanto el espectro de FTIR como el de
RMN de protón.
En la Figura 6.14 se muestra la comparación de los espectros de FTIR del FSOC y del
polímero obtenido (P-FSOC) al polimerizar el ortoespirocarbonato. Se observa claramente la
formación de un carbonilo a 1745 cm-1 que corresponde a un carbonato. También es notoria la
presencia de una banda ancha centrada a 3445 cm-1 característica para compuestos grupos
hidroxilos. También apareció una banda ancha a 1252 cm-1 para el estiramiento C-O. Por otro
lado desaparecieron las bandas características para los ortoespirocarbonatos anteriormente
mencionadas.

66
Figura 6.14. Comparación de los espectros de FTIR del FSOC y del polímero obtenido después de fotopolimerizar el FSOC.
En la Figura 6.15 se muestra también el espectro de RMN de 1H para el polímero obtenido de
la fotopolimerización del FSOC. En este se puede ver los picos entre 7.0 y 8.0 ppm para los
protones aromáticos del fluoreno. En el intervalo entre 4.25 y 4.75 ppm se observan una serie
de señales que corresponden a los protones de los carbonos unidos al grupo carbonato del
poliétercarbonato obtenido. Entre 3.5 y 4.0 ppm se observan otras señales que corresponden a
los protones de los carbonos unidos a los grupos éter del polímero. También se corrobora la
presencia de grupos hidroxilo terminales ya que se observa un pico ancho centrado a 1.8 ppm.
76
4,6
2
10
36
,80
12
52
,50
13
96
,29
14
47
,65
17
45
,512
87
0,1
9
30
65
,34
34
45
,36
0
10
20
30
40
%T
57
4,6
1
75
4,3
5
10
41
,94
11
08
,70
11
90
,87
13
70
,61
14
47
,65
15
20
25
30
35
40
%T
1000 2000 3000 4000 Wavenumbers (cm-1)
FSOC
P-FSOC
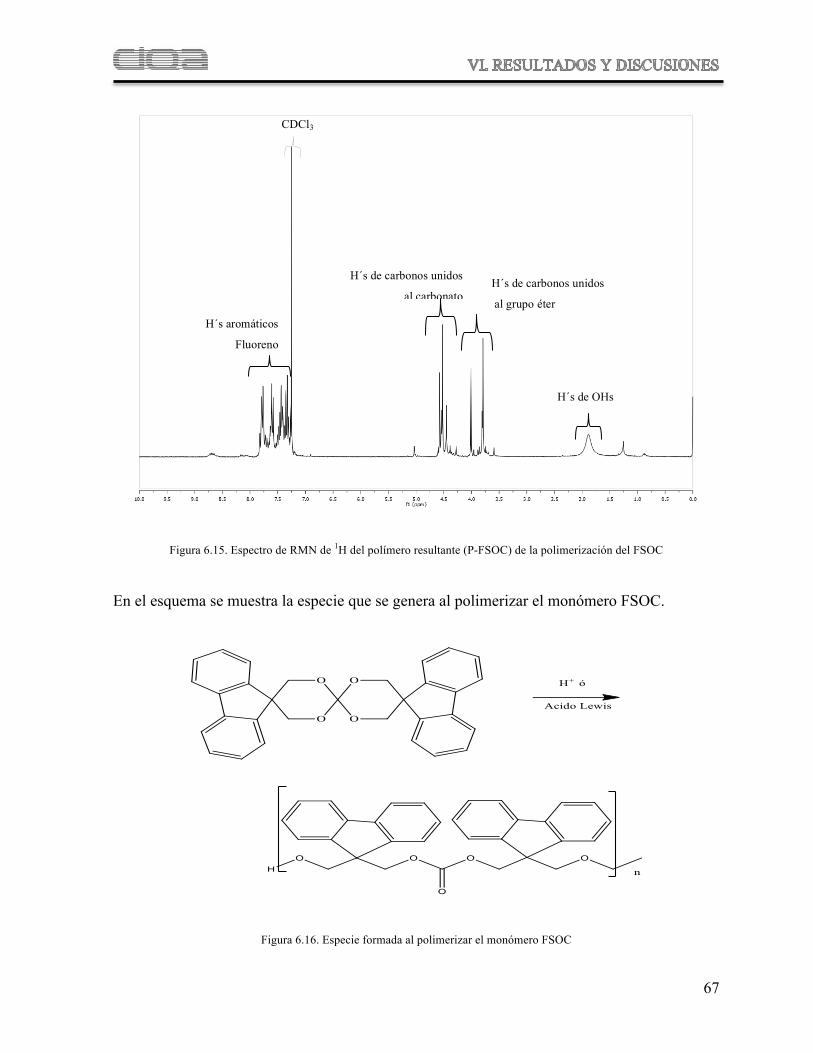
67
Figura 6.15. Espectro de RMN de 1H del polímero resultante (P-FSOC) de la polimerización del FSOC
En el esquema se muestra la especie que se genera al polimerizar el monómero FSOC.
Figura 6.16. Especie formada al polimerizar el monómero FSOC
H´s aromáticos Fluoreno
CDCl3
H´s de OHs
H´s de carbonos unidos al carbonato
H´s de carbonos unidos al grupo éter

68
El mecanismo mediante el cual procede esta reacción se muestra en la Figura 6.17. Una vez
que se fotoliza la sal de yodonio como resultado de la absorción de luz UV y se genera el
súper ácido correspondiente, éste protona cualquiera de los oxígenos del grupo
ortoespirocarbonato. Al estar activado este monómero, puede ser rápidamente atacado por otra
molécula de FSOC en la cual los electrones sin compartir de cualquiera de los oxígenos del
grupo ortoespirocarbonato atacan al carbono adyacente a uno de los oxígenos de la primera
molécula activada, promoviendo la doble apertura de anillo, generando en el proceso un grupo
hidroxilo terminal y un grupo carbonato. Una vez que la segunda molécula ataca a la primera
entonces esta molécula se activa a su vez generando una molécula con un ion oxonio el cual
será atacado nuevamente por una tercera molécula de FSOC y así sucesivamente hasta que se
consume todo el monómero.
Figura 6.17. Mecanismo de polimerización del monómero FSOC
!
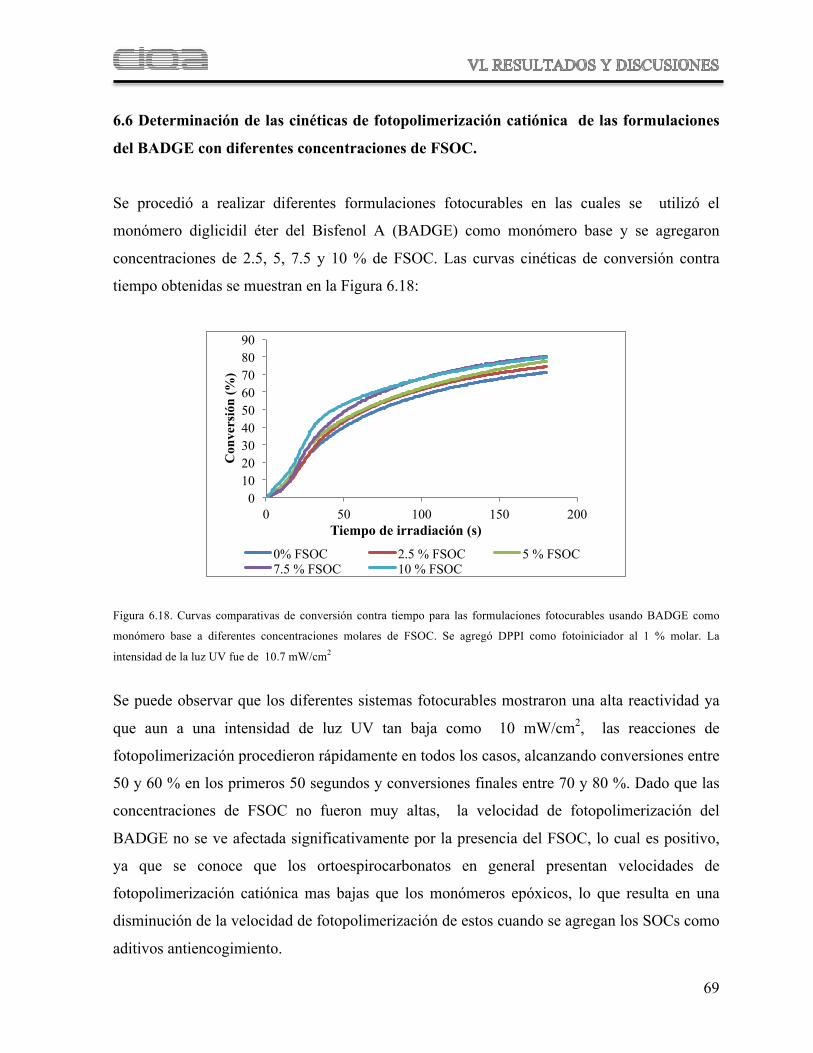
69
6.6 Determinación de las cinéticas de fotopolimerización catiónica de las formulaciones
del BADGE con diferentes concentraciones de FSOC.
Se procedió a realizar diferentes formulaciones fotocurables en las cuales se utilizó el
monómero diglicidil éter del Bisfenol A (BADGE) como monómero base y se agregaron
concentraciones de 2.5, 5, 7.5 y 10 % de FSOC. Las curvas cinéticas de conversión contra
tiempo obtenidas se muestran en la Figura 6.18:
Figura 6.18. Curvas comparativas de conversión contra tiempo para las formulaciones fotocurables usando BADGE como
monómero base a diferentes concentraciones molares de FSOC. Se agregó DPPI como fotoiniciador al 1 % molar. La
intensidad de la luz UV fue de 10.7 mW/cm2
Se puede observar que los diferentes sistemas fotocurables mostraron una alta reactividad ya
que aun a una intensidad de luz UV tan baja como 10 mW/cm2, las reacciones de
fotopolimerización procedieron rápidamente en todos los casos, alcanzando conversiones entre
50 y 60 % en los primeros 50 segundos y conversiones finales entre 70 y 80 %. Dado que las
concentraciones de FSOC no fueron muy altas, la velocidad de fotopolimerización del
BADGE no se ve afectada significativamente por la presencia del FSOC, lo cual es positivo,
ya que se conoce que los ortoespirocarbonatos en general presentan velocidades de
fotopolimerización catiónica mas bajas que los monómeros epóxicos, lo que resulta en una
disminución de la velocidad de fotopolimerización de estos cuando se agregan los SOCs como
aditivos antiencogimiento.
0 10 20 30 40 50 60 70 80 90
0 50 100 150 200
Con
vers
ión
(%)
Tiempo de irradiación (s)
0% FSOC 2.5 % FSOC 5 % FSOC 7.5 % FSOC 10 % FSOC

70
En este caso al agregar el FSOC al 2.5 % se encontró que la velocidad inicial de
fotopolimerización no se ve afectada y solo se registra un ligero incremento en la conversión
final de 74 a 77%. Conforme se fue aumentando la concentración de FSOC de 5 a 7.5% se
observa el mismo patrón de comportamiento ya que la velocidad de fotopolimerización es
muy similar en los tres casos y solo se observa de nuevo un ligero aumento en la conversión
(79 y 80% respectivamente). Al aumentar la concentración de FSOC al 10% la velocidad de
fotopolimerización es similar en los primeros 25 segundos pero después de este tiempo la
polimerización se lleva a cabo ligeramente más rápido que en los casos anteriores. Sin
embargo no se observó un aumento en el porcentaje de conversión final ya que se obtuvo un
80% de conversión.
Como ya se mencionó antes, los SOCs al polimerizar generan poliétercarbonatos los cuales
son especies que presentan una alta movilidad e inducen un efecto de plastificación cuando
copolimerizan con otros monómeros como los epóxicos disminuyendo la Tg del polímero
base.26 Para este caso específico, el FSOC al polimerizar produce un poliétercarbonato con
grupos fluorenilo voluminosos tal como se mostró anteriormente en la Figura 6.16.
La flexibilidad impartida por los poliétercarbonatos derivados de los SOCs retrasa la gelación
o el punto de solidificación de los monómeros epóxicos al polimerizar, por lo que de esta
manera es posible alcanzar conversiones ligeramente más altas que cuando no se usan los
SOCs.37
Dado que el FSOC y el BADGE polimerizan por apertura de anillo catalizada por ácido fuerte,
esperaríamos obtener copolímeros de tipo poliéter (poliéter carbonato). Sin embargo se ha
reportado que algunos SOCs no polimerizan tan rápidamente como los monómeros epóxicos y
por lo tanto tienden a homopolimerizar.65 Pero existen otros reportes en los que se confirma
que compuestos de tipo SOC copolimerizan cuando se hacen reaccionar con monómeros
epóxicos.66-68 En nuestro caso es difícil determinar si hay copolimerización ya que las
concentraciones del FSOC son relativamente bajas y sería difícil ver una segunda Tg para una
sección tan pequeña del copolímero. Sin embargo como se mostrará en la sección de análisis
por DSC solo se observa una inflexión muy marcada para la Tg en los casos de las

71
formulaciones con FSOC y no se observa claramente una segunda Tg aun en el caso de la
concentración con 10% de FSOC por lo que podemos inferir que se produce copolimerización
de ambos monómeros FSOC y BADGE.
Figura 6.19. Copolímero de FSOC y BADGE
Entonces, derivado de los resultados obtenidos por RT-FTIR, se puede concluir que la
presencia del FSOC a diferentes concentraciones en formulaciones con BADGE tiene un
efecto positivo ya que no afecta adversamente la velocidad de fotopolimerización del BADGE
y además incrementa la conversión final del mismo en proporción directa a la concentración
del FSOC.
Con el fin de aumentar la reactividad del sistema se procedió a realizar un estudio en el cual se
incrementó la concentración de fotoiniciador catiónico DPPI utilizando una concentración de
FSOC del 10%. Los resultados obtenidos se muestran en la Figura 6.20. Se observa que no
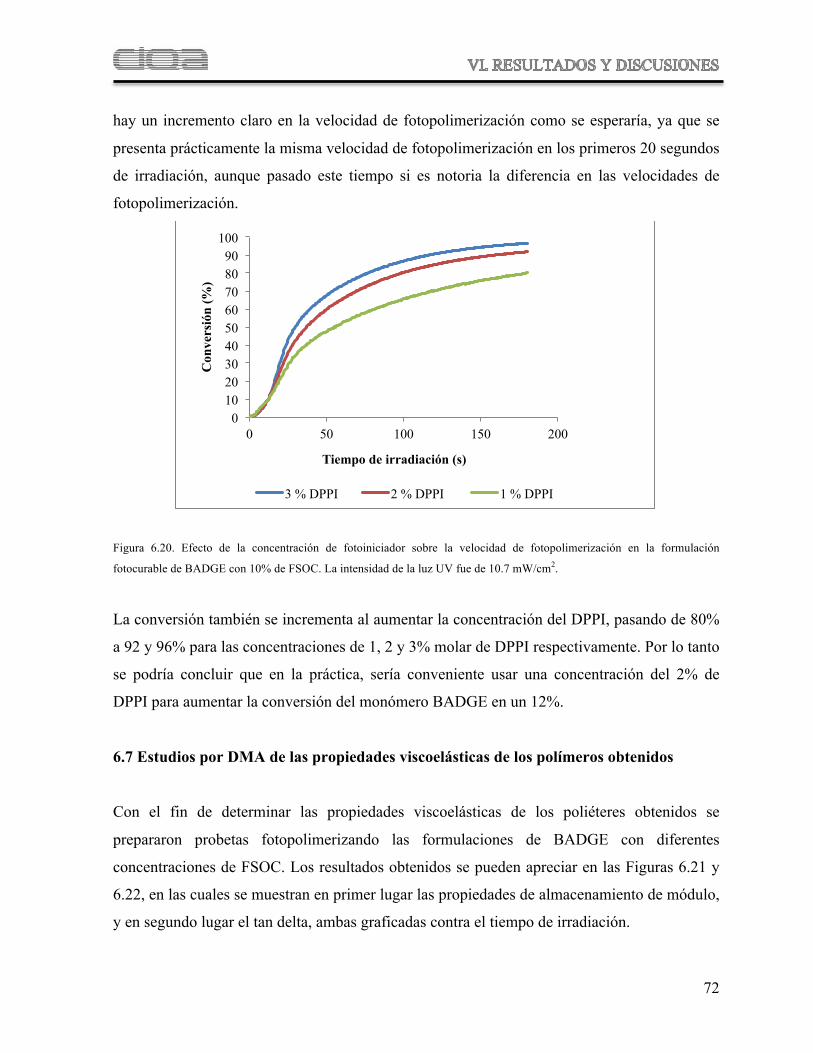
72
hay un incremento claro en la velocidad de fotopolimerización como se esperaría, ya que se
presenta prácticamente la misma velocidad de fotopolimerización en los primeros 20 segundos
de irradiación, aunque pasado este tiempo si es notoria la diferencia en las velocidades de
fotopolimerización.
Figura 6.20. Efecto de la concentración de fotoiniciador sobre la velocidad de fotopolimerización en la formulación
fotocurable de BADGE con 10% de FSOC. La intensidad de la luz UV fue de 10.7 mW/cm2.
La conversión también se incrementa al aumentar la concentración del DPPI, pasando de 80%
a 92 y 96% para las concentraciones de 1, 2 y 3% molar de DPPI respectivamente. Por lo tanto
se podría concluir que en la práctica, sería conveniente usar una concentración del 2% de
DPPI para aumentar la conversión del monómero BADGE en un 12%.
6.7 Estudios por DMA de las propiedades viscoelásticas de los polímeros obtenidos
Con el fin de determinar las propiedades viscoelásticas de los poliéteres obtenidos se
prepararon probetas fotopolimerizando las formulaciones de BADGE con diferentes
concentraciones de FSOC. Los resultados obtenidos se pueden apreciar en las Figuras 6.21 y
6.22, en las cuales se muestran en primer lugar las propiedades de almacenamiento de módulo,
y en segundo lugar el tan delta, ambas graficadas contra el tiempo de irradiación.
0 10 20 30 40 50 60 70 80 90
100
0 50 100 150 200
Con
vers
ión
(%)
Tiempo de irradiación (s)
3 % DPPI 2 % DPPI 1 % DPPI
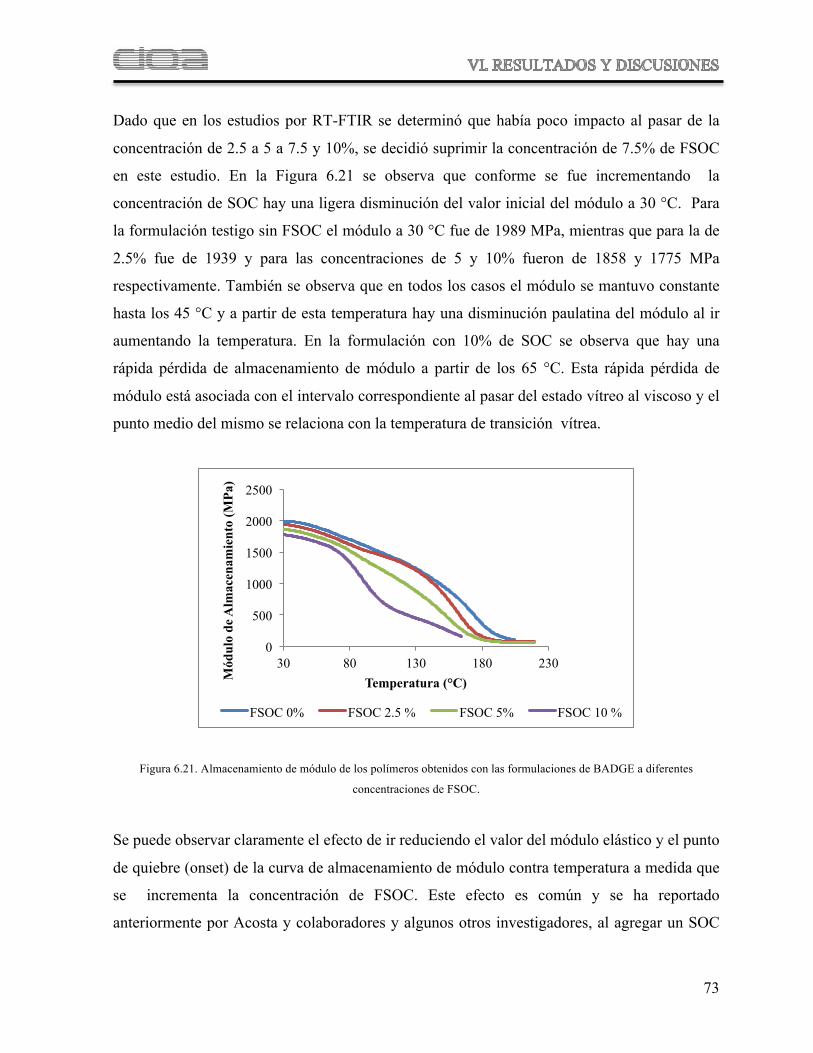
73
Dado que en los estudios por RT-FTIR se determinó que había poco impacto al pasar de la
concentración de 2.5 a 5 a 7.5 y 10%, se decidió suprimir la concentración de 7.5% de FSOC
en este estudio. En la Figura 6.21 se observa que conforme se fue incrementando la
concentración de SOC hay una ligera disminución del valor inicial del módulo a 30 °C. Para
la formulación testigo sin FSOC el módulo a 30 °C fue de 1989 MPa, mientras que para la de
2.5% fue de 1939 y para las concentraciones de 5 y 10% fueron de 1858 y 1775 MPa
respectivamente. También se observa que en todos los casos el módulo se mantuvo constante
hasta los 45 °C y a partir de esta temperatura hay una disminución paulatina del módulo al ir
aumentando la temperatura. En la formulación con 10% de SOC se observa que hay una
rápida pérdida de almacenamiento de módulo a partir de los 65 °C. Esta rápida pérdida de
módulo está asociada con el intervalo correspondiente al pasar del estado vítreo al viscoso y el
punto medio del mismo se relaciona con la temperatura de transición vítrea.
Figura 6.21. Almacenamiento de módulo de los polímeros obtenidos con las formulaciones de BADGE a diferentes
concentraciones de FSOC.
Se puede observar claramente el efecto de ir reduciendo el valor del módulo elástico y el punto
de quiebre (onset) de la curva de almacenamiento de módulo contra temperatura a medida que
se incrementa la concentración de FSOC. Este efecto es común y se ha reportado
anteriormente por Acosta y colaboradores y algunos otros investigadores, al agregar un SOC
0
500
1000
1500
2000
2500
30 80 130 180 230
Mód
ulo
de A
lmac
enam
ient
o (M
Pa)
Temperatura (°C)
FSOC 0% FSOC 2.5 % FSOC 5% FSOC 10 %
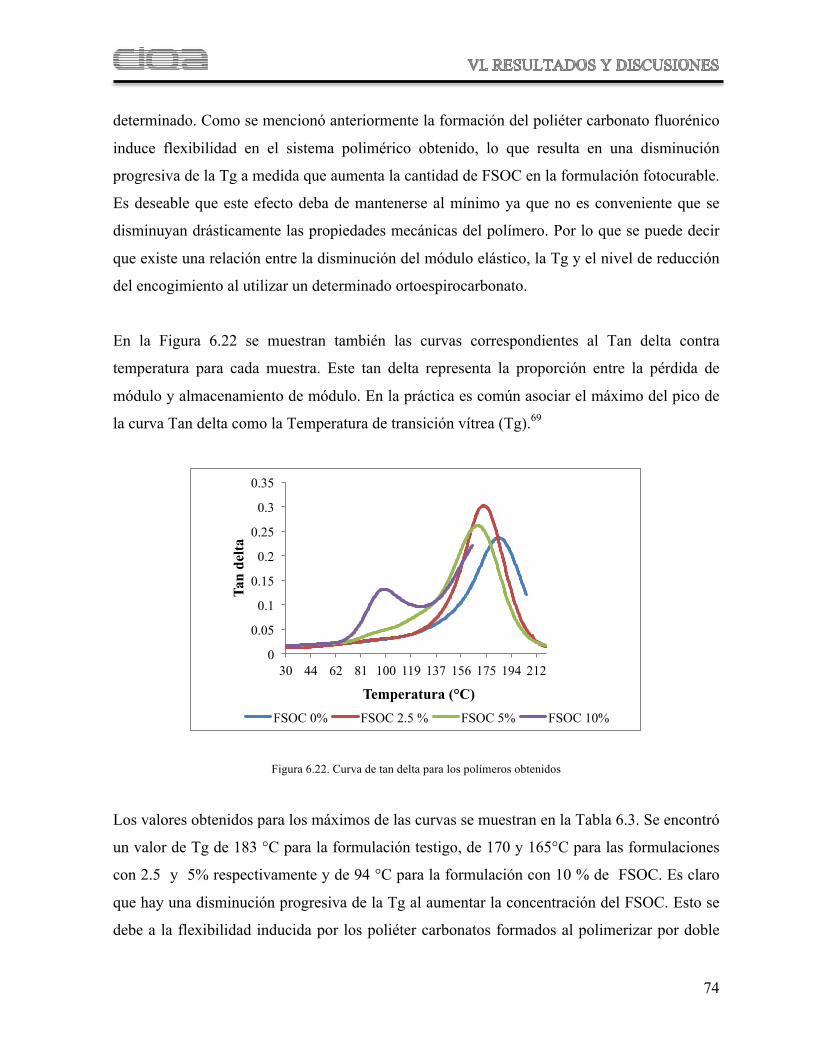
74
determinado. Como se mencionó anteriormente la formación del poliéter carbonato fluorénico
induce flexibilidad en el sistema polimérico obtenido, lo que resulta en una disminución
progresiva de la Tg a medida que aumenta la cantidad de FSOC en la formulación fotocurable.
Es deseable que este efecto deba de mantenerse al mínimo ya que no es conveniente que se
disminuyan drásticamente las propiedades mecánicas del polímero. Por lo que se puede decir
que existe una relación entre la disminución del módulo elástico, la Tg y el nivel de reducción
del encogimiento al utilizar un determinado ortoespirocarbonato.
En la Figura 6.22 se muestran también las curvas correspondientes al Tan delta contra
temperatura para cada muestra. Este tan delta representa la proporción entre la pérdida de
módulo y almacenamiento de módulo. En la práctica es común asociar el máximo del pico de
la curva Tan delta como la Temperatura de transición vítrea (Tg).69
Figura 6.22. Curva de tan delta para los polímeros obtenidos
Los valores obtenidos para los máximos de las curvas se muestran en la Tabla 6.3. Se encontró
un valor de Tg de 183 °C para la formulación testigo, de 170 y 165°C para las formulaciones
con 2.5 y 5% respectivamente y de 94 °C para la formulación con 10 % de FSOC. Es claro
que hay una disminución progresiva de la Tg al aumentar la concentración del FSOC. Esto se
debe a la flexibilidad inducida por los poliéter carbonatos formados al polimerizar por doble
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
30 44 62 81 100 119 137 156 175 194 212
Tan
delta
Temperatura (°C) FSOC 0% FSOC 2.5 % FSOC 5% FSOC 10%

75
apertura de anillo del FSOC. Adicionalmente, la disminución más pronunciada de la Tg al
agregar el 10% de FSOC también puede atribuirse al efecto estérico del FSOC ya que esta es
una molécula voluminosa. Se ha reportado que entre más voluminoso sea el SOC es más
importante la disminución en la Tg.70
Tabla 6.3. Temperaturas de transición vítrea (Tg) obtenidas por DMA para los polímeros
formulados con FSOC
Muestra Tg de los polímeros
obtenidos (°C)
BADGE 183
BADGE + 2.5 % FSOC 170
BADGE + 5 % FSOC 165
BADGE + 7.5 % FSOC ---------
BADGE + 10 % FSOC 94
6.8 Determinación de la Tg por DSC de las formulaciones BADGE-FSOC
En las Figuras 6.23 a, b, c, d y e., se muestran los termogramas obtenidos para los poliéteres
derivados de las formulaciones fotocurables de BADGE con diferentes concentraciones de
FSOC.
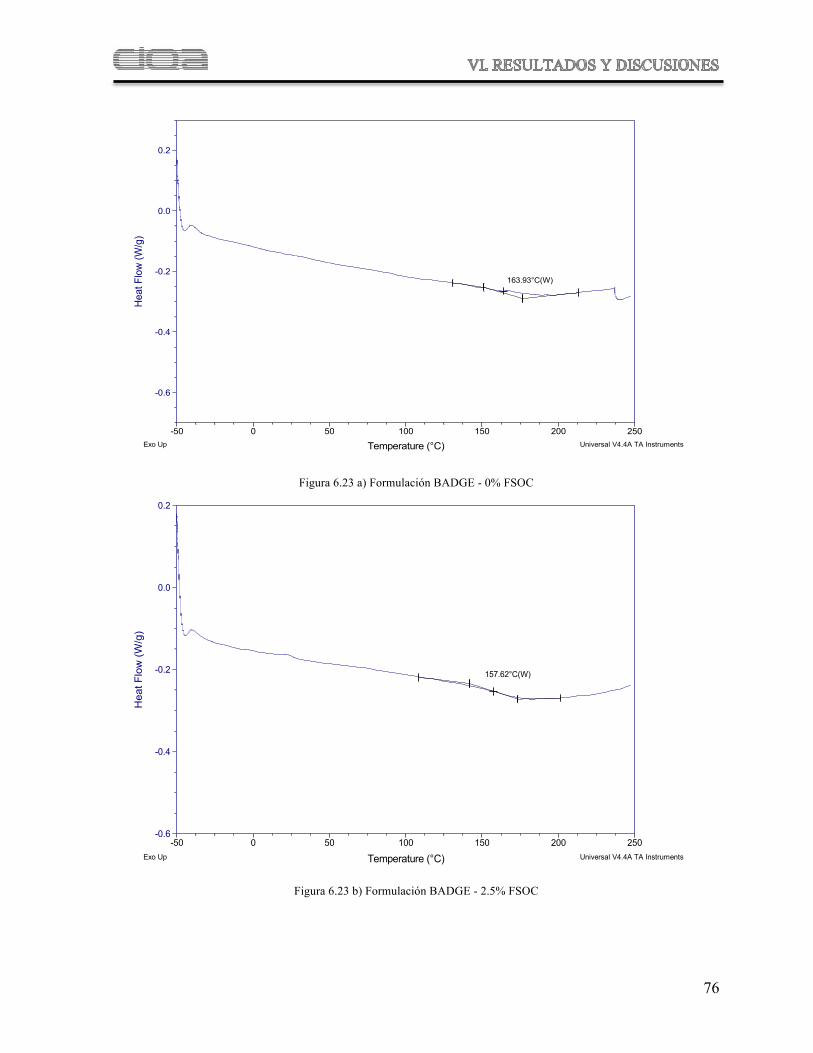
76
Figura 6.23 a) Formulación BADGE - 0% FSOC
Figura 6.23 b) Formulación BADGE - 2.5% FSOC
163.93°C(W)
-0.6
-0.4
-0.2
0.0
0.2H
eat F
low
(W/g
)
-50 0 50 100 150 200 250
Temperature (°C)
Sample: 0% FSOCSize: 8.9000 mgMethod: CAL/CALComment: R=10C/MIN ATM= N2
DSCFile: A:\M1220FSO.FR0Operator: G. MENDEZRun Date: 16-May-2011 10:43Instrument: 2920 MDSC V2.3A
Exo Up Universal V4.4A TA Instruments
157.62°C(W)
-0.6
-0.4
-0.2
0.0
0.2
Hea
t Flo
w (W
/g)
-50 0 50 100 150 200 250
Temperature (°C)
Sample: 2.5% FSOCSize: 10.5000 mgMethod: CAL/CALComment: R=10C/MIN ATM= N2
DSCFile: A:\M12225FS.R25Operator: G. MENDEZRun Date: 23-May-2011 13:47Instrument: 2920 MDSC V2.3A
Exo Up Universal V4.4A TA Instruments
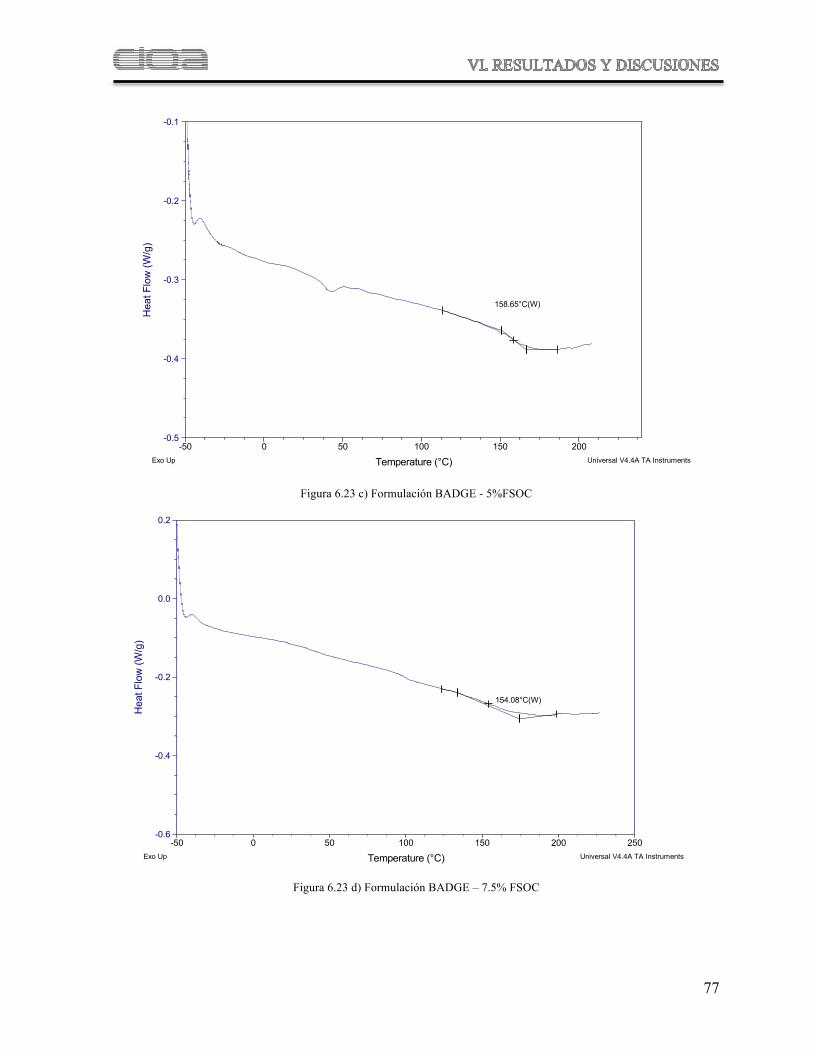
77
Figura 6.23 c) Formulación BADGE - 5%FSOC
Figura 6.23 d) Formulación BADGE – 7.5% FSOC
158.65°C(W)
-0.5
-0.4
-0.3
-0.2
-0.1
Hea
t Flo
w (W
/g)
-50 0 50 100 150 200
Temperature (°C)
Sample: 5% FSOCSize: 12.3000 mgMethod: CAL/CALComment: R=10C/MIN ATM= N2
DSCFile: A:\M1225FSO.F5ROperator: G. MENDEZRun Date: 16-May-2011 17:13Instrument: 2920 MDSC V2.3A
Exo Up Universal V4.4A TA Instruments
154.08°C(W)
-0.6
-0.4
-0.2
0.0
0.2
Hea
t Flo
w (W
/g)
-50 0 50 100 150 200 250
Temperature (°C)
Sample: 7.8% FSOCSize: 12.3000 mgMethod: CAL/CALComment: R=10C/MIN ATM= N2
DSCFile: A:\M12275FS.75FOperator: G. MENDEZRun Date: 16-May-2011 12:47Instrument: 2920 MDSC V2.3A
Exo Up Universal V4.4A TA Instruments
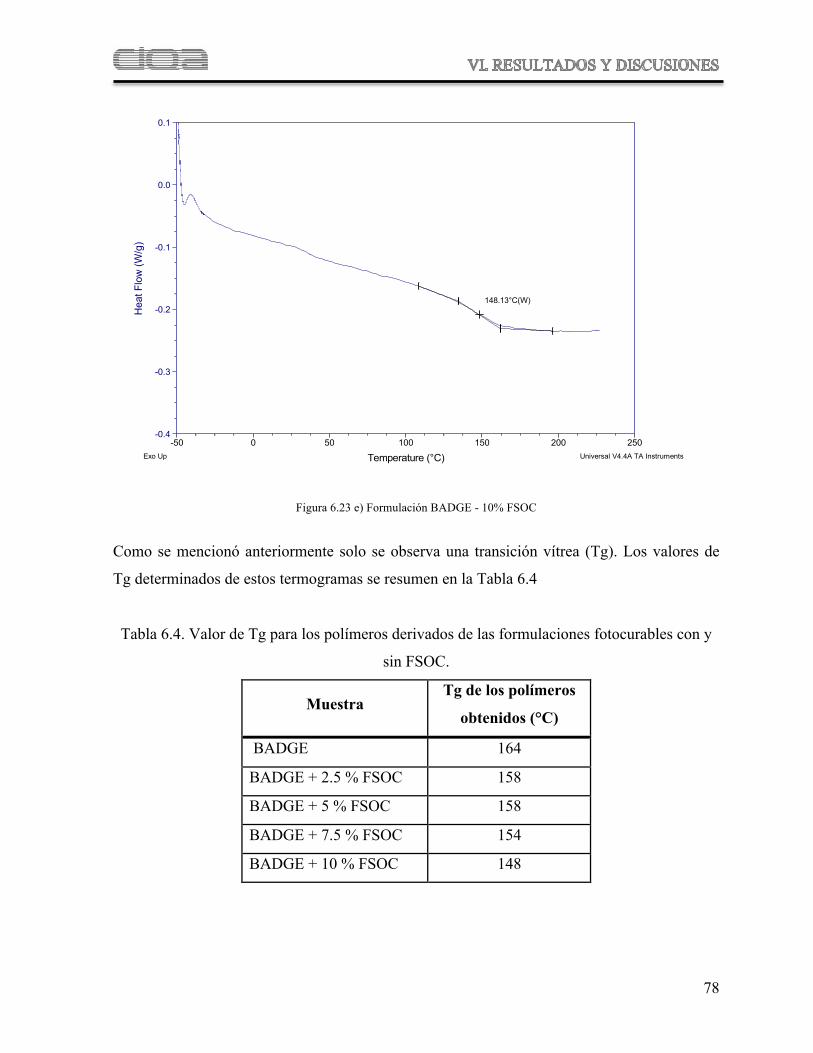
78
Figura 6.23 e) Formulación BADGE - 10% FSOC
Como se mencionó anteriormente solo se observa una transición vítrea (Tg). Los valores de
Tg determinados de estos termogramas se resumen en la Tabla 6.4
Tabla 6.4. Valor de Tg para los polímeros derivados de las formulaciones fotocurables con y
sin FSOC.
Muestra Tg de los polímeros
obtenidos (°C)
BADGE 164
BADGE + 2.5 % FSOC 158
BADGE + 5 % FSOC 158
BADGE + 7.5 % FSOC 154
BADGE + 10 % FSOC 148
148.13°C(W)
-0.4
-0.3
-0.2
-0.1
0.0
0.1
Hea
t Flo
w (W
/g)
-50 0 50 100 150 200 250
Temperature (°C)
Sample: 10% FSOCSize: 10.3000 mgMethod: CAL/CALComment: R=10C/MIN ATM= N2
DSCFile: A:\M1221OFS.F10Operator: G. MENDEZRun Date: 17-May-2011 11:22Instrument: 2920 MDSC V2.3A
Exo Up Universal V4.4A TA Instruments

79
Al igual que en los valores de Tg determinados por DMA se observa una disminución
progresiva al ir incrementando la concentración de FSOC en la formulación fotocurable. Se ha
reportado que es normal encontrar una diferencia de entre 20 a 40 °C de diferencia entre la Tg
obtenida por DMA y la obtenida por DSC. Esto se atribuye el efecto de la frecuencia.71
6.9 Determinación del % de encogimiento de las formulaciones fotocurables de BADGE
con FSOC
En la Tabla 6.5 se muestran los valores de encogimiento resultantes de la determinación de la
densidad de la formulación fotocurable y de la densidad de las probetas fotocuradas a
diferentes proporciones de FSOC.
Tabla 6.5. Valores de % de encogimiento encontrados para las diferentes formulaciones
BADGE-FSOC.
Muestra % de encogimiento
BADGE -4.77
BADGE + 2.5 % FSOC -2.42
BADGE + 5 % FSOC -1.94
BADGE + 7.5 % FSOC -1.56
BADGE + 10 % FSOC 0.19
Se encontró que al ir incrementando la concentración de FSOC hay una disminución
progresiva del encogimiento. El encogimiento del sistema fotocurable testigo sin FSOC
exhibió un nivel de encogimiento de -4.77%, el cual se encuentra dentro del intervalo
reportado para polímeros derivados de monómeros epóxicos que es de -4 a -6%. Se puede
observar que a la concentración del 10% de FSOC no solo se eliminó totalmente el
encogimiento sino que incluso se llegó a observar un ligero nivel de expansión de 0.19%.
Como se mencionó anteriormente el comportamiento antiencogimiento del FSOC se atribuye
por un lado a la formación del poliéter carbonato derivado de la doble apertura de anillo del

80
FSOC, y por otro lado al volumen estérico inducido por los grupos voluminosos fluorénicos
localizados en el polímero poliéter carbonato, lo cual impide la compactación adecuada de las
cadenas poliméricas.
De esta manera podemos decir en base a los resultados obtenidos que el agente
antiencogimiento preparado en este trabajo de tesis mostró una alta eficiencia en eliminar el
encogimiento en la fotopolimerización catiónica del monómero BADGE, que es uno de los
más usados a nivel industrial. En trabajos anteriormente reportados por Sangermano y
Acosta37,38 en los que se sintetizaron SOCs derivados de la glicerina y evaluados en resinas
epóxicas se encontró que aunque hubo una reducción considerable del encogimiento, no se
eliminó totalmente, aun a concentraciones hasta del 20% del SOC. Por lo tanto el FSOC
mostró una eficiencia superior a la de otros SOCs reportados anteriormente.
6.10 Estudio de estabilidad térmica por TGA de los polímeros preparados con BADGE Y
FSOC
Se decidió estudiar la estabilidad térmica de los polímeros sintetizados en este estudio dado
que se conoce que los policarbonatos pueden sufrir degradación tanto termoxidativa como
fotooxidativa. La presencia de grupos carbonato puede acelerar la degradación de una cadena
polimérica.72
En este caso se midió la estabilidad térmica usando la técnica de análisis termogravimétrico
(TGA). Los resultados se muestran en la Figura 6.24. Se puede observar que todas las
muestras incluído el blanco, mostraron excelente estabilidad térmica soportando temperaturas
de hasta 350 °C antes de que empiece el proceso de pérdida en peso debido a la degradación
del material polimérico. Se observa también que en todos los casos el proceso de degradación
se lleva a cabo en dos etapas, la primera en la cual el polímero alcanza temperaturas alrededor
de 350-360 °C sin sufrir una aparente pérdida en peso, a partir de la cual se observa una rápida
disminución de peso de alrededor del 80-85%, y la segunda etapa a partir de los 450 °C se
observa que el residuo obtenido después de la degradación térmica es altamente estable
soportando temperaturas de hasta 600 °C sin observarse degradación aparente. Sin embargo a
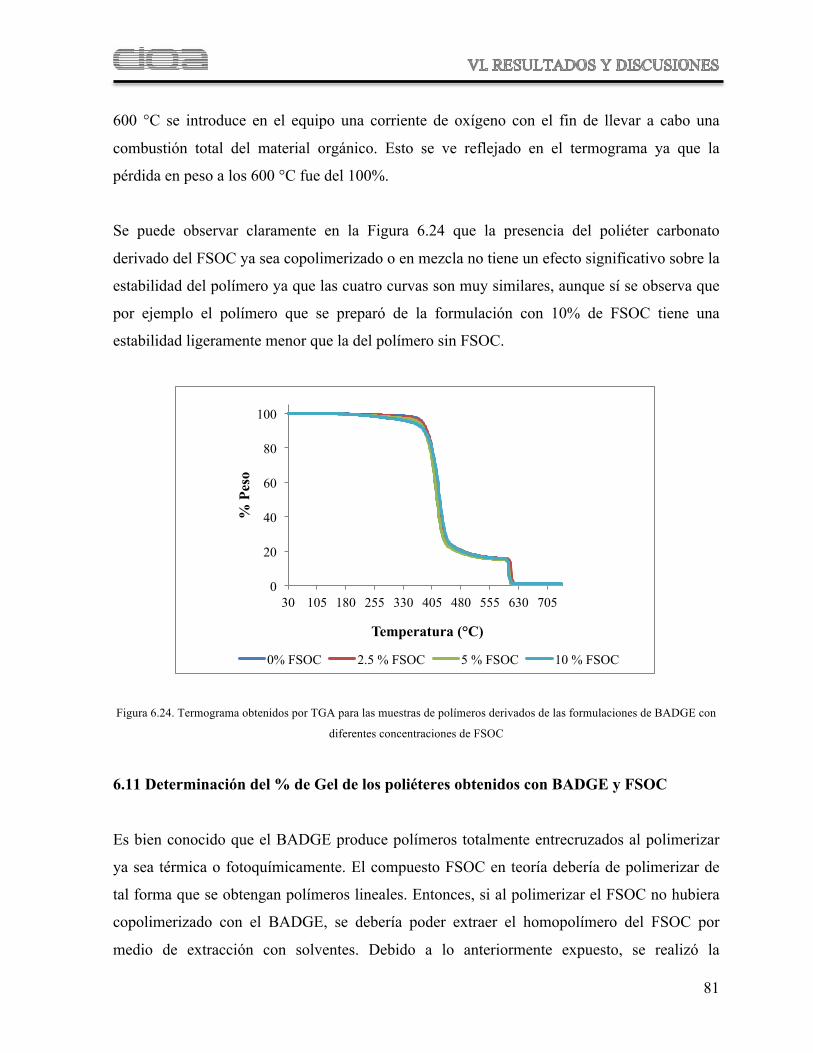
81
600 °C se introduce en el equipo una corriente de oxígeno con el fin de llevar a cabo una
combustión total del material orgánico. Esto se ve reflejado en el termograma ya que la
pérdida en peso a los 600 °C fue del 100%.
Se puede observar claramente en la Figura 6.24 que la presencia del poliéter carbonato
derivado del FSOC ya sea copolimerizado o en mezcla no tiene un efecto significativo sobre la
estabilidad del polímero ya que las cuatro curvas son muy similares, aunque sí se observa que
por ejemplo el polímero que se preparó de la formulación con 10% de FSOC tiene una
estabilidad ligeramente menor que la del polímero sin FSOC.
Figura 6.24. Termograma obtenidos por TGA para las muestras de polímeros derivados de las formulaciones de BADGE con
diferentes concentraciones de FSOC
6.11 Determinación del % de Gel de los poliéteres obtenidos con BADGE y FSOC
Es bien conocido que el BADGE produce polímeros totalmente entrecruzados al polimerizar
ya sea térmica o fotoquímicamente. El compuesto FSOC en teoría debería de polimerizar de
tal forma que se obtengan polímeros lineales. Entonces, si al polimerizar el FSOC no hubiera
copolimerizado con el BADGE, se debería poder extraer el homopolímero del FSOC por
medio de extracción con solventes. Debido a lo anteriormente expuesto, se realizó la
0
20
40
60
80
100
30 105 180 255 330 405 480 555 630 705
% P
eso
Temperatura (°C)
0% FSOC 2.5 % FSOC 5 % FSOC 10 % FSOC

82
determinación del porcentaje de gel para determinar si hubo material soluble ya fuera un
homopolímero del FSOC o monómero sin reaccionar. La muestra de polímero se sometió a
una extracción con cloroformo por espacio de 24 h a temperatura ambiente encontrándose
resultados mostrados en la Tabla 6.6.
Se encontró que en todos los casos el porcentaje de gel fue casi de 100% por lo que se puede
deducir de estos resultados que tanto el monómero BADGE como el FSOC, bajo las
condiciones de reacción empleadas, copolimerizaron consumiéndose totalmente ambos
monómeros. Al correlacionar estos datos con los obtenidos por RT-FTIR podríamos
argumentar que aunque en esos resultados se obtuvieron conversiones de alrededor del 80%,
esas mediciones se realizaron en un intervalo de 180 segundos, mientras que cuando se
realizaron las probetas en el horno de luz UV para llevar a cabo la determinación de % de gel,
el tiempo de irradiación fue de 15 minutos. También hay que hacer notar que en el caso de la
determinación de cinéticas por RT-FTIR, la intensidad de la luz UV fue de 10 mW/cm2,
mientras que la lámpara del horno en que se prepararon las probetas tiene una intensidad de
300 W. Por lo tanto las condiciones de mayor tiempo e intensidad de luz UV debieron de
haber promovido conversiones entre 95 y 98%. Otra explicación adecuada, es que hay que
considerar que algunos grupos funcionales en este caso grupos epóxicos del BADGE, pueden
quedar atrapados en la red entrecruzada tridimensional por lo que aun y cuando en la
determinación de las cinéticas de fotopolimerización la conversión no sea casi del 100%, el
monómero se ha consumido al 100% y por lo tanto no hay material que pueda ser extraído.
Tabla 6.6. Valores de % de gel obtenidos con BADGE y FSOC.
Muestra % de gel
BADGE 99.7
BADGE + 2.5 % FSOC 99.6
BADGE + 5 % FSOC 98.4
BADGE + 7.5 % FSOC 99.5
BADGE + 10 % FSOC 99.6

83
6.12 Estudio de Fotopolimerización de monómeros epóxicos formulados con FSOC
utilizando luz visible
Como parte de un convenio de cooperación con el Profesor. Marco Sangermano del
Politecnico de Torino, en Turín Italia, se decidió realizar un estudio para evaluar la eficiencia
del compuesto FSOC en polímeros obtenidos al fotopolimerizar monómeros epóxicos con luz
visible. Actualmente, existe una tendencia a utilizar la luz visible para llevar a cabo
fotopolimerizaciones a nivel industrial. Además, se podría pensar que el compuesto FSOC
podría utilizarse como agente antiencogimiento para la aplicación de resinas dentales en las
cuales se utiliza luz visible derivada de lámparas de halógeno ó de lámparas de LED de luz
azul. Aunque la luz UV es más energética y puede resultar en fotopolimerizaciones más
rápidas, con mayor conversión y mayor productividad en procesos fotocurables, también
presenta desventajas sobre todo la generación de ozono y por motivos de seguridad de los
operarios.
Por esta razón el autor de esta tesis realizó una estancia de investigación de un mes en el
Politécnico de Torino. En esta estancia se buscó diseñar formulaciones fotocurables utilizando
tanto el BADGE como el monómero 3,4-Epoxiciclohexil metil, 3´, 4´-epoxiciclohexan
carboxilato (3,4-EP) para ser fotopolimerizados cationicamente usando el compuesto FSOC
como agente antiencogimiento.
Se empezó por utilizar el mismo monómero BADGE con diferentes concentraciones de
FSOC y DPPI y se irradió con una lámpara de tipo LED con máximo de absorción a 415 nm.
Se encontró que no hubo polimerización y esto se debe a que la sal de yodonio como se
mencionó anteriormente tiene un intervalo de absorción entre 200 y 300 nm. Entonces debido
a que no hubo absorción de energía por la sal de yodonio, no ocurrió la fotopolimerización.
Se decidió entonces agregar un fotosensibilizador que en este caso fue la canforquinona la cual
tiene una absorción de 0.1294 al pico de 244 nm y de 0.0499 al pico de 466 nm (ver Figura
6.25), en conjunto con la sal de diariliodonio utilizando el mismo monómero BADGE.
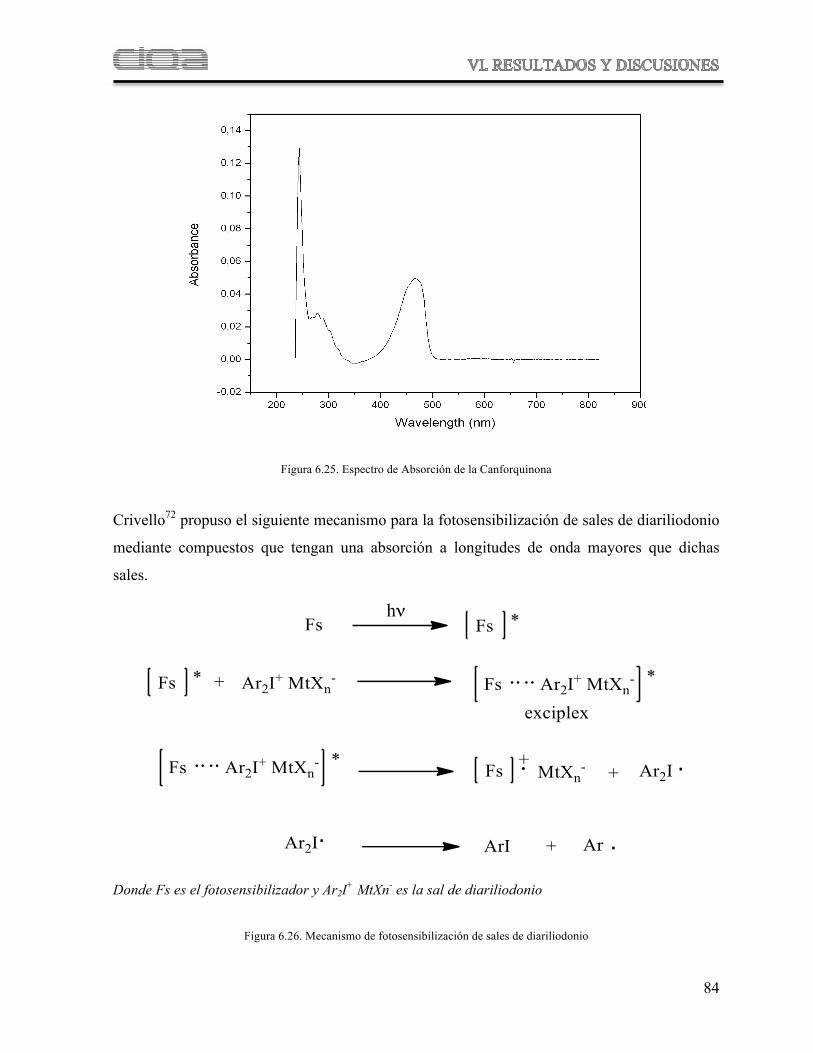
84
Figura 6.25. Espectro de Absorción de la Canforquinona
Crivello72 propuso el siguiente mecanismo para la fotosensibilización de sales de diariliodonio
mediante compuestos que tengan una absorción a longitudes de onda mayores que dichas
sales.
Donde Fs es el fotosensibilizador y Ar2I+ MtXn- es la sal de diariliodonio
Figura 6.26. Mecanismo de fotosensibilización de sales de diariliodonio
!
! !
h"

85
La fotosensibilización procede por transferencia de electrones; la cual involucra la absorción
de luz por el fotosensibilizador para originar la correspondiente especie excitada [Fs]*. Esta
especie reacciona con la sal de yodonio para generar un complejo de tipo exciplex. Después,
dentro de este complejo ocurre una transferencia de electrón del fotosensibilizador a la sal de
diariliodonio, reduciendo ésta a un radical Ar2I.. Esta especie a su vez se descompone
produciendo yodobenceno y un radical arilo, lo que hace que la fotosensibilización sea
irreversible. El radical catión generado puede por si mismo iniciar la fotopolimerización
catiónica o esta especie puede generar un super ácido al reaccionar ya sea con impurezas o
especies ácidas.
Se encontró que tampoco usando este sistema de iniciación polimerizó el monómero BADGE
por lo que se decidió cambiar entonces de monómero utilizando el 3,4-EP.
6.13 Determinación de las cinéticas de fotopolimerización catiónica de las formulaciones
3,4-EP con diferentes concentraciones de FSOC.
Se realizaron pruebas preliminares y se encontró que mediante el sistema de fotoiniciación
canforquinona-sal de yodonio, sí se llevaba a cabo la fotopolimerización del monómero 3,4-
EP, obteniendo materiales sólidos. Se determinaron las cinéticas de fotopolimerización con un
sistema discontinuo de irradiación, irradiando la muestra por intervalos de 20 segundos. El
procedimiento consistió en determinar el espectro de FTIR de la formulación fotocurable antes
de ser irradiada con la luz azul determinando la absorbancia de los picos de interés, en este
caso el pico a 790 cm-1 que corresponde al pico de los grupos epóxidos. Después la
formulación fotocurable se adherió a una pastilla de silicio (transparente en la región de IR)
para su posterior irradiación con la luz del LED el cual tiene una intensidad de 600 mW/cm2.
La muestra se irradió por 20 segundos y después se determinó nuevamente la absorbancia del
pico anteriormente mencionado. Esta operación se repitió hasta completar 120 segundos. Las
curvas de conversión contra tiempo obtenidas para las formulaciones con diferentes
concentraciones de FSOC se muestran en la Figura 6.27 y se calcularon usando la siguiente
fórmula:
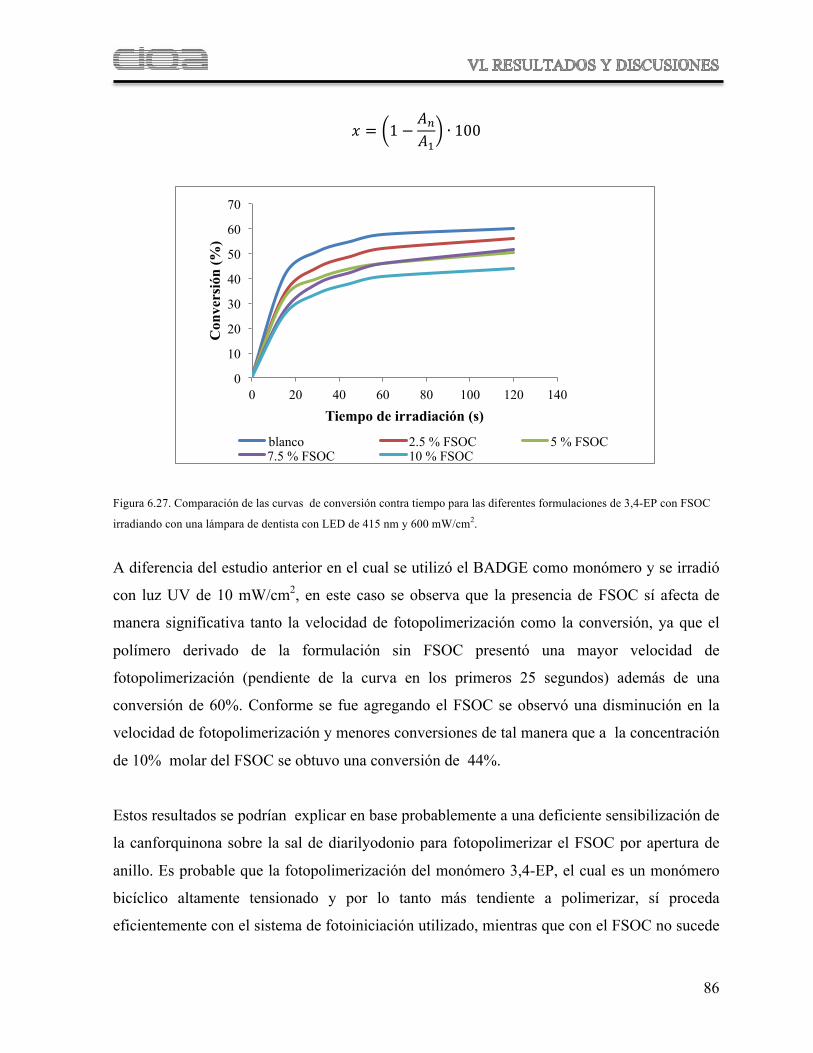
86
! ! !! !!!!! !""
Figura 6.27. Comparación de las curvas de conversión contra tiempo para las diferentes formulaciones de 3,4-EP con FSOC
irradiando con una lámpara de dentista con LED de 415 nm y 600 mW/cm2.
A diferencia del estudio anterior en el cual se utilizó el BADGE como monómero y se irradió
con luz UV de 10 mW/cm2, en este caso se observa que la presencia de FSOC sí afecta de
manera significativa tanto la velocidad de fotopolimerización como la conversión, ya que el
polímero derivado de la formulación sin FSOC presentó una mayor velocidad de
fotopolimerización (pendiente de la curva en los primeros 25 segundos) además de una
conversión de 60%. Conforme se fue agregando el FSOC se observó una disminución en la
velocidad de fotopolimerización y menores conversiones de tal manera que a la concentración
de 10% molar del FSOC se obtuvo una conversión de 44%.
Estos resultados se podrían explicar en base probablemente a una deficiente sensibilización de
la canforquinona sobre la sal de diarilyodonio para fotopolimerizar el FSOC por apertura de
anillo. Es probable que la fotopolimerización del monómero 3,4-EP, el cual es un monómero
bicíclico altamente tensionado y por lo tanto más tendiente a polimerizar, sí proceda
eficientemente con el sistema de fotoiniciación utilizado, mientras que con el FSOC no sucede
0
10
20
30
40
50
60
70
0 20 40 60 80 100 120 140
Con
vers
ión
(%)
Tiempo de irradiación (s)
blanco 2.5 % FSOC 5 % FSOC 7.5 % FSOC 10 % FSOC

87
así. De esta manera se explicaría también que a diferencia del estudio con luz UV en el cual
aumentó la conversión al agregar el FSOC, en este caso hubo una disminución de la
conversión del monómero base.
Para investigar si se estaba llevando a cabo la fotosensibilización de la sal de yodonio por la
canforquinona utilizando luz azul de una lámpara de LED, se preparó una solución en
cloroformo de 200 mg de FSOC con 5 mg de DPPI y 5 mg de canforquinona y después se
irradió con la lámpara LED por 15 minutos. La solución obtenida se analizó por
espectroscopía de FTIR y se comparó contra el espectro obtenido del experimento de
fotopolimerización del FSOC utilizando DPPI y luz UV para iniciar la polimerización. Los
resultados se muestran en la Figura 6.28.
Es claro al observar el espectro que sí está ocurriendo la polimerización por apertura de anillo,
ya que se está formando el poliétercarbonato y esto indica que la fotosensibilización por la
canforquinona sí se está llevando a cabo. Sin embargo, se puede ver que el espectro derivado
de la fotosensibilizacion con canforquinona esta en menor concentración que el obtenido por
luz UV. Es probable entonces que la cantidad de super ácido generado por la fotólisis de la
sal de diariliodonio, es menor que en el caso de cuando se usó la luz UV. Al haber menos
cantidad de ácido y debido a la menor reactividad del FSOC en comparación con el 3,4-EP,
debe haber una competición para reaccionar con el super ácido generado, pero dado que el
FSOC debe de polimerizar más lentamente y con menor conversión, esto afecta directamente a
la reactividad total del sistema fotocurable del 3,4-EP con FSOC.
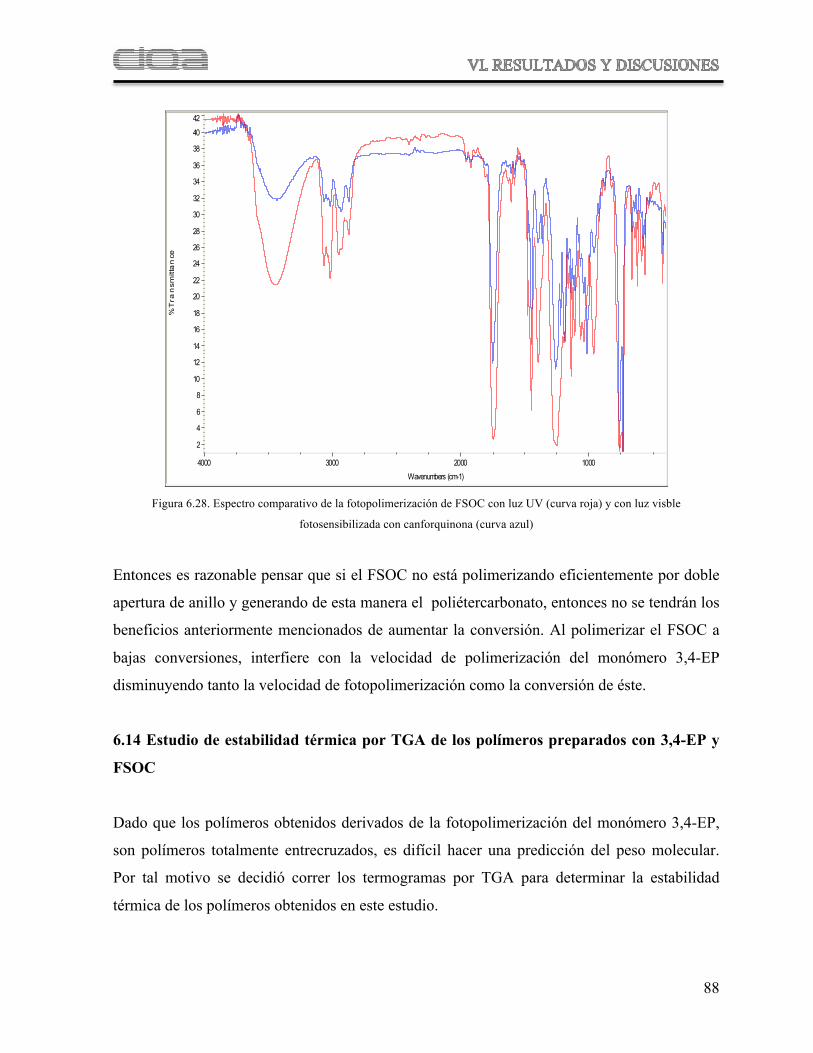
88
Figura 6.28. Espectro comparativo de la fotopolimerización de FSOC con luz UV (curva roja) y con luz visble
fotosensibilizada con canforquinona (curva azul)
Entonces es razonable pensar que si el FSOC no está polimerizando eficientemente por doble
apertura de anillo y generando de esta manera el poliétercarbonato, entonces no se tendrán los
beneficios anteriormente mencionados de aumentar la conversión. Al polimerizar el FSOC a
bajas conversiones, interfiere con la velocidad de polimerización del monómero 3,4-EP
disminuyendo tanto la velocidad de fotopolimerización como la conversión de éste.
6.14 Estudio de estabilidad térmica por TGA de los polímeros preparados con 3,4-EP y
FSOC
Dado que los polímeros obtenidos derivados de la fotopolimerización del monómero 3,4-EP,
son polímeros totalmente entrecruzados, es difícil hacer una predicción del peso molecular.
Por tal motivo se decidió correr los termogramas por TGA para determinar la estabilidad
térmica de los polímeros obtenidos en este estudio.
2
4
6
8
10
12
14
16
18
20
22
24
26
28
30
32
34
36
38
40
42
%T
ran
smitt
an
ce
1000 2000 3000 4000 Wavenumbers (cm-1)

89
En la Figura 6.29 se muestran los resultados de las diferentes curvas obtenidas para los
polímeros derivados de las formulaciones de 3,4-EP con 0, 2.5, 5, 7.5 y 10% molar de FSOC.
Se puede observar que en este caso la estabilidad térmica de los polímeros en todos los casos
fue muy similar. Sin embargo, se encontró que los poliéteres derivados del monómero 3,4-EP
son menos estables que los del BADGE ya que soportan temperaturas de 175 °C antes de que
el polímero empiece a degradarse con la consecuente pérdida en peso del polímero. A
diferencia de los polímeros del BADGE en los que la degradación se llevó solo en dos etapas,
en el caso del 3,4-EP se llevó a cabo en cuatro etapas. Esto podría explicarse asignando las
diferentes curvas de degradación a zonas con mayor o menor grado de entrecruzamiento de tal
manera que las zonas del polímero que tuvieran menor grado de entrecruzamiento se
degradarían a temperaturas más bajas y las zonas con mayor grado de entrecruzamiento lo
harían a temperaturas más altas.
Figura 6.29. Comparación de las curvas de pérdida en peso contra temperatura de los polímeros derivados de las
formulaciones con diferentes concentraciones de FSOC

90
Dado que tanto la curva del blanco como la de los polímeros con FSOC son prácticamente
iguales podríamos deducir que el FSOC no interfiere negativamente con la estabilidad térmica
del poliéter obtenido en la fotopolimerizacion del monómero epóxico 3,4-EP.
6.15 Determinación del % de encogimiento de las formulaciones fotocurables de 3,4-EP
con FSOC
Al igual que en el caso del BADGE se realizaron las medidas del cambio de volumen al pasar
del estado viscoso de la formulación fotocurable al estado sólido del poliéter obtenido. En la
Tabla 6.7 se muestran los resultados obtenidos. Se encontró que el valor de encogimiento del
polímero sin FSOC fue de -4.92%, lo cual como ya se mencionó anteriormente está dentro del
intervalo esperado para polímeros obtenidos por fotopolimerización catiónica. Al introducir un
2.5% de FSOC en la formulación fotocurable esto resulta en una reducción en el encogimiento
del 17% con respecto al encogimiento del polímero sin el SOC. Al incrementar a 5% la
concentración de FSOC, la reducción del encogimiento es casi del 50% mientras que a la
concentración del 7.5% de FSOC la reducción del encogimiento es del 80%. Finalmente, a la
concentración del 10% de FSOC se observa no solo una eliminación total del encogimiento
sino que se obtuvo un ligero nivel de expansión de 0.78%. Estos resultados son análogos a los
obtenidos para el BADGE fotopolimerizando con luz UV ya que también se encontró un
ligero nivel de expansión al usar la concentración de 10 % de FSOC.
Aunque los resultados pudieran parecer conflictivos con respecto a los obtenidos en las curvas
cinéticas de conversión contra tiempo, al preparar las probetas necesarias para realizar el
análisis de cambio de volumen estas fueron primero irradiadas con luz azul proveniente de una
lámpara de LED y después se introdujeron en un horno a 80 °C por una hora, lo que
coadyuvaría a que la polimerización por doble apertura de anillo se llevara a cabo con mayor
conversión, promoviendo de esta manera que se redujera el encogimiento en la polimerización
del monómero 3,4-EP, lo cual se puede constatar que se llevó a cabo eficientemente
alcanzando inclusivo una ligera expansión
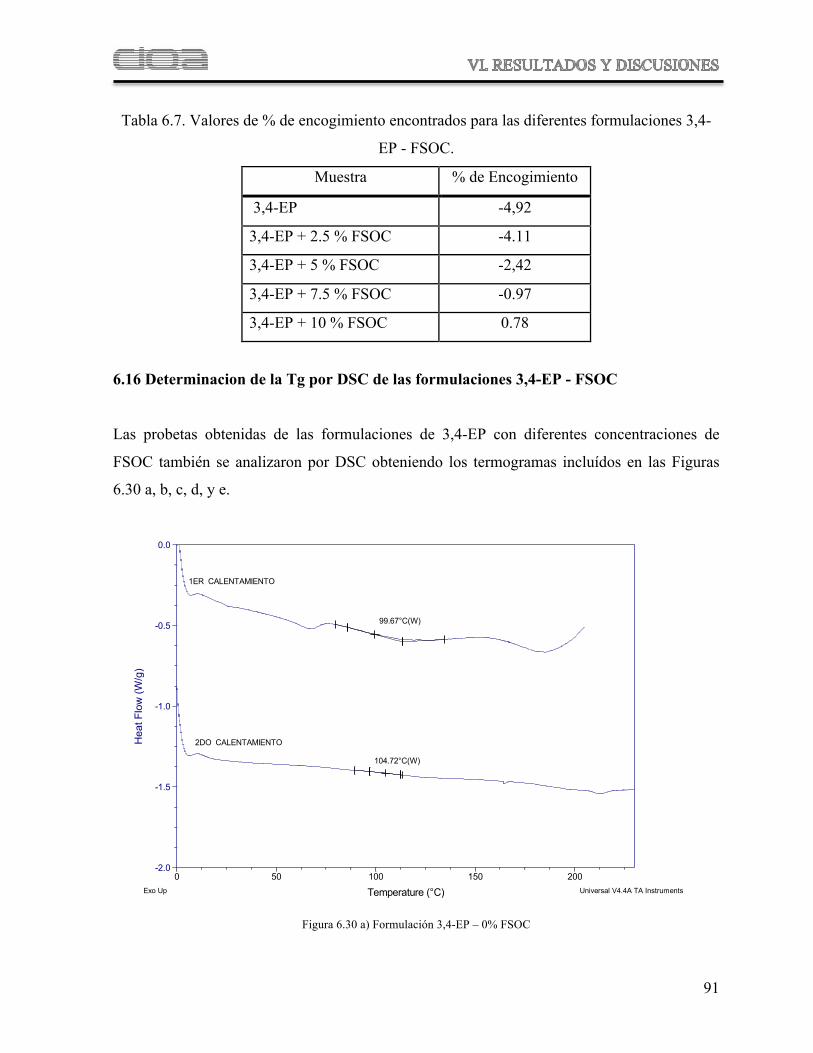
91
Tabla 6.7. Valores de % de encogimiento encontrados para las diferentes formulaciones 3,4-
EP - FSOC.
Muestra % de Encogimiento
3,4-EP -4,92
3,4-EP + 2.5 % FSOC -4.11
3,4-EP + 5 % FSOC -2,42
3,4-EP + 7.5 % FSOC -0.97
3,4-EP + 10 % FSOC 0.78
6.16 Determinacion de la Tg por DSC de las formulaciones 3,4-EP - FSOC
Las probetas obtenidas de las formulaciones de 3,4-EP con diferentes concentraciones de
FSOC también se analizaron por DSC obteniendo los termogramas incluídos en las Figuras
6.30 a, b, c, d, y e.
Figura 6.30 a) Formulación 3,4-EP – 0% FSOC
!
99.67°C(W)
104.72°C(W)
1ER CALENTAMIENTO
2DO CALENTAMIENTO
-2.0
-1.5
-1.0
-0.5
0.0
Hea
t Flo
w (W
/g)
0 50 100 150 200
Temperature (°C)
Sample: 0% FSOCSize: 9.5000 mgMethod: CAL/ENF/CALComment: R=10C/MIN ATM= N2 50 ML/MIN
DSCFile: A:\M2860FSO.S00Operator: G. MENDEZRun Date: 25-Aug-2011 17:09Instrument: 2920 MDSC V2.3A
Exo Up Universal V4.4A TA Instruments
92
Figura 6.30 b) Formulación 3,4-EP – 2.5% FSOC
Figura 6.30 c) Formulación 3,4-EP – 5% FSOC
!
101.11°C(W)
97.37°C(W)2DO CALENTAMIENTO
1ER CALENTAMIENTO
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
Hea
t Flo
w (W
/g)
0 50 100 150 200
Temperature (°C)
Sample: 2.5% FSOCSize: 7.7000 mgMethod: CAL/ENF/CALComment: R=10C/MIN ATM= N2 50 ML/MIN
DSCFile: A:\M286FO25.F25Operator: G. MENDEZRun Date: 26-Aug-2011 15:54Instrument: 2920 MDSC V2.3A
Exo Up Universal V4.4A TA Instruments
!
61.19°C(W)
95.53°C(W)
105.24°C(W)
ENFRIAMIENTO
1ER CALENTAMIENTO
2DO CALENTAMIENTO
-0.5
0.0
0.5
1.0
1.5
Hea
t Flo
w (W
/g)
0 50 100 150 200
Temperature (°C)
Sample: 5% FSOCSize: 9.9000 mgMethod: CAL/ENF/CALComment: R=10C/MIN ATM= N2 50 ML/MIN
DSCFile: A:\M286FOSO.F50Operator: G. MENDEZRun Date: 26-Aug-2011 14:25Instrument: 2920 MDSC V2.3A
Exo Up Universal V4.4A TA Instruments
93
Figura 6.30 d) Formulación 3,4-EP – 7.5% FSOC
Figura 6.30 e) Formulación 3,4-EP – 10% FSOC
!
92.73°C(W)
58.90°C(W)
101.33°C(W)
ENFRIAMIENTO
1ER CALENTAMIENTO
2DO CALENTAMIENTO
-0.3
0.2
0.7
1.2
1.7
Hea
t Flo
w (W
/g)
0 50 100 150 200
Temperature (°C)
Sample: 7.5% FSOCSize: 10.2000 mgMethod: CAL/ENF/CALComment: R=10C/MIN ATM= N2 50 ML/MIN
DSCFile: A:\M286FOS7.F75Operator: G. MENDEZRun Date: 26-Aug-2011 12:28Instrument: 2920 MDSC V2.3A
Exo Up Universal V4.4A TA Instruments
!
90.69°C(W)
87.32°C(W)
2DO CALENTAMIENTO
1ER CALENTAMIENTO
-0.4
-0.2
0.0
0.2
0.4
0.6
0.8
Heat
Flo
w (W
/g)
0 50 100 150 200
Temperature (°C)
Sample: 10% FSOCSize: 9.0000 mgMethod: CAL/ENF/CALComment: R=10C/MIN ATM= N2 50 ML/MIN
DSCFile: A:\M286FOS1.F10Operator: G. MENDEZRun Date: 26-Aug-2011 10:54Instrument: 2920 MDSC V2.3A
Exo Up Universal V4.4A TA Instruments

94
Los resultados de las Tg´s obtenidas se resumen en la Tabla 6.8. Se encontró una Tg de 104.7
°C para el poliéter obtenido sin FSOC, lo cual concuerda con el valor de 105 °C reportado
anteriormente en un estudio en el cual se obtuvo este mismo polímero derivado del 3,4-EP y
al cual se agregaron posteriormente diferentes concentraciones de SOC DIOL. En la Tabla 6.8
se puede observar como al ir agregando mayores concentraciones de FSOC hay una
disminución progresiva de la Tg. Sin embargo, en este caso la disminución no es tan notoria al
agregar el 10% de FSOC como en el caso del BADGE, ya que la Tg en esta caso fue de 87.32
°C lo que solo representa una disminución de 17.4 °C.
Tabla 6.8. Valor de Tg para los polímeros derivados de las formulaciones fotocurables de 3,4-
EP con y sin FSOC.
Muestra Tg de los polímeros
obtenidos (°C)
3,4-EP 104.72
3,4-EP + 2.5 % FSOC 97.37
3,4-EP + 5 % FSOC 95.53
3,4-EP + 7.5 % FSOC 92.73
3,4-EP + 10 % FSOC 87.32
En este caso no fue posible realizar los análisis de DMA durante la estancia en Italia debido al
poco tiempo que se tuvo (1 mes). Sin embargo, la disminución en los valores de Tg corroboran
definitivamente la presencia del FSOC, por lo que se debería tener una situación análoga a los
resultados obtenidos con luz UV, en los cuales hay una disminución del módulo y del
máximo del pico de tan delta.
6.17 Determinación del % de Gel de los poliéteres obtenidos con 3,4-EP y FSOC
Al igual que en el caso en el que se usó la luz UV y el monómero BADGE en este caso
también se determinó el porciento de gel para determinar cuánto material soluble se
encontraba presente en las muestras poliméricas obtenidas al fotopolimerizar con luz azul el

95
monómero 3,4-EP, con diferentes concentraciones de FSOC. Los resultados se muestran en la
Tabla 6.9.
Se observa que en todos los casos el porcentaje de material entrecruzado fue casi del 100%, lo
que nos indica que no había material extraíble en solventes, tales como monómeros residuales
u homopolímeros lineales.
Tabla 6.9. Valores de % de gel obtenidos con 3,4-EP y FSOC.
Muestra % de Gel
3,4-EP 100
3,4-EP + 2.5 % FSOC 100
3,4-EP + 5 % FSOC 99.9
3,4-EP + 7.5 % FSOC 99.9
3,4-EP + 10 % FSOC 99.9

96
7. CONCLUSIONES
1. Se sintetizó el FSOC con un rendimiento del 93% mediante una reacción de
transesterificación al hacer reaccionar el diol FDiOH con el tetraetil ortocarbonato.
2. Se logró incrementar la conversión del monómero BADGE al fotopolimerizar este en
presencia del FSOC (2.5-10% molar), desde un 74% para la formulación sin FSOC hasta un
80% con 10 % molar de FSOC. Esto se debe a la flexibilidad inducida por la formación del
poliéter carbonato originado por la doble apertura de anillo del FSOC, produciendo un retraso
en el punto de gelación que permite alcanzar mayores conversiones.
3. La presencia del FSOC en las formulaciones fotocurables origina polímeros cuya Tg va
disminuyendo conforme se aumenta su concentración debido al efecto de flexibilización. En
este caso la Tg (determinada por DMA) disminuyó desde 183 °C para el blanco hasta 94°C
para el polímero con 10% de FSOC.
4. El FSOC demostró una alta eficiencia como agente antiencogimiento ya que no solo
eliminó totalmente el encogimiento que fue de -4.77% para el polímero sin FSOC, sino que
alcanzó un ligero nivel de expansión de 0.19% para el polímero con 10% de FSOC.
5. La presencia del poliétercarbonato derivado del FSOC no propició una disminución
significativa de la estabilidad térmica en el poliéter derivado del BADGE.
6. Se encontró por luminescencia que el FSOC puede actuar como fotosensibilizador, pero a
altas concentraciones de DPPI, lo cual lo hace inviable en la práctica.
7. También se logró desarrollar una formulación fotocurable utilizando el monómero 3,4-EP
como monómero base con diferentes concentraciones de FSOC fotopolimerizando con luz
azul de una lámpara de LED. Se usó canforquinona y DPPI como sistema de fotoiniciación.

97
8. A diferencia del caso en el que se polimerizó el BADGE formulado con el FSOC
utilizando la luz UV, en el cual al aumentar la concentración del FSOC en la formulación, se
aumentaba la conversión, cuando se agregó el FSOC a las formulaciones con el monómero
3,4-EP en las que se usó el sistema de iniciación canforquinona-DPPI, al aumentar la
concentración del FSOC se observó una disminución de la conversión del monómero base.
Esto se atribuyó a que la fotólisis de la sal de yodonio promovida con luz azul y sensibilizada
por la canforquinona no es tan eficiente como la fotólisis directa de la sal de yodonio con luz
UV.
9. En resumen, el FSOC tiene un efecto positivo sobre la fotopolimerización del BADGE al
utilizar luz UV ya que elimina el encogimiento y aumenta la conversión final del polímero,
mientras que en la fotopolimerización del 3,4-EP con luz visible aunque la energía provista
por la lámpara de luz azul no es tan energética y por tanto provoca una deficiente
polimerización del FSOC, las características inherentes del FSOC pueden eliminar el
encogimiento y lograr aun un ligero nivel de expansión al someter la muestra de poliéter a un
postratamiento térmico de 120 °C por 1 hora.

98
8. REFERENCIAS
1. Bailey W.J. J. Macromol. Sci. Chem.; 1975, A9 (5): 849-865
2. Bailey, W. J.; Iwama, H.; Tsushima, R.; J. Polym. Sci. Symp. 1976, 56, 117
3. Sadhir, R.K.; Luck, R.M.; Expanding Monomers: Synthesis, characterization and Applications,
CRC Press, Boca Raton, FL, 1992.
4. Bailey, W.J.; Sun, R.L. Katsuki, T.; Endo, H.; Iwama, H.; Tsushima, R.; Saigou, K.; Bitritto,
M.M., Ring-opening polymerization with expansion in volume, in Ring-opening polymerization,
ACS Symp, #59, Saegusa T.; Goethals, E.Eds., American Chemical Society, Washington, DC
1977, chap.4
5. Odian, G.; Principles of polymerization, Wiley, New York, 2004
6. Wade, L.G.; Quimica Organica, Pearson Prentice Hill, Madrid, 2004, cap. 26
7. Nichols, F.S.; Flowers, R.G.; Ind Eng Chem 1950, 42, 292-299
8. Bailey, W.J.; Sun, R.L.; Am. Chem, Soc, div. Polym. Chem Prepr 1972, 13 (1), 400-402
9. Tobolsky, A.V.; Leonard, F.; Roeser, G.P.; J Polym Sci 1948, 3, 604-608
10. Sangermano, M.; Bongiovanni, R.; Malucelli, G. Priola, A.; in "New developments in cationic
photopolymerization: process and properties", in Horizons in Polymer Research, R.K. Bregg, Ed.,
Nova Science Publisher Inc. New York, 2006 p. 61
11. Moszner, N.; Salz, U.; Composites for Dental Restoratives in Polymers for dental and
orthopedic applications, ed Shaalaby, S.W. and Salz, U.; CRC Press, Boca Raton 2007
12. Lee, H.; Neville, K.; Handbook of epoxy resins, Mc Graw-Hill, New York, 1982, cap.14

99
13. Dombrowski, J.R., Caldwell, J.R., US Pat 3 929 863, 1975
14. Satoyuki, C; Toshikazu, T., ; Takeshi, E.; Macromolecules , 1992, 25(2), 625-8.
15. Canadell, J.; Mantecon, A.; Cadiz, V. J. Polym Sci, Part A: Polym Chem, 2006, 44(16),
4722-4730.
16. Sangermano, M.; Malucelli, G.; Delleani, G.; Priola, A.; Polymer International, 2007, (56),
1224-1229
17. Takata T., Endo T. Prog. Polym. Sc. 1993, 18, 839-870
18. Brady R.F., J. Macromol. Sc. Rev. Macromol. Chem. Phys. C, 1980, 32, 135
19. Takata T., Endo T, Macromolecules, 1988, 21, 900
20. Endo, T.; Bailey, W.J.; J Polym Sci., Polym Lett Ed., 1975, 13, 193.
21. Smith R.E., Pinzino C.S., Chappelow C.C., Holden A.J., Kostoryz E.C., Guthrie J.R.,Yourtee
D.M., Eick J.D., J. Appl. Polym. Sc., 2004, 92, 62-71
22. Bailey, W.J.; Sun, R.L.; Katsuki, T.; Endo, H.; Iwama, H.; Tsushima, R.; Saigo, K.;
Bitritto, M.M.; In ring Opening Polymerization, ACS Symp 1977, No 59, Saegusa, T.; Goethals,E.,
eds; American Chemical Society; Washington, D.C., p.53.
23. Cervellera, R.; Ramis, X.; Salla, J. M.; Serra, A.; Mantecon, A. Polymer, 2005,
46:18, 6878-6887.
24. Zhou, Z.; Jin, B.; He, P; J Appl Polym Sci 2002, 84:7, 1457-1464.
25. Bailey, W.J.; J. Elastoplast., 1973, 5, 142,

100
26. Takata, T. Endo, T. Expanding Monomers: Synthesis, characterization and Applications, CRC
Press, Boca Raton, FL, 1992. Cap. 3.
27. Rokicki, G.; Prog. Polym Sci. 2000, 259-342
28. Endo, T.; Bailey, W.J.; J Polym Sci Polym Lett Ed. 1975, 13, 193.
29. Endo, T.; Okawara, M.; Synthesis, 1984, 837-845
30. Takata, T.; Ariga, T.; Endo, T.; Macromolecules, 1992, 25, 3829-3833
31. Liaw, B.R.; Li, Ma; Polymer 1998, 39, 2951-2955
32. Sakai, S.; Kiyohara, Y. ; Itoh, K. Ishii, Y. Organometal. Chem Synth 1970, 35, 2347-52
33. Sakai, S.; Kobayashi, Y.; Ishii, Y. Chem Commun 1970, 235
34. Sakai, S. Asai, Y.; Kiyohara, Y.itoh, K, Ishii, Y.; Organometal. Chem Synth 1970, 1, 45-52
35. Sangermano, M.; Acosta, R.; Puente, B.A.; Berlanga, M.L.; Garcia, A.E.; Guerrero, R..
European Polymer Journal, 2008, 44, 1046-1052.
36. Sangermano, M.; Giannelli, S.; Acosta Ortiz, R.; Berlanga Duarte, M.L.; Rueda Gonzalez,
A.K.; Garcia Valdez, A.E.; J. Appl Polym Sci, 2009, 112, 1780-1787.
37. Acosta, R.; Berlanga, M.L.; Savage, A.G.; Sangermano, M.; Garcia, A.E.: Polymer
International, 2010, 59, 680-685
38. Sangermano, M.; Berlanga, M. L.; Acosta, R.; Savage, A.G.; Garcia, A.E.; Reactive &
Functional Polymers, 2010, 70(2), 98-102
39. Bailey, W.J., Amone, M.J., polymer Preprints, Proc. Div Polym Sci Am Chem Soc p.28, 1987

101
40. Kido, N.; Matsumura, S.; Inada, H.; Endo, T. Jpn. Kokai Tokkyo Koho , 1992, JP
04066583 A 19920302
41. Takata, T., Endo, T.; Macromolecules, 1988, 21, 900
42. Komatsu, S.; Takata, T., Endo, T.; Macromolecules, 1991, 24, 2132
43. Endo, T.; Sato, H.; Takata, T., Macromolecules, 1987, 20, 1416
44. Komatsu, S.; Takata, T., Endo, T.; Polym Preprints Jpn, 1990, 39, 1532
45. Brunelle, D.J.; Ring Opening Polymerization, mechanisms, catalyst, structure and utility,
Hanser Publishers, Munich, Viena, New York, Barcelona, 1993
46. Penczek, S., Kubisa, P.; Cationic Ring-opening polymerization in Ring Opening
Polymerization, ed. Brunelle, D.J.; Hanser Publishers, Munich, Viena, New York, Barcelona,
1993, cap. 2
47. Allen, N.S. Photopolymerization and Photoimaging Science and Technology, Elsevier Applied
Science Publishers, London, 1989
48. Crivello, J.V., Dietliker, K; Chemistry & Technology of UV & EB formulation for coatings,
inks & paints, Vol 3 Ed P.K.T. Oldring SITA Technology Ltd. London, 1991, p. 327
49. Itoh, H.; Kameyama, A.; Nishikubo, T.; J. Polym Sci: Part A: Polym Chem, 1996, 34, p.217.
50. Crivello, J.V.; UV Curing: Science and Technology; Pappas, S.P. Ed.; Technology Marketing
Corporation Stamford, Conn, 1978, p.24
51. Crivello, J.V., Dietliker, K; Chemistry & Technology of UV & EB formulation for coatings,
inks & paints, Vol 3 Ed P.K.T. Oldring SITA Technology Ltd. London, 1991, p. 327

102
52. Rodríguez, M. R.; Neumann, M. G. J Polym Sci, Part A: Polym Chem , 2000, Volume Date
2001, 39(1), 46-55
53. Crivello, J.V.; Lee, J.L.; J Polym Sci, Part A: Polym Chem, 1989, 27, 3951-3968
54. Crivello, J.V.; US Pat 3 981 897 Sept 21 1976.
55. Smith, G.H.; US Pat 4 394 403 jul 19 1983, Belgium Pat 828 841 Nov7 1975
56. Crivello, J.V.; J. Polym Sci: Part A: Polym Chem, 1999, 37, 4241-4254
57. Dektar, J.L; Hacker, N.P.; J. Org Chem 1990, 55, 639.
58. Decker, D.; Moussa, K.; Makromol Chem, 1990, 191, 963.
59. Decker, C.; Moussa, K.; Eur Polymer. J. 1990, 26, 393
60. Sepe, M.P.; Dynamic Mechanical Analysis for Plastic engineering, Plastic design library,
Norwich, NY, 1998
61. Crivello, J.V.; Lee, J.L.; J Polym Sci, Part A: Polym Chem, 1989, 27, 3951-3968
62. Andrasik, S. J.; Belfield, K.D.; Bondar, M.V.; Hernandez, F.E.; Morales, A.R.; Przhonska,
O.V.; Yao, S.. ChemPhysChem (2007), 8(3), 399-404.
63. Belfield, K.D.; Bondar, M.V.; Przhonska, O.V. Journal of Fluorescence (2006), 16(1), 111-
117)
64. http://en.wikipedia.org/wiki/Fluorene
65. Bluestein, C.; Expanding Monomers in Radiation Curing in Polymer Science and Technology,
Practical Aspects and Applications, vol 4, ed. J.P. Fouassier, Kluwer Academic publishers,
Belfast, 1993

103
66. Takata, T, Endo, T. Polymer Preprints Jpn, 1988, 37, 241
67. Pigott, M.R.; Pshnov, T.; Polym Mater Sci Eng 1986; 54, 18,
68. Piggot, M.R.; Woo, M.S.; Polym Compos 1986, 7 (3) 182
69. http://en.wikipedia.org/wiki/Dynamic_mechanical_analysis
70. Sadhir R.K; Luck, R.., Expanding monomers, synthesis, characterization and applications, Eds
Rajender, K.Sadir, Russell M.Luck, CRC Press, Baton Rouge, 1992 cap. 5
71. Nielsen, L.E.; Mechanical Properties of Polymers and composites, Marcel Dekker, New York,
1994.
72. McNeill, I.C.; Rincon, A., Polymer Degradation and Stability, 1993, 13-19
73. Gomurashvili, Z.; Crivello, J.V., Macromolecules, 2002, 35, 2962-2969
74. Amy Grace Savage Gómez, tesis de maestría, CIQA, 2009
75. Ghera, E.; Sprinzak, Y.; Reactions of active Methylene compounds in pyridine solution,
Contribution from the Daniel Sieff Research Institute, Rehovoth, Israel, 1959
Top Related