







Idiomas
Páginas
Jurídico


DDP-4 INHIBIDORES
Los inhibidores de DPP-4, la proliferación, neogénesis e inhibición de la apoptosis en las células beta.Las incretinas enterohormonas capaces de incrementar la secreción insulínica de manera glucosa-dependiente.
Péptido semejante al glucagón tipo1 (Glucagon Like Peptide-1 o GLP-1)
Péptido insulinotrópico dependiente de glucosa (Glucose-depen-dent insulinotropic peptideo GIP)

Producido en las células L del intestino
A partir de proglucagón, tiene un poderoso efecto secretagogo de la insulina y estimula, asimismo, de manera notable, la bio-síntesis de insulina.
También inhibe la secreción de glucagón
Reduce:
Producción hepática de glucosa,
La velocidad de vaciamiento gástrico,
El apetito
La ingestión calórica.
Diabéticos tipo 2, la actividad del GLP-1 con frecuencia se encuentra reducida y la infusión continua del GLP-1mejora la función secretora de insulina y deprime la secreción de glucagón
GLP-1

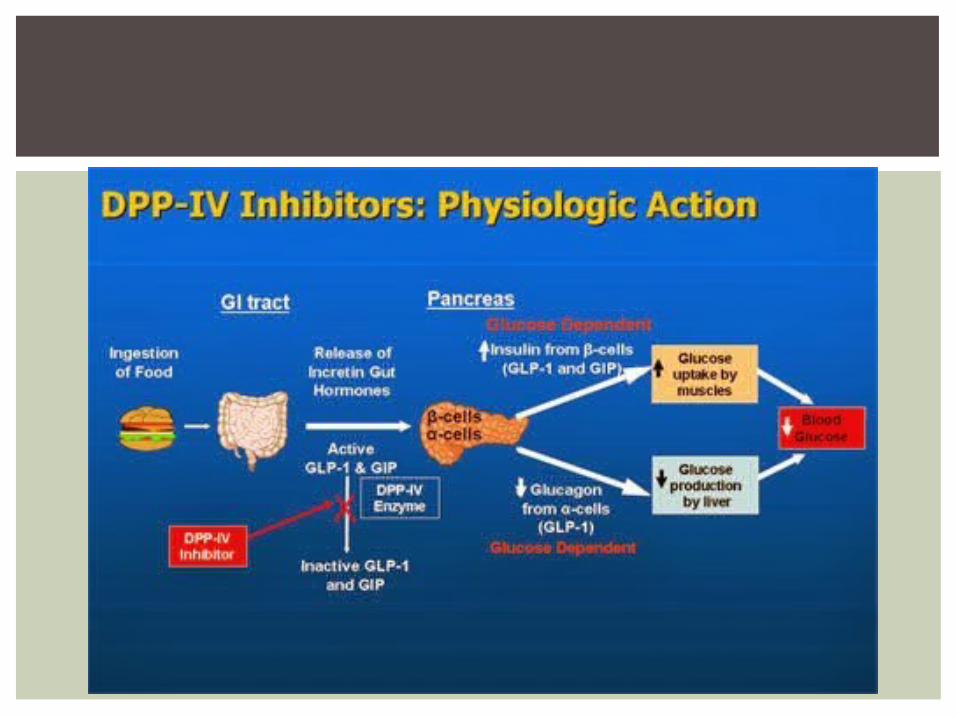
Sin embargo, tanto el GLP-1 como el GIP tienen una vida media plasmática sumamente corta.
Poco práctico su uso terapéutico en diabetes tipo 2.
En efecto, ambos péptidos se inactivan en pocos
minutos, por la enzima dipeptidil peptidasa-4 (DPP-4), de manera que no es posible que su administración produzca un efecto sostenido, a menos que se utilicen en infusión continua.

Para el desarrollo de una terapia eficaz basada en las incretinas como blanco farmacológico se han empleado dos estrategias:
1)el desarrollo de análogos del GLP-1 resistentes a la degradación por la DPP-4;
2)el desarrollo de inhibidores de la DPP-4, para elevar las concentraciones endógenas del GLP-1.

La DPP-4 pertenece a una amplia familia de peptidasas, con diversas funciones biológicas.
Entre las que figuran: DPP-8, DPP-9, la proteína de activación de
fibroblastos (FAP), QPP (Quiescent Cell Proline Peptidase), la ami-nopeptidasa P y la prolidasa.
Alteración en la funcionalidad de los linfocitos T, alopecia, trombocitopenia, reticulocitopenia y esplenomegalia en ratas, y diarrea sanguinolenta en perros.
SELECTIVIDAD

De los agentes actualmente disponibles en el mercado, la sitagliptina muestra la mayor selectividad farmacodinámica
La sitagliptina es más de 2,600 veces más selectiva para DPP-4 que para DPP-8 o DPP-9

En ratones obesos, una inhibición de la DPP-4 mayor del 80% de su actividad normal eleva dos a tres veces los niveles posprandiales de GLP-1.
Los datos de ensayos clínicos confirman esta relación en humanos.
Reducen la actividad enzimática de manera dosis dependiente.
INHIBICIÓN ENZIMÁTICA: NATURALEZA, DURACIÓN Y DOSIS-
DEPENDENCIA
La duración de la acción inhibitoria
1)la vida media
plasmática del fármaco;
2)la naturaleza
de la interacción
entre el fármaco y la enzima.
8 a 15 horas para
sitagliptina 2 a 3 horas
para vildagliptina

La sitagliptina se comporta como un inhibidor competitivo
La inhibición media de la actividad plasmática de la DPP-4 en 24 horas es >80% para dosis iguales o superiores a 50 mg.
Vildagliptina es un sustrato de la DPP-4 que produce degradación parcial del fármaco y muestra lenta disociación de su ligadura con la enzima.
La biodisponibilidad oral de la sitagliptina es de alrededor del 87%.
El tiempo para alcanzar la concentración máxima es de una a cuatro horas, tiempo que no se
modifica con la comida.
La biodisponibilidad oral de la vildagliptina también es cercana al 85%
[]max entre una y dos horas.
La absorción de la vildagliptina se afecta poco por las comidas, lo que puede reducir la concentración máxima (Cmax) en 20% y retrasar el Tmaxa dos
horas y media.
FARMACOCINÉTICA

La sitagliptina muestra una ligadura proteica cercana al 38%; la de la vildagliptina se aproxima al 10%.
El volumen de distribución de la sitagliptina en estado estacionario, tras una dosis intravenosa de 100 mg, se acerca a 198 litros.
El volumen de distribución de la vildagliptina en estado de equilibrio, tras su administración endovenosa, se acerca a 70 litros, lo que sugiere también una distribución extravascular.
DISTRIBUCIÓN

80% de la dosis administrada de sitaglipina se elimina sin cambios, orina.
Citocromo p450
METABOLIZACIÓN Y EXCRECIÓN

80% de la dosis administrada de sitaglipina se elimina sin cambios, orina.
Citocromo p450
La secreción tubular de la sitagliptina parece estar mediada por los transportadores:
OAT3 (Organic Anion Transporter-3) –el más importante–
OATP4C1 (Organic Anion Transporting Polypeptide 4C1)
MDR1 Pgp (Multidrug Resistance P-glycoprotein).

100 mg via oral dia
Renal impairmentCrCl >50 mL/min: Dose adjustment not necessaryCrCl 30-50 mL/min: 50 mg PO qDayCrCl <30 mL/min: 25 mg PO qDay
Hepatic impairment
Mild to moderate impairment: Dose adjustment not necessary
Severe impairment: Not studied
Embarazo Categoria B
SITAGLIPTINA DOSIS

1-10% Nasopharyngitis (5%) Diarrhea (4%) Headache (3.6%) Constipation (3%) Peripheral edema
(2%) Nausea (2%) Pharyngitis (1%) Osteoarthritis (1%) URI (1%)
<1% Hypersensit ivity reactions such as
anaphylaxis, angioedema, rash, urticaria, cutaneous vasculit is , and exfol iative skin condit ions ( including Stevens- Johnson syndrome)
Hepatic enzyme elevations Acute pancreatit is, including fatal
and nonfatal hemorrhagic and necrotizing pancreatit is
Constipation Vomiting Worsening renal function,
including acute renal fai lure (sometimes requiring dialysis)
Arthralgia Myalgia Pain in extremity Back pain Postmarketing Reports Pruritus
EFECTOS ADVERSOS

Hipersensibilidad
Precaución falla renal, hepática, o cardiaca.
No diabéticos con cetoácidosis; sin efecto
No use en DM1
Cuidado cuando se combina con CYP3a4/5 inhibidores.
Asocia pancreatitis aguda, hemorrágico y necrotizante,
Uso concomitante con secretagogos riesgo hipoglucemia.
angioedema
CONTRAINDICACIONES

Mechanism of Action Inhibidor de DPP4, incrementa la duración de la
acción de hormonas incretinas. Incretinas, aumentan liberación de insulina de
células pancreáticas BETA, disminuyen excreción de glucagón de Células ALPHA
AbsorptionBioavailability: 87%Peak plasma time: 1-4 hr.

DistributionProtein bound: 38%
MetabolismLimited; primarily via CYP3A4 and CYP2C8
EliminationHalf-life, terminal: 12.4 hrExcretion: Urine (87%), feces (13%)

2.5- 5 mg via oral/ dia
Modificaciones de dosis:Puede necesitar reducir dosis de sulfonilurea u otros
agentes secretores de insulina.Combinan con inhibidores fuertes de CYP450 3ª4/5 2.5
mg.
CrCl ≥50 mL/min: No dose adjustment required CrCl <50 mL/min: Not to exceed 2.5 mg PO qDay ESRD requiring hemodialysis: Not to exceed 2.5 mg PO
qDay administered postdialysis.
Embarazo familia B
SAXAGLIPTINA

1-10% (selected) Urinary tract infection (7%) Headache (7%) Hypersensitivity-related events (<4%; eg, urticaria, facial
edema) Peripheral edema (<4%; increased incidence when
coadministered with thiazolidinediones) Upper respiratory tract infection (3%) Gastroenteritis (2%) Hypoglycemia (1.6%) Frequency Not Defi ned Increased creatinine phosphokinase Increased creatinine Idiopathic thrombocytopenic purpura rash

HipersensibilidadNecesidad de ajuste en enfermedad renalDecrementar dosis en combinacion con fuertes
inhibidores de CYP450 3ª4/5Riesgo pancreatico Edema Hipoglicemia cuando se administra en combinacion
con otros hipoglucemiantesFalla cardiaca

Mechanism of ActionDipeptidyl peptidase IV
(DPP-4) inhibition that results in increased incretin hormones and enhanced glycemic control
AbsorptionPeak plasma time: 2hr
(saxagliptin); 4 hr (5-hyroxy saxagliptin)
MetabolismHepatic by CYP450 3A4/5 to active metabolite (50% potency of parent compound)
EliminationHalf-life (elimination): 2.5 hr (saxagliptin); 3.1 hr (5-hydroxy saxagliptin)
Renal clearance: 7.2 L/hr
Excretion: Urine (75%); feces (22%)
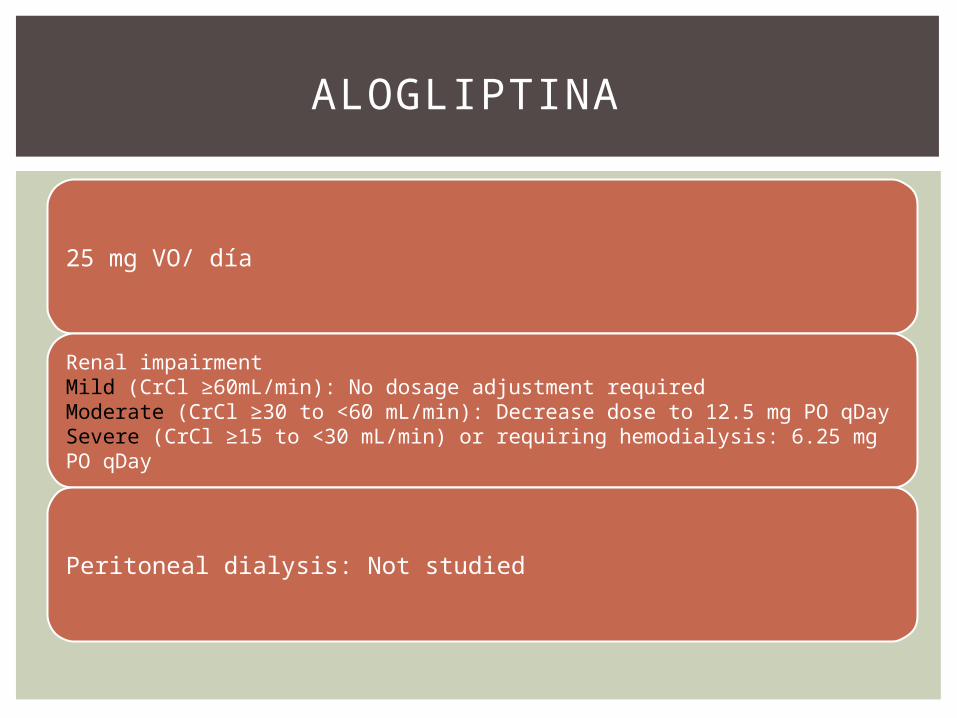
25 mg VO/ día
Renal impairmentMild (CrCl ≥60mL/min): No dosage adjustment requiredModerate (CrCl ≥30 to <60 mL/min): Decrease dose to 12.5 mg PO qDaySevere (CrCl ≥15 to <30 mL/min) or requiring hemodialysis: 6.25 mg PO qDay
Peritoneal dialysis: Not studied
ALOGLIPTINA

Hipoglicemia 1.5-35% >combinado con insulina1-10%NasofaringitisCefalea Infeccion de vias respiratorias altas<1%Hipersensibilidadpancreatitis
EFECTOS ADVERSOS
Top Related