







Idiomas
Páginas
Jurídico


Genotipos y fenotipos en enfermedades con defectos en la reparación del daño al DNA
Graciela Spivak
Biology Department, Stanford University, Stanford CA 94305, USA

Ejemplos de enfermedades hereditarias con defectos en reparación del DNA
G. Spivak©

Ejemplos de genes de reparación del DNA cuyas mutacionescausan enfermedades en humanos
G. Spivak©

Clinical features XP CS TTD UVSS
Dermatological:
Quemaduras de sol agudas + + +/– +
Telangiectasia + + +/– +
Pecas + –/+ +/– +
Tumores de piel + – – –
Other organs & systems:
Anormalidades oculares – + + –
Defectos neurologicos + + + –
Reflejos anormales + + – –
Inmadurez sexual + + + –
Enanismo caquectico – + + –
Defectos oseos, osteoporosis – + + –
Defectos del pelo, uñas – – + –
Xeroderma pigmentosum (XP) Síndrome de Cockayne (CS)
Síndrome UV-sensitivo (UVSS) Tricotiodistrofia (TTD)
Enfermedades Humanas con defectos en NER
G. Spivak©

La mielina forma la sustancia blanca. Es un
fosfolípido que aisla los axones, incrementando la
velocidad de los impulsos eléctricos.
Dismielinización (mielina anormal) occurre en CS,
TTD y en el síndrome autoinmune Aicardi-Goutières.
sustancia blanca sustancia gris
Las células neurales forman la sustancia gris.
La pérdida de neuronas en NER-deficiente XP
puede ser causada por acumulación de lesiones en
el DNA, como la purinas cíclicas y productos de la
peroxidación de lípidos, reparados por NER.
Neurodegeneración en XP puede ser debida a
defectos en NER.
Neurodegeneración en CS no es debida a
defectos en la reparación del DNA por NER.
Causas de neurodegeneración

XERODERMA PIGMENTOSUM (XP)
Group GGR TCR Protein function
A – – Verification
B – – TFIIH helicase, ATPase
C – + Recognition, initial opening of the strands (complex
with hRAD23b, centrin1)
D – – TFIIH helicase
E – + Recognition
F – – Endonuclease (complex with ERCC1)
G – – Endonuclease, stabilizes TFIIH
Variant + + Translesion DNA polymerase
• Fotosensitividad elevada o severa
• Piel reseca, tipo pergamino
• Muy alta incidencia de cancer de piel, ojos, punta de la lengua
• Incidencia elevada de tumores internos
• ~20 % de los pacientes sufren degeneración neurológica progresiva
XP
clá
sic
a

Fenotipos en pacientes con XP
La severidad depende de:
• La mutación(s) (homocigótica or heterocigótica, dominioy/o función afectada)
• El contexto genético:– Sistema inmune– Capacidad antioxidante
• El medio ambiente (protección solar, polución, etc.)

Mutaciones en XPD, una helicasa en el complejo TFIIH
(mapa cortesia de Miria Stefanini)
XP32NH2
arg616glnXP32NH1
Mutaciones del codón 683
causan solo XP, y afectan
al 80% de los pacientes
XP-D
Mutaciones del codón 616
resultan en proteina
inactiva; homocigotas son
probablemente letales.
El fenotipo de los
heterocigotas depende del
otro alelo.

Pacientes XP-D con mutaciones R683Q (ArgGln) y síntomas ligeros
XP32NHheterocigotaR683Q/R616Q
XP62TA, XP32TA, XP29TAhomocigotas R683Q
Quemaduras de sol & pigmentación;No/poco cancerNo/tardía neuropatía
Falik-Zaccai et al., Environmental Molecular Mutagenesis, 53:505-514, 2012
a b
d ec
proteina inactiva

Pacientes XP-D con mutaciones R683W (ArgTrp) en un alelo
AS552 & AS553R683W/I455del (–1 aa)BSC, melanoma, neurodegeneración, fallecidos a 35 & 39 años.
XP29BER683W/Q542X (truncado)>300 canceres de piel, neurodegeneration, muerte a los 37 años.
XP34BE, XP35BER683W/199insPP (+2 aa)Protección solar: pigmentación mínimaFunción neural normal
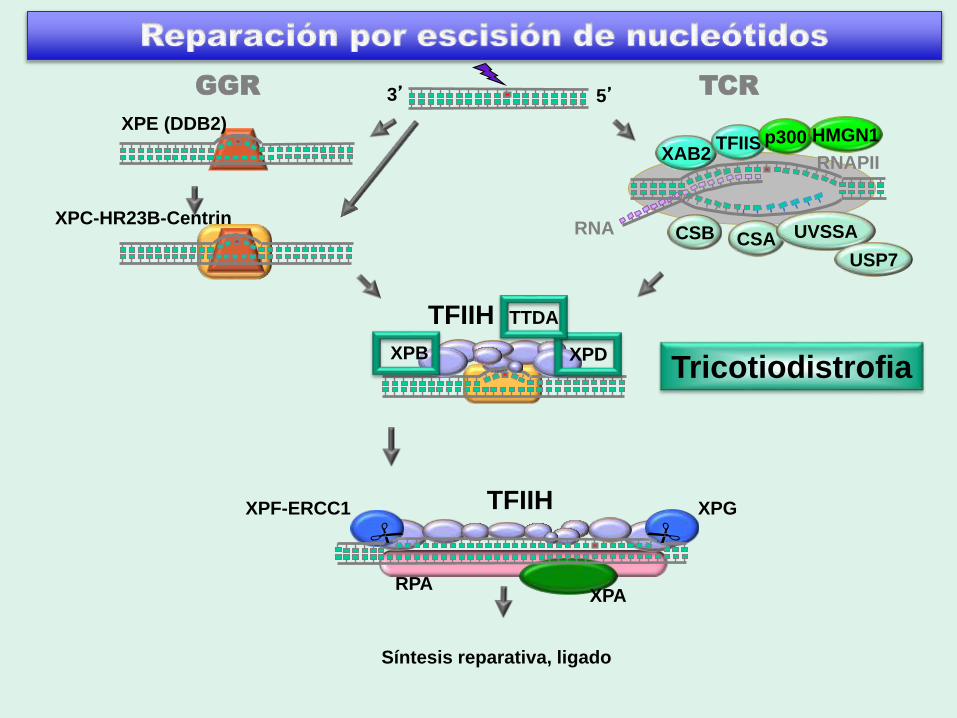
Figure 1

3’ 5’
RPA
XPF
XPC-HR23B-Centrin
XPE (DDB2)
TFIIH
XPA
XPG
RNA
RNAPII
CSACSB UVSSA
XAB2TFIIS
Síntesis reparativa, ligado
GGR TCR
TFIIH
USP7
p300 HMGN1
XPB XPDXeroderma
pigmentosum
ERCC1

Síndrome de Cockayne
Criterios requeridos para el diagnóstico:
Falta de crecimiento, baja estatura (<5to percentil)
Disfunción neurológica progresiva, leucodistrofia
(degeneración progresiva de la sustancia blanca)
Microcefalia
Criterios adicionales (por lo menos 5):
Sensibilidad a la luz solar, quemaduras
Neuropatía periferal desmielinizante
Retinopatía pigmentaria y/o cataratas
Pérdida del oído sensorineural
Enanismo caquéctico
Ojos hundidos
Postura encorvada
Progeria (envejecimiento prematuro)
Corta vida
Otros síntomas: Defectos del andar, contracturas, espasticidad, temblores
Caries dentales
Defectos de circulación (manos y pies fríos), temperatura corporal baja
Problemas alimentarios - Dormir con ojos abiertos
Calcificaciones en los ganglios basales
Anormalidades del hígado; enzimas hepáticas elevadas
Hipertensión - picazón severa - osteoporosis - hiper-reflexia
Ian - falleció a los 5 años
Brandon - falleció a los 6 años

La clasificación en CS depende de los síntomas clínicos:
Desarrollo Comienzo
Prenatal síntomas Progreso Supervivencia
•CS tipo I normal primer año mediano 10-20 años
•CS tipo II anormal inmediato rápido < 7 años
•CS tipo III normal tardío lento >30 años
Grupos de complementación:
•CS-A
•CS-B
•XPB-CS
•XPD-CS Aspecto combinado de CS y XP
•XPG-CS
•XPF-CS
•ERCC1
Mutaciones en CSA o CSB pueden resultar en CS tipos I, II o III
No hay diferencias fenotípicas entre pacientes de
CS con mutaciones en CSA o en CSB

3’ 5’
RPA
XPF
XPC-HR23B-Centrin
XPE (DDB2)
TFIIH
XPA
XPG
RNA
RNAPII
CSACSB UVSSA
XAB2TFIIS
Síntesis reparativa, ligado
GGR TCR
TFIIH
USP7
p300 HMGN1
XPB XPDCockayne
syndrome
ERCC1

Tricotiodistrofia Síntomas similares a CS
Diagnóstico:• Pelo quebradizo, deficiente en
azufre
• La microscopía polarizada
muestra bandas alternadas (cola
de tigre)
• Estructura anormal de la hebra
capilar
Grupos de complementación:
• XPB
• XPD
• TTD-A
• TTDN1
• ?
• ?
fotosensibles, NER–
componentes de TFIIH
no fotosensibles, NER+
función desconocida
Adaptado de K. H. Kraemer et al., Neuroscience 145:1388-1396 (2007)
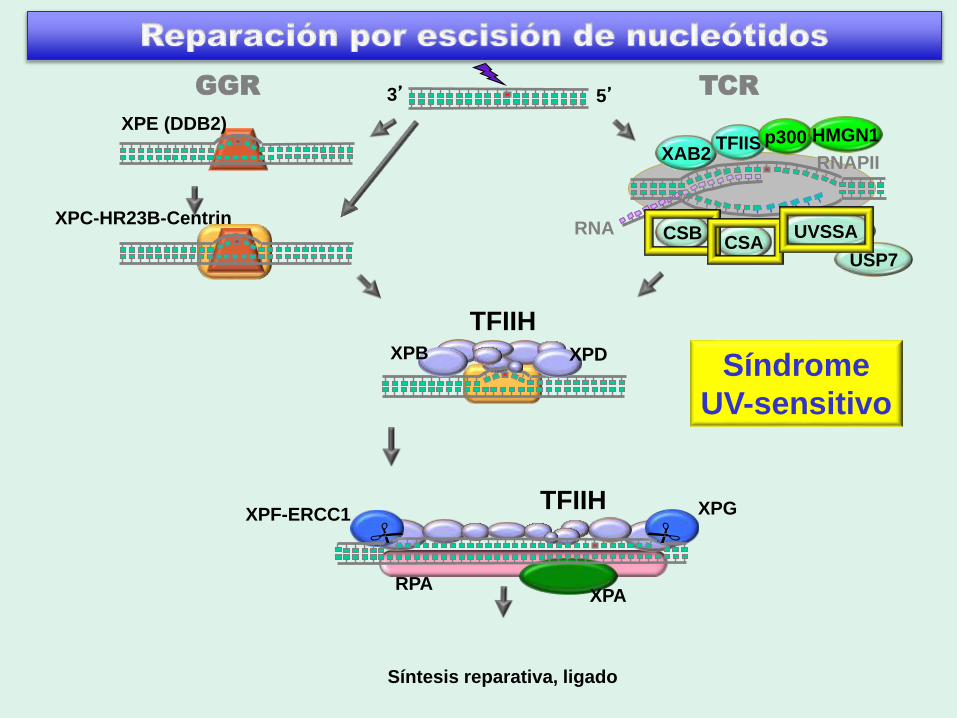
3’ 5’
RPA
XPF-ERCC1
XPC-HR23B-Centrin
XPE (DDB2)
TFIIH
XPA
XPG
RNA
RNAPII
CSACSB UVSSA
XAB2TFIIS
Síntesis reparativa, ligado
GGR TCR
TFIIH
USP7
p300 HMGN1
XPB XPDTricotiodistrofia
TTDA

Síndrome UV-sensitivo
G. Spivak©
8 pacientes 6 Japoneses
1 Israelita
1 Frances
3 grupos de complementación UVSS-A: mutados en KIAA1530 (UVSSA)
UVSS-CSB: mutados en CSB (ERCC6)
UVSS-CSA: mutados en CSA (ERCC8)
Características:
Quemaduras de sol severas
Hiperpigmentación en areas expuestas al sol
Neurología y desarrollo normales
NO cancer
3’ 5’
RPA
XPF-ERCC1
XPC-HR23B-Centrin
XPE (DDB2)
TFIIH
XPA
XPG
RNA
RNAPII
CSACSB UVSSA
XAB2TFIIS
GGR TCR
TFIIH
USP7
p300 HMGN1
XPB XPDSíndrome
UV-sensitivo
Síntesis reparativa, ligado

¿Por qué los pacientes con CS o UVSS son sensitivos a la luz solar pero no son propensos al
cáncer?
• La falta de TCR causa muerte celular por apoptosis
• Las células que sobreviven poseen GGR normal: pocas mutaciones
Por contraste, en pacientes con xerodermapigmentosa:
• GGR es deficiente: las células sobrevivientes adquieren muchasmutaciones que pueden resultar en cáncer.
G. Spivak©

XP / T
TD
Xerodermapigmentosum
Xerodermapigmentosum with
neurological anormalities
XP / CS
Tricothiodistrophy
Cock
ayne
Synd
rome
UVSS
COFS
Synd
rome
XPA
XPC
XPF
XPE
XPV
XPG
CSA
TTD-A
Adapted from K. H. Kraemer et al., Neuroscience 145:1388-1396 (2007)
UVSSA
Genes y síndromes
TTDN1
?
?
?

XPB
CSA
CSB
KIAA1530???
Cockayne syndrome
UV-sensitive syndrome
XPD
XPG
XPF
ERCC1

lesión
RNAPIIo
mRNA
RNAPIIa
promotor
CSB
CSB
2. Elongación
CSB
CSB
3. Recluta CSA
ubiquitinación de RNAPII
esencial para TCR
P
CSB participa en muchas funciones celulares
nucleosoma
1. Restorar RNAPII competente para
iniciación
4. Remodelador de cromatina
CSA
CSB
5. Localización nucleolar,
transcripción de rDNA por RNAPI
6. Estimulación de BER
7. Reparación de complejos topo I-DNA
8. Mitocondrias
Ub

DSC COFS
CS
CSCS
CS
CSCS
CS
CSCS
CS CS
CS
CS
CS
UVSS
CS
C-terminusN-terminus
CS
CS*
CS**
CS**
UVS1KO, CS3AM: C308 G308 = stop codon 77
KPSX6 (mild, adult-onset): 304-307 stop codon 82
CS548VI, CS539VI (severe): promoter, intron 1
CS*
CS**
>71 mutaciones identificadas en CSB
UVSS
No proteina CSB
CS
Laugel et al., Human Mutation 2009; http://www.umd.be/CSB/

Known mutations in the CSA gene
CS type II (severe)CS type I (moderate)
UVSS
CS, type not determined
Laugel et al., Human Mutation 2009; http://www.umd.be/CSA/
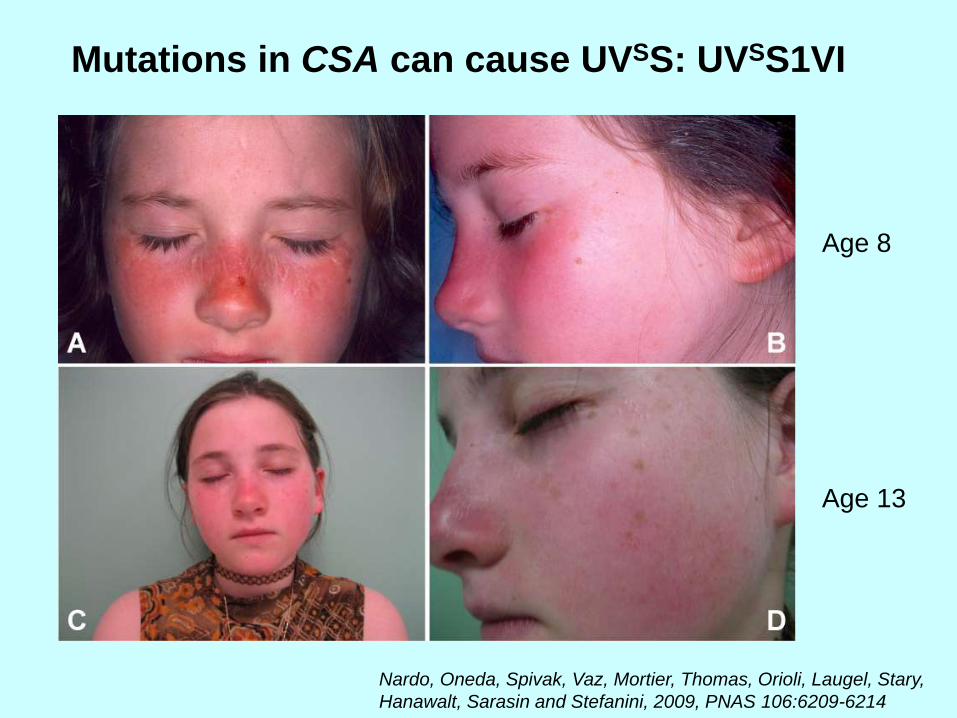
Mutations in CSA can cause UVSS: UVSS1VI
Age 8
Age 13
Nardo, Oneda, Spivak, Vaz, Mortier, Thomas, Orioli, Laugel, Stary,
Hanawalt, Sarasin and Stefanini, 2009, PNAS 106:6209-6214

WD 1 WD 2 WD 3 WD 4 WD 5 WD 6 WD 7
GHYKTVDCCVFQSNFQELYSGSRDCNILAWV
GHYKTVDCCVFQSNFQELYSGSRDCNILACV
CSA es una proteina de motivos WD-repeat
La paciente UVS1VI es homocigótica
para la transversión G->T, que resulta
en la substitución trp (W) to cys (C) en
el séptimo motivo WD-repeat.
Ejemplo de una hélice de 7 paletas
típica de proteinas de WD-repeat
(W: tryptofano; D: acido aspartico).
Cada motivo repetido forma una paleta
que puede contactar otras proteinas.

Ciertas mutaciones afectan la función de CSA o CSB en TCR de
lesiones causadas por la radiación UV y aductos abultados,
pero ambas proteínas juegan un papel adicional y separable que
determina el fenotipo.
Conclusiones
G. Spivak©

G. Spivak©
A qué se deben las diferencias fenotípicas entre CS y UVSS?

Respuestas celulares a la UV
Spivak et al., DNA Repair 2006
normal XPA, B, XPC, CS UVSS
D, F, G E
Supervivencia + - - - -
NER global + - - + +
TCR de fotolesiones + - + - -

Supervivencia al daño oxidativo
0.1
1
10
100
0 5 10 15 20
% S
urviva
l
KBrO3, mM
0.1
1
10
100
0 25 50 75 100
% S
urviva
l
Menadione, microM
Normal
Normal
WTUVSS
CS-A
CS-ACS-B
UVSS
WT

Reactivación en células huéspedes
b-galactosidasa
pCMVb
24-48 h
lacZ
Luz UV
G. Spivak©
Tratamientos
químicos

REACTIVACIÓN EN CÉLULAS HUÉSPEDES
Lesiones/4.5kb gen lacZ
0 1 2 3 4
Tg
0 2 4 6 8
8-oxoG
0 5 10 15 20
sitios AP
0 5 10 15 20
SSB
0 10 20 301
10
100
UV (CPD)
Normal
UVSS
CS-B
CS-A
% a
cti
vid
ad
b-g
ala
cto
sid
asa
G. Spivak©

Respuestas celulares a la UV
Spivak et al., DNA Repair 2006
normal CS UVSS
Supervivencia + - -NER global + + +
TCR de fotolesiones + - -
Reactiv. cél. huésp. UV + - -
Respuestas celulares al daño oxidativo
Supervivencia + - +
Reactiv. cél. huésp. oxid. + - +

Hipótesis:
•Las células de CS son deficientes en el procesamiento de lesiones
oxidativas en las cadenas de DNA transcriptas.
•Durante períodos de actividad metabólica
intensa, la generación de lesiones oxidativas
resulta en muerte celular masiva, creando
deficiencias en tejidos y órganos esenciales.
•Las células de UVSS poseen mecanismos normales para transcribir genes
que contienen lesiones oxidativas.
Incremento en
apoptosis
durante el
desarrollo?
¿Por qué los pacientes con CS tienen problemas neurológicos o de desarrollo?
¿Por qué los pacientes con UVSS no los tienen?
G. Spivak©
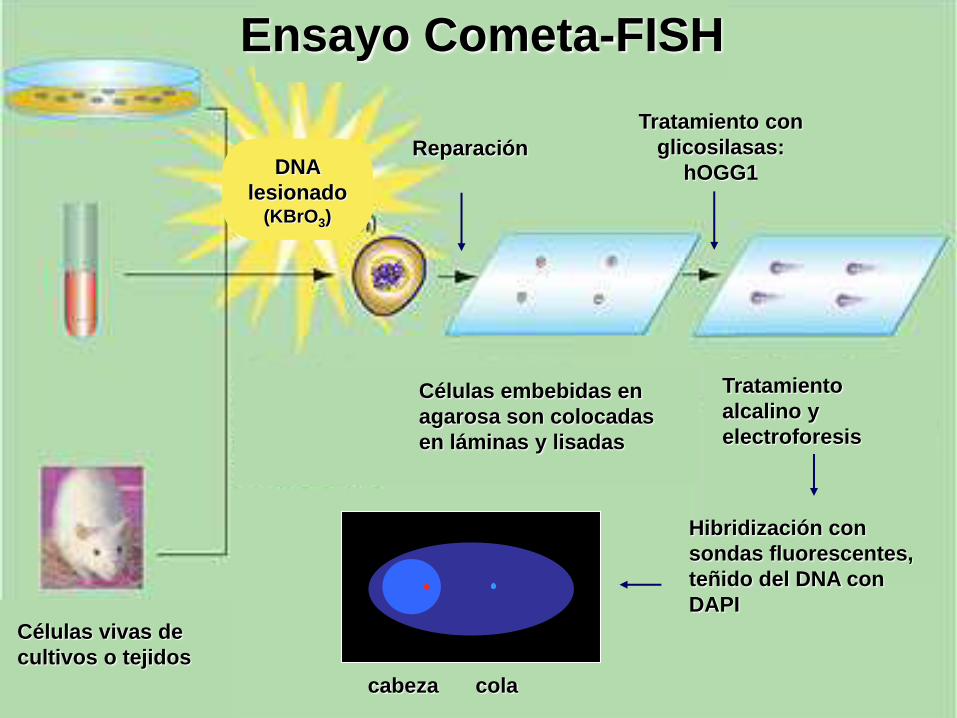
Tratamiento con
glicosilasas:
hOGG1
Tratamiento
alcalino y
electroforesis
Hibridización con
sondas fluorescentes,
teñido del DNA con
DAPI
Reparación
Ensayo Cometa-FISH
Células embebidas en
agarosa son colocadas
en láminas y lisadas
Células vivas de
cultivos o tejidos
cabeza cola
DNA
lesionado(KBrO3)
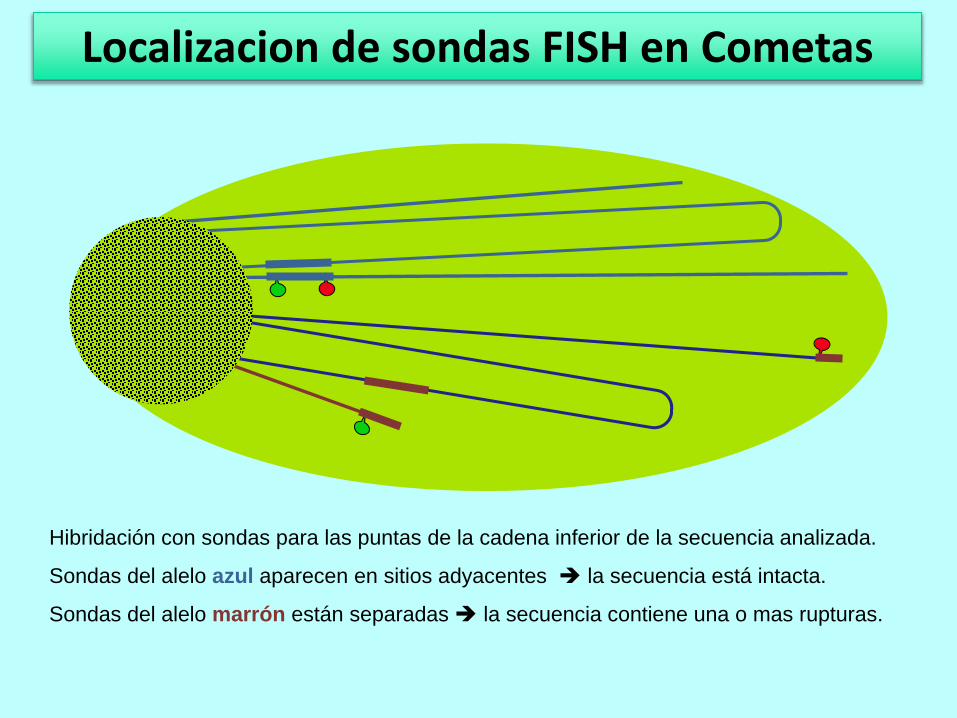
Hibridación con sondas para las puntas de la cadena inferior de la secuencia analizada.
Sondas del alelo azul aparecen en sitios adyacentes la secuencia está intacta.
Sondas del alelo marrón están separadas la secuencia contiene una o mas rupturas.
MAR
Localizacion de sondas FISH en Cometas
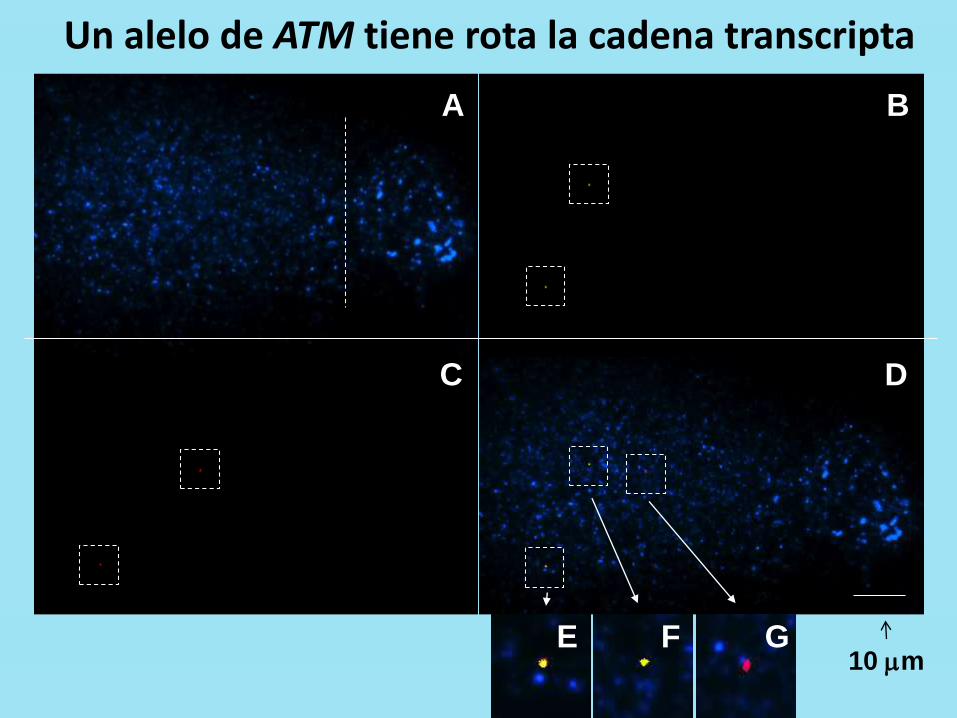
A B
C D
E F G
Un alelo de ATM tiene rota la cadena transcripta
10 mm

Reparación de 8-oxoG acoplada a la transcripción y global
Repair Time (Hr)
Normal CSB
Repair Time (Hr)
UVSSA
Repair Time (Hr) Repair Time (Hr)
WT with α-Amanitin
TCR – ATM gene
Non-transcribed strand
Transcribed strand
Fibroblastos humanos expuestos a 50 mM KBrO3 por una hora
Normal CSB
Repair Time (Hr) Repair Time (Hr)
UVSSA WT with α-Amanitin
Repair Time (Hr) Repair Time (Hr)
GGR
With OGG1
Without OGG1

Enzimas necesarias para la TCR de 8-oxoG
Repair Time (Hr)
Normal OGG1 Knockdown
Repair Time (Hr)
TCR – ATM gene
Non-transcribed strand
Transcribed strand
Repair Time (Hr)
XPA
Normal
Repair Time (Hr) Repair Time (Hr)
GGR
With OGG1
Without OGG1
OGG1 Knockdown XPA
Repair Time (Hr)

Modelo: conversación entre caminos de reparación
reparaciónglobal via BER
hOGG1 glycosilasa
AP endonucleasa, dRpaae
sitio AP
mella de 1 nucleótido
8-oxoG

Modelo: conversación entre caminos de reparación
genomic repair via BER
Reclutamiento de factores de TCR
RNAP removida o retrocede;reclutamiento de factores de NER
reparación completatranscripción continúa
RNAPII
CSACSB UVSSAXAB2TFIIS
USP7
p300HMGN1
RPA
TFIIH
XPA
XPF-ERCC1 XPG
Transcripciónbloqueada
hOGG1 glycosylase
AP endonuclease, dRpase
AP site
single nucleotide gap
8-oxoG✖
✖
✖

1. Diferencias en TCR de lesiones oxidativas
2. Diferencias en traspaso de lesiones oxidativas por las
RNAP
3. Diferencias en funcionamiento de las mitocondrias
G. Spivak©
A qué se deben las diferencias fenotípicas entre CS y
UVSS?
A qué se debe la hipersensibilidad de CS a la
oxidación?

600
400
200
Control MB + VL
0 1 2 3 10 0 1 2 3 10
RO
Transcripción en vitro
Time,
minutes
EcoRI
AdMLP
HindIII
Run off
productos
troncados
RNA 630 nt
+ extracto nuclear de HeLa
+ NTPs
+ 32P-CTP
azul de metileno
luz visible
8-oxoG

Funciones de CSA y CSB en mitocondrias
mitofagia = autofagia de mitocondrias
control de calidad por eliminación de organelas envejecidas o dañadas
proteinas
agregadasneurodegeneración
Síntomas de deficiencia mitocondrial:
Enanismo, atrofia cerebral y cerebelar, neuropatía periferal,
leucodistrofia, sordera, ataxia, temblores, cataratas, etc.

G. Spivak©
A qué se deben las diferencias fenotípicas entre CS y
UVSS?
1. Traspaso por polimerasas de RNA
2. Función, estructura y autofagia de mitocondrias
Lineas de investigación:
Top Related