







Idiomas
Páginas
Jurídico


Provided by
Histiocitosis
Lead contributors:
Jorge Luis Braier, MD Julio Goldberg, MD
Guillermo Chantada, MD Diego Rosso, MD
Hospital JP Garrahan Buenos Aires, Argentina
Agradecimientos:
Los Doctores Amalia Laterza, Bettina Cervini y Daniel Alderete contribuyeron
aportando datos y fotografías de pacientes
A. Introduccion
Las enfermedades histiocitarias se caracterizan por la acumulación
anormal de células pertenecientes al sistema monocítico-macrofágico (células
dendríticas y macrófagos) Ellas incluyen una amplia variedad de enfermedades
de difícil clasificación y tratamiento, que afectan a niños y adultos.
Las histiocitosis representan un amplio espectro de enfermedades
definidas por el compromiso patológico de las células con funciones específicas
en la fagocitosis y la presentación de antígenos. Hay muchas similitudes entre
los monocitos, macrófagos, histiocitos y células dendríticas (CD), siendo difícil
diferenciarlos morfológicamente.

Page 2 of 56
Las enfermedades histiocitarias se definen en general por la presencia de
las células predominantes en cada una de ellas siguiendo criterios
anatomopatológicos e inmunohistoquímicos bien establecidos.1-3
El uso de la tipificación del inmunofenotipo por inmunocitoquímica o por
citometría de flujo de la superficie celular con el objeto de definir los
diferentes tipos de superficie de estas células, ayuda a diferenciar los tipos
celulares, pero factores clínicos, así como atipías citológicas o citogenéticas o
procedimientos moleculares, pueden ser necesarios para llegar a un
diagnóstico final.
La célula de Langerhans (CL), es una célula presentadora de antígenos
por excelencia del sistema inmune y ha sido encontrada en piel y otras
localizaciones. En otros órganos, salvo en la córnea y el cerebro, las células
interdigitantes dendríticas, como las CL actúan como células presentadoras de
antígenos. Cuando cualquiera de estas células incorpora el antígeno y migran
hacia los ganglios linfáticos, su morfología cambia y ellas se identifican como
células indeterminadas. Durante este estadio, estas células tienen el mismo tipo
de marcadores de superficie que las CL pero carecen de gránulos de Birbeck.4,5
Favara, Jaffe y colaboradores han publicado clasificaciones de las
enfermedades histiocitarias basadas en que ellas están relacionadas a las
células dendríticas, monocitos/macrófagos y enfermedades malignas.3 Las
enfermedades relacionadas con las células dendríticas incluyen la Histiocitosis
de células de Langerhans (HCL), caracterizada por la presencia de CL situada en
la unión de la dermis con la epidermis en la piel, así como en pulmones,
ganglios linfáticos, bazo o médula ósea y hueso. La coloración de estas células
con anti CD 207 (anti-langerina) ha aportado una herramienta específica para
identificar la proteína que constituye los gránulos de Birbeck que se ven por
microscopía electrónica.6 Sin embargo, también se ha hallado que los
histiocitos de la HCL se originan de células dendríticas mieloides y no en
células de Langerhans de la piel.7 Las coloraciones inmunohistoquímicas con
anti CD 207 y anti CD 1 a son consideradas patognomónicas en el diagnóstico

Page 3 of 56
de la HCL. Otros antígenos como el S 100 o el HLA-DR son característicos pero
no específicos para las CL.8 La Histiocitosis maligna verdadera ha
evolucionado a un diagnóstico de una entidad más específica después de haber
excluido casos de linfoma anaplásico a células grandes y otros linfomas a
células grandes con inmunofenotipo único.3 El tercer grupo de enfermedades de
células dendríticas derivan del dendrocito dérmico / intersticial que se colorea
con anticuerpos anti CD 68, Fascina y factor XIIIa que son encontrados en
pacientes con enfermedad de Erdheim-Chester (EEC)9, 10 y Xantogranuloma
Juvenil (XGJ). Estas células no expresan CD1a ni CD 207. La función
macrofágica exuberantemente anormal es fundamental en la fisiopatología de
la Linfohistiocitosis Hemofagocítica (LHH). Otra entidad descripta es la
enfermedad de Rosai-Dorfman o también conocida como Histiocitosis
Sinusoidal con linfoadenopatía masiva.11 El hallazgo histopatológico típico en
esta enfermedad es la presencia de linfocitos intactos en el citoplasma de los
macrófagos (emperipolesis), que deben ser encontrados para su diagnostico.12
A. References 1Favara BE, Jaffe R. The histopathology of Langerhans cell histiocytosis. Br J Cancer. Suppl 1994; 23: S17-23. 2Jaffe R. Pathology of histiocytosis X. Perspect Pediatr Pathol. 1987; 9: 4-47. 3Jaffe R WL, Facchetti F. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H. . Tumors derived from Langerhans cells. Lyon: IARC Press; 2008. 4Mierau GW. Intranuclear Birbeck granules in Langerhans cell histiocytosis. Pediatr Pathol. 1994; 14: 1051-1054. 5Favara BE, Feller AC, Pauli M et al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol. 1997; 29: 157-166. 6Lau SK, Chu PG, Weiss LM. Immunohistochemical expression of Langerin in Langerhans cell histiocytosis and non-Langerhans cell histiocytic disorders. Am J Surg Pathol. 2008; 32: 615-619. 7Allen CE, Li L, Peters TL et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J Immunol. 2010; 184: 4557-4567. 8Ye F, Huang SW, Dong HJ. Histiocytosis X. S-100 protein, peanut agglutinin, and transmission electron microscopy study. Am J Clin Pathol. 1990; 94: 627-631.

Page 4 of 56
9Kenn W, Eck M, Allolio B et al. Erdheim-Chester disease: evidence for a disease entity different from Langerhans cell histiocytosis? Three cases with detailed radiological and immunohistochemical analysis. Hum Pathol. 2000; 31:734-739. 10Dickson BC, Pethe V, Chung CT et al. Systemic Erdheim-Chester disease. Virchows Arch. 2008; 452: 221-227. 11Carbone A, Passannante A, Gloghini A et al. Review of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) of head and neck. Ann Otol Rhinol Laryngol. 1999; 108:1095-1104. 12Iyer VK, Handa KK, Sharma MC. Variable extent of emperipolesis in the evolution of Rosai Dorfman disease: Diagnostic and pathogenetic implications. J Cytol. 2009; 26:111-116.
B. Histiocitosis de Celulas de Langerhans
B.1 Definición e Historia Paul Langerhans describió inicialmente las células epidérmicas
multilobuladas con prolongaciones pseudopódicas que son conocidas como
Células de Langerhans después de haber realizado, en 1868, una coloración de
un preparado de piel con oro coloidal. El creyó en un principio que estas células
eran neuronas debido a la apariencia de ramificaciones dentríticas que
presentaban. En 1973, Christian Nezelof y colegas, utilizaron el microscopio
electrónico para evaluar las biopsias de esta enfermedad, en ese momento
conocida como Histiocitosis X (HX).1 Ellos encontraron los gránulos
pentalaminares de Birbeck en las células de HX que eran idénticos a los
descriptos previamente en las células epidérmicas de Langerhans y por lo tanto
decidieron redenominar esta enfermedad HX como Histiocitosis de Células de
Langerhans (HCL). La CL ha sido considerada como elemento central de una
condición común que ha sido descripta con varios nombres, incluyendo la HX,
elGranuloma Eosinófilo, la enfermedad de Letterer Siwe, la enfermedad de
Hand-Schuller-Christian y la Reticuloendoteliosis difusa. La HCL es actualmente
la nomenclatura apropiada para los desórdenes caracterizados por la presencia
de células de Langerhans CD 207 +. Se piensa que la HCL es el resultado de la

Page 5 of 56
proliferación de CL redondeadas, inmunofenotípicamente y funcionalmente
inmaduras junto con eosinófilos, macrófagos y linfocitos y en ocasiones, células
gigantes multinucleadas. Existe una controversia si la HCL es debida a una
transformación maligna o a una disregulación inmune de la célula de
Langerhans. En cualquier caso, el pronóstico clínico ha mejorado con agentes
quimioterápicos con actividad contra células malignas o inmunológicamente
activadas.
Aunque esta enfermedad ya se conoce hace ya aproximadamente un
siglo, su fisiopatología sigue siendo un enigma y su tratamiento es inespecífico.
B.2 Epidemiología La incidencia de HCL ha sido estimada en 2 a 10 casos por millón de
niños hasta la edad de 15 años.2 La proporción varón/mujer es cercana a 1.
Aunque la enfermedad puede presentarse a cualquier edad, puede ser
congénita3 o presentarse en adultos,4,5 el pico de incidencia se presenta entre
1 y 3 años de edad. Los pacientes con compromiso unisistémico son mayores
que los que presentan compromiso multisistémico. Se han reportado raramente
casos en gemelos idénticos y no idénticos con comienzo temprano de HCL.6
Hay también raramente reportados casos en hermanos o múltiples casos en una
familia.7 No hay evidencias de una mutación génica específica en la HCL. Se
encontró una débil asociación entre HCL y exposición a solventes en los padres,
e infecciones perinatales.
B.3 Etiología y Patogenia La HCL tiene características de transformación neoplásica como de
disregulación inmunológica, lo que causa un debate sobre su etiología.

Page 6 of 56
B.4 Displasia / Enfermedad Maligna Los estudios que sugerían clonalidad en la HCL fueron publicados en
1994. En este estudio se encontraron proliferaciones monoclonales en las
biopsias de lesiones provenientes de cuadros clínicos unisistémicos o
multisistémicos de HCL8 aunque luego se vio que la HCL en adultos no es
monoclonal. La pérdida de heterozigocidad en el locus de los posibles genes
supresores tumorales ha sido reportada en 2 estudios, pero en un estudio
reciente no se pudieron encontrar deleciones o amplificaciones en el DNA de
las células CD 207+.
B.5 Disregulación Inmunológica La histopatología de las lesiones y las manifestaciones clínicas sugieren
la participación de citoquinas en su patogenia.9,10 Las citoquinas muchas de
ellas llamadas “interleuquinas” que originalmente se denominaron linfoquinas,
son proteínas o glicoproteínas, que regulan el sistema inmune y participan en
las comunicaciones intercelulares.
En su mayoría son productos de secreción de linfocitos y monocitos, que
regulan el crecimiento, diferenciación e inmunocompetencia de las células de la
progenie hematopoyética entre otras muchas acciones .
En las lesiones óseas por células de Langerhans se produce interleuquina
(IL) 1. Ésta estaría involucrada en la destrucción ósea. Además, la IL-1 parece ser
una de las moléculas necesarias para la diferenciación y migración de la célula
de Langerhans normal de la piel hacia el ganglio linfático.11 El factor de
necrosis tumoral alfa (TNFα), también conocido como “caquexina” es una
citoquina polipeptídica que participa en la resistencia a infecciones y tumores
malignos, actuando como inmunoestimulante y mediador de la respuesta
inflamatoria.12 Las células hematopoyéticas progenitoras (CD34+) cultivadas en

Page 7 of 56
presencia del factor estimulante de colonias granulocítico-macrofágico (GM-CSF)
y el TNFα, se diferencian a células de Langerhans.13 Se documentó la presencia
de IL-1α y TNFα en lesiones de histiocitosis.10 Las concentraciones séricas de
TNFα en pacientes con histiocitosis (HCL) se hallaron significativamente más
elevados que en controles sanos.14
Las concentraciones séricas de IL-1Ra (antagonista natural de la IL-1) en
pacientes con histocitosis (HCL) se hallaron significativamente más elevados
que en controles. El incremento del IL-1Ra señala y categoriza la inflamación
presente en los pacientes y de sus consecuencias negativas (ej. Fibrosis) sobre
los diferentes órganos.
Las CL de la HCL tienen algunas características similares a las células
dendítricas inmaduras así como a las células dendríticas activadas. El
tranforming growth factor beta (TGF-beta) así como la IL-10 han sido reportadas
como responsables de inhibir la maduración de las Cl en la HCL.15
En la HCL, ningún estímulo inmunológico pudo ser definido. Los intentos
de determinar una causa viral no han tenido éxito.16 Existe un aumento de las
células T reguladoras en las lesiones de HCL así como expansión de estas
células inmunológicas inhibitorias en la sangre periférica de pacientes con
enfermedad activa, aunque la etiología y la significación de estas observaciones
no han sido aclaradas. Se han encontrado niveles elevados de otros reguladores
del sistema inmunológico y del metabolismo óseo, como la Osteoprotegerina
(OPG) en el plasma de pacientes con HCL activa. Los niveles de OPG son
elevados en pacientes con enfermedad multisistémica y disminuyen con la
respuesta al tratamiento.17,18
B.6 Anatomía Patológica Las lesiones típicas de HCL están compuestas por células de Langerhans
(CL), células intermedias e interdigitantes de linaje dendrítico, linfocitos T,
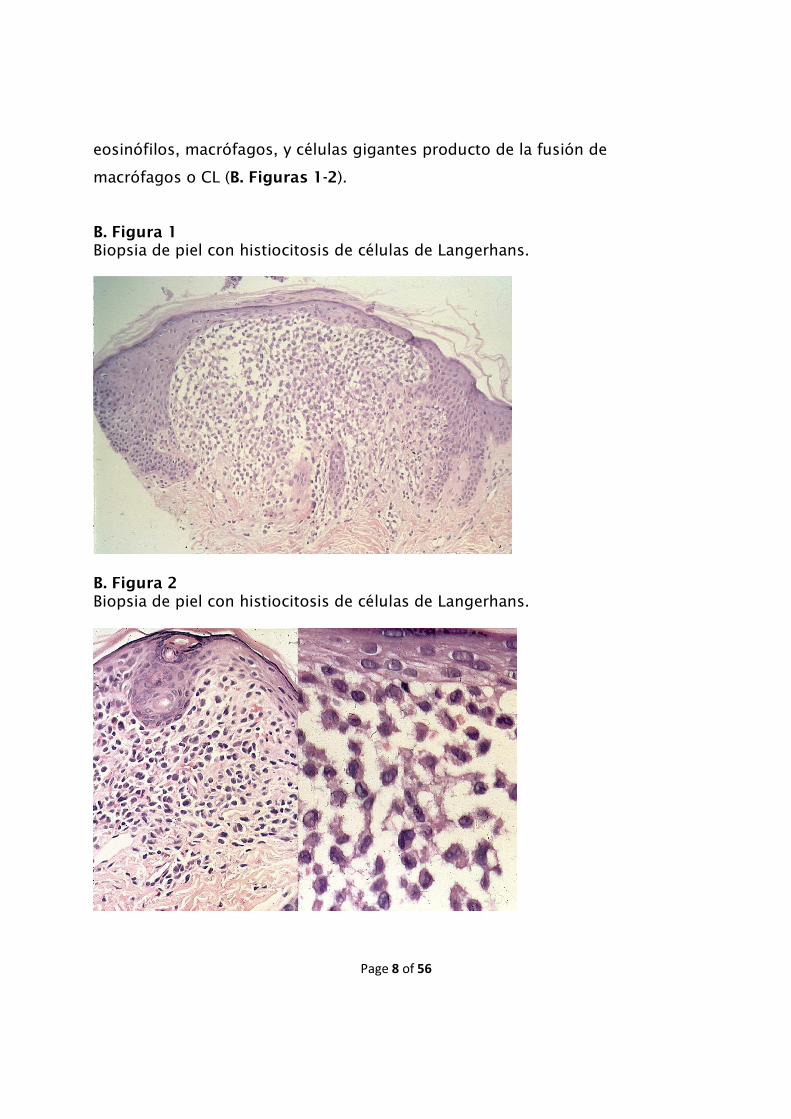
Page 8 of 56
eosinófilos, macrófagos, y células gigantes producto de la fusión de
macrófagos o CL (B. Figuras 1-2).
B. Figura 1 Biopsia de piel con histiocitosis de células de Langerhans.
B. Figura 2 Biopsia de piel con histiocitosis de células de Langerhans.
Page 9 of 56
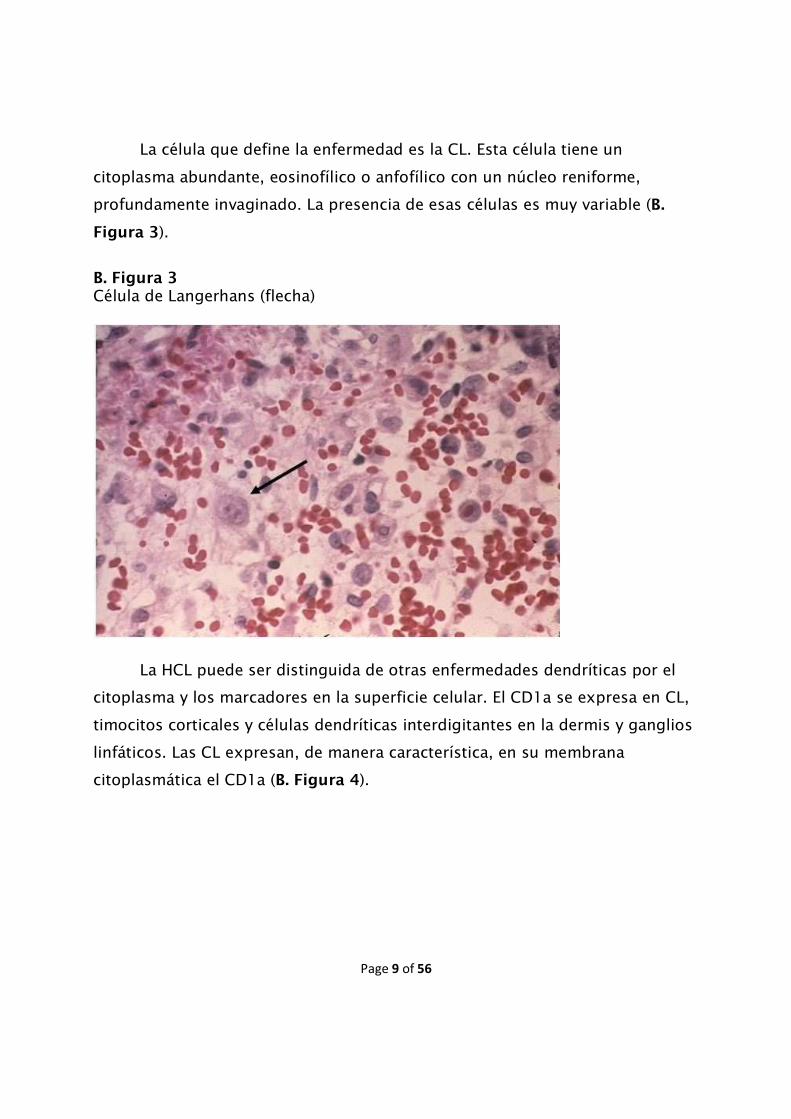
La célula que define la enfermedad es la CL. Esta célula tiene un
citoplasma abundante, eosinofílico o anfofílico con un núcleo reniforme,
profundamente invaginado. La presencia de esas células es muy variable (B.
Figura 3).
B. Figura 3 Célula de Langerhans (flecha)
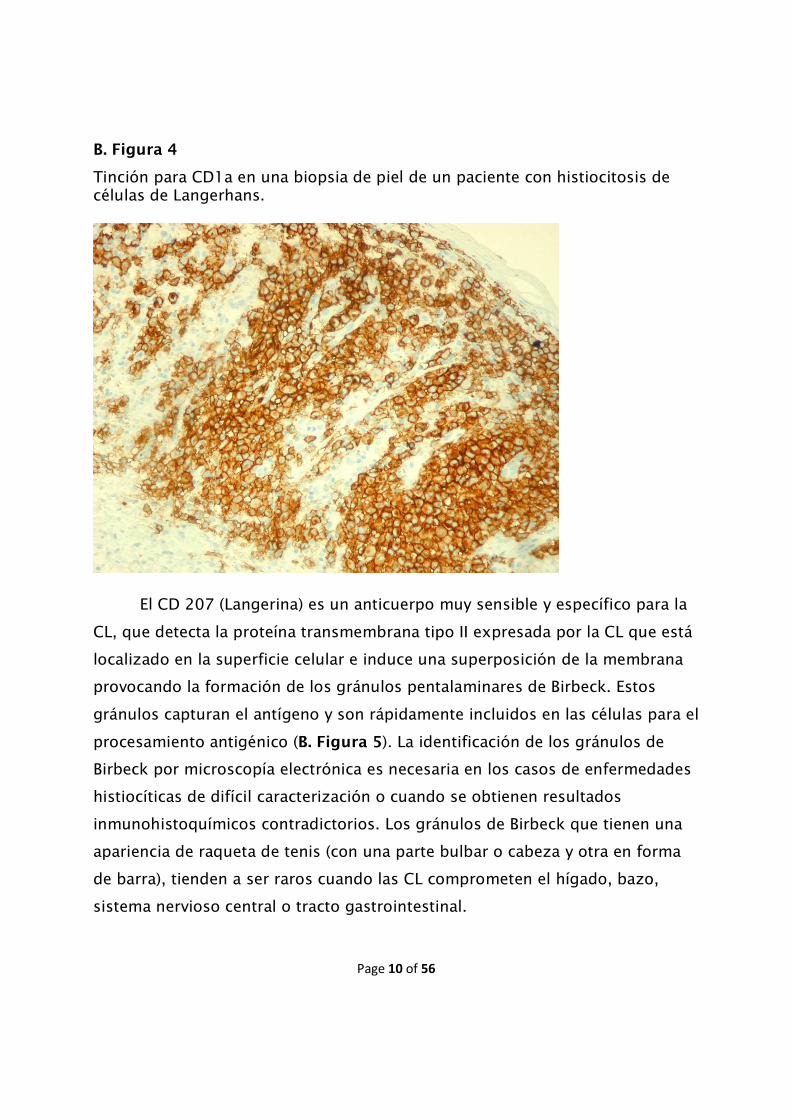
La HCL puede ser distinguida de otras enfermedades dendríticas por el
citoplasma y los marcadores en la superficie celular. El CD1a se expresa en CL,
timocitos corticales y células dendríticas interdigitantes en la dermis y ganglios
linfáticos. Las CL expresan, de manera característica, en su membrana
citoplasmática el CD1a (B. Figura 4).
Page 10 of 56
B. Figura 4
Tinción para CD1a en una biopsia de piel de un paciente con histiocitosis de células de Langerhans.
El CD 207 (Langerina) es un anticuerpo muy sensible y específico para la
CL, que detecta la proteína transmembrana tipo II expresada por la CL que está
localizado en la superficie celular e induce una superposición de la membrana
provocando la formación de los gránulos pentalaminares de Birbeck. Estos
gránulos capturan el antígeno y son rápidamente incluidos en las células para el
procesamiento antigénico (B. Figura 5). La identificación de los gránulos de
Birbeck por microscopía electrónica es necesaria en los casos de enfermedades
histiocíticas de difícil caracterización o cuando se obtienen resultados
inmunohistoquímicos contradictorios. Los gránulos de Birbeck que tienen una
apariencia de raqueta de tenis (con una parte bulbar o cabeza y otra en forma
de barra), tienden a ser raros cuando las CL comprometen el hígado, bazo,
sistema nervioso central o tracto gastrointestinal.

Page 11 of 56
B. Figura 5 Microscopía electrónica mostrando un gránulo de Birbeck (flecha).
B. References 1Nezelof C, Guibert C. [Histiocytosis X; Nosology and Prognosis. Study Based on the Observed Course of 52 Personal Cases]. Arch Fr Pediatr. 1963; 20: 1063-1103. 2Salotti JA, Nanduri V, Pearce MS et al. Incidence and clinical features of Langerhans cell histiocytosis in the UK and Ireland. Arch Dis Child. 2009; 94: 376-380. 3Vade A, Hayani A, Pierce KL. Congenital histiocytosis X. Pediatr Radiol. 1993; 23: 181-182. 4Gadner H. Treatment of adult-onset Langerhans cell histiocytosis--is it different from the pediatric approach? Ann Oncol. 2010; 21: 1141-1142. 5Arico M, Girschikofsky M, Genereau T et al. Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer. 2003; 39: 2341-2348. 6Patel AB, Renge RL. Langherhans cell histiocytosis in monozygotic twins. Indian Pediatr. 2001; 38: 788-791. 7Hsu TS, Komp DM. Clinical features of familial histiocytosis. Am J Pediatr Hematol Oncol. 1981; 3: 61-65. 8Willman CL, Busque L, Griffith BB et al. Langerhans'-cell histiocytosis (histiocytosis X)--a clonal proliferative disease. N Engl J Med. 1994; 331: 154-160. 9Kannourakis G, Abbas A. The role of cytokines in the pathogenesis of Langerhans cell histiocytosis. Br J Cancer Suppl. 1994; 23: S37-40. 10Egeler RM, Favara BE, van Meurs M et al. Differential In situ cytokine profiles of Langerhans-like cells and T cells in Langerhans cell histiocytosis: abundant expression of cytokines relevant to disease and treatment. Blood. 1999; 94: 4195-4201.

Page 12 of 56
11Heufler C, Koch F, Schuler G. Granulocyte/macrophage colony-stimulating factor and interleukin 1 mediate the maturation of murine epidermal Langerhans cells into potent immunostimulatory dendritic cells. J Exp Med. 1988; 167: 700-705. 12Tracey KJ, Cerami A. Tumor necrosis factor: a pleiotropic cytokine and therapeutic target. Annu Rev Med. 1994; 45: 491-503. 13Caux C, Dezutter-Dambuyant C, Schmitt D, Banchereau J. GM-CSF and TNF-alpha cooperate in the generation of dendritic Langerhans cells. Nature. 1992; 360: 258-261. 14Rosso DA, Ripoli MF, Roy A et al. Serum levels of interleukin-1 receptor antagonist and tumor necrosis factor-alpha are elevated in children with Langerhans cell histiocytosis. J Pediatr Hematol Oncol. 2003; 25: 480-483. 15de Graaf JH, Tamminga RY, Dam-Meiring A et al. The presence of cytokines in Langerhans' cell histiocytosis. J Pathol. 1996; 180: 400-406. 16Mierau GW, Wills EJ, Steele PO. Ultrastructural studies in Langerhans cell histiocytosis: a search for evidence of viral etiology. Pediatr Pathol. 1994; 14: 895-904. 17Rosso DA, Karis J, Braier JL et al. Elevated serum levels of the decoy receptor osteoprotegerin in children with langerhans cell histiocytosis. Pediatr Res. 2006; 59: 281-286. 18Ishii R, Morimoto A, Ikushima S et al. High serum values of soluble CD154, IL-2 receptor, RANKL and osteoprotegerin in Langerhans cell histiocytosis. Pediatr Blood Cancer. 2006; 47: 194-199.
C. Características Clínicas Las manifestaciones clínicas de la HCL son muy variables, cuadros
clínicos leves e incluso con regresiones espontáneas, crónicos recurrentes, o
diseminados y potencialmente mortales.
Los signos clínicos más frecuentes que se presentan en la HCL involucran
a la piel o a los huesos usualmente con una lesión dolorosa. Los pacientes
pueden también tener entre otros síntomas: fiebre, pérdida de peso, diarrea,
edema, disnea, polidipsia y poliuria.
Los pacientes con HCL se dividen por categorías de alto y bajo riesgo,
según el compromiso específico del órgano. Los pacientes pueden presentar la
enfermedad en un sitio (unifocal) o en múltiples sitios (multifocal). Las
decisiones terapéuticas están basadas de acuerdo al compromiso o no de de
órganos de bajo o alto riesgo o si la HCL presenta una forma unisistémica o
multisistémica. Los pacientes pueden tener una HCL con cualquier combinación
Page 13 of 56
de compromisos de piel, huesos, ganglios linfáticos y aún así ser considerados
como de bajo riesgo.
La enfermedad multisistémica presenta compromiso de dos o más
órganos o sistemas: hígado, bazo, pulmones, sistema hematopoyético (riesgo)
o huesos, piel, ganglios linfáticos, sistema endocrinológico (bajo riesgo) y
Sistema nervioso Central (SNC).
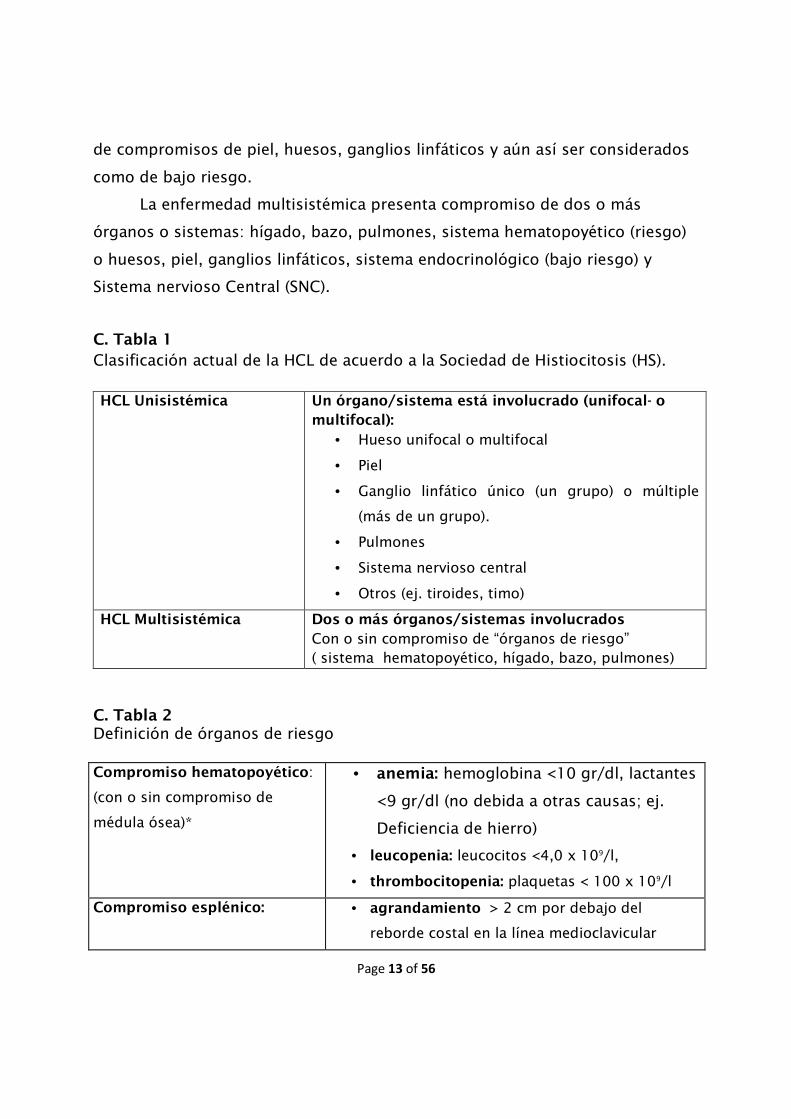
C. Tabla 1
Clasificación actual de la HCL de acuerdo a la Sociedad de Histiocitosis (HS). HCL Unisistémica
Un órgano/sistema está involucrado (unifocal- o
multifocal):
• Hueso unifocal o multifocal
• Piel
• Ganglio linfático único (un grupo) o múltiple
(más de un grupo).
• Pulmones
• Sistema nervioso central
• Otros (ej. tiroides, timo)
HCL Multisistémica
Dos o más órganos/sistemas involucrados
Con o sin compromiso de “órganos de riesgo” ( sistema hematopoyético, hígado, bazo, pulmones)
C. Tabla 2 Definición de órganos de riesgo
Compromiso hematopoyético:
(con o sin compromiso de
médula ósea)*
• anemia: hemoglobina <10 gr/dl, lactantes
<9 gr/dl (no debida a otras causas; ej.
Deficiencia de hierro)
• leucopenia: leucocitos <4,0 x 109/l,
• thrombocitopenia: plaquetas < 100 x 109/l
Compromiso esplénico: • agrandamiento > 2 cm por debajo del
reborde costal en la línea medioclavicular

Page 14 of 56
Compromiso hepático: • agrandamiento > 3 cm por debajo del
reborde costal en la linea medioclavicular y/o
• disfunción hepática (ej. hipoproteinemia
<55gr/l, hipoalbuminemia <25gr/l no debida a
otras causas) y/o
• diagnóstico histopatológico
Compromiso pulmonar: • cambios típicos en la tomografía computada de
alta resolución (Si está disponible, con
tomografía computada con multidetector a baja
dosis) y/o
• diagnóstico histopatológico / citológico
*El compromiso de médula ósea se define por la presencia de células CD1a + en sus muestras. El significado clínico y pronóstico de su presencia, no está aun comprobado. Sin embargo, un estudio mostró que el compromiso pulmonar sin otro
compromiso en órganos de riesgo, no tuvo un pronóstico desfavorable.1
• “Sitios especiales”- Son lesiones en un sitio anatómico funcionalmente
crítico (ej. apófisis odontoide, lesion vertebral con invasión intraespinal
de partes blandas en donde la progresión de la enfermedad y los riesgos
de un intento de tratamiento local pueden causar un riesgo inmediato al
paciente. Una enfermedad localizada en un sitio especial puede justificar
un tratamiento sistémico. Las lesiones vertebrales sin compromiso de
partes blandas (ej. Vertebra plana) no están incluidas dentro del grupo de
“sitios especiales”
• “Compromiso óseo craneofacial” - lesiones en órbita, temporal,
mastoides, esfenoides, región zigomática, etmoides, maxilar superior,
senos paranasales, o base de cráneo, con extensión intracraneal de
partes blandas.

Page 15 of 56
• Compromiso de la region ocular- proptosis, exoftalmía o lesiones en las
órbitas, región zigomática o hueso esfenoidal.
• Compromiso de la región auditiva- otitis media, otorrea o lesiones en el
hueso temporal (mastoides, peñasco)
• Compromiso de la región oral lesiones en encías, hueso del paladar,
maxilares superior e inferior
• “Lesiones de riesgo para el SNC” -Existen datos preliminares que
sugieren que el compromiso del cráneo (excluyendo la calota) predispone
al desarrollo de diabetes insípida (DI). En particular, aquellos pacientes
con lesiones en las regiones orbitarias, auditivas y orales tienen un
riesgo significativamente mayor de desarrollar DI. Este riesgo es todavía
mayor cuando la enfermedad persiste en actividad por un largo período o
si se reactiva.2
C.1 Compromiso Específico de Órganos C.1.1 Piel Las lesiones de piel pueden ser la única evidencia de la enfermedad o formar
parte de un compromiso sistémico. Los lactantes pueden tener una lesión cutánea
seborreica descamativa en cuero cabelludo a menudo confundida con la dermatitis
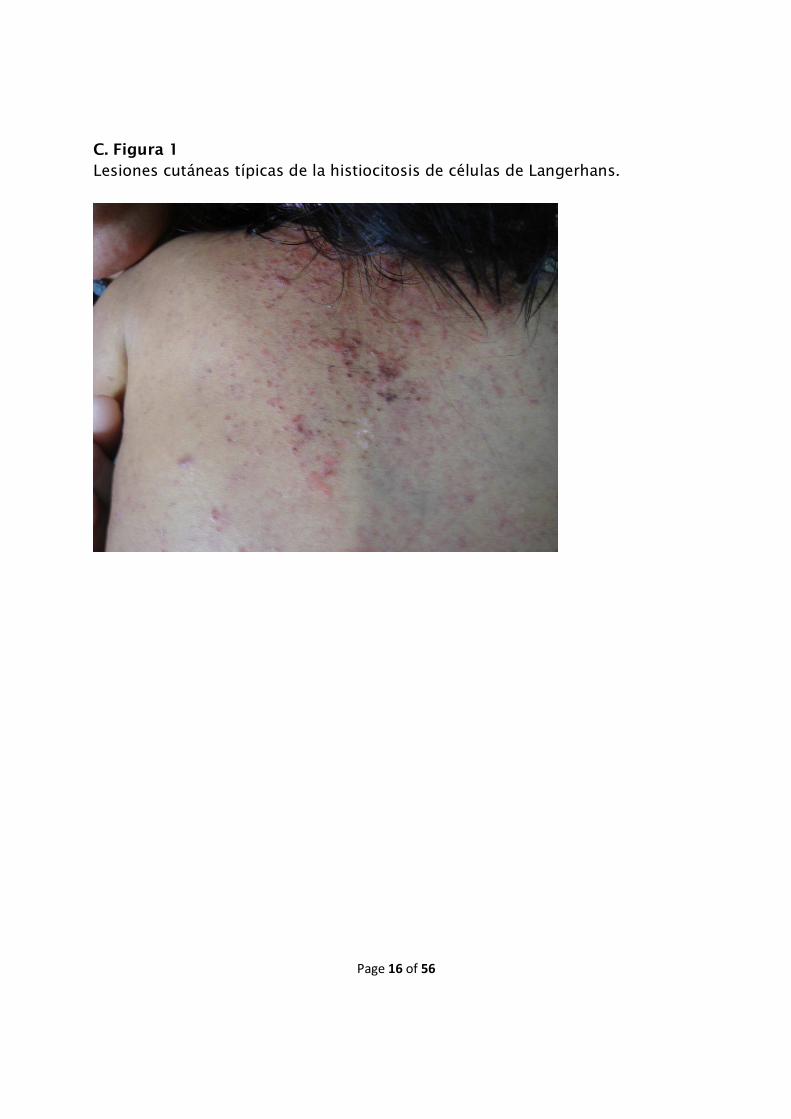
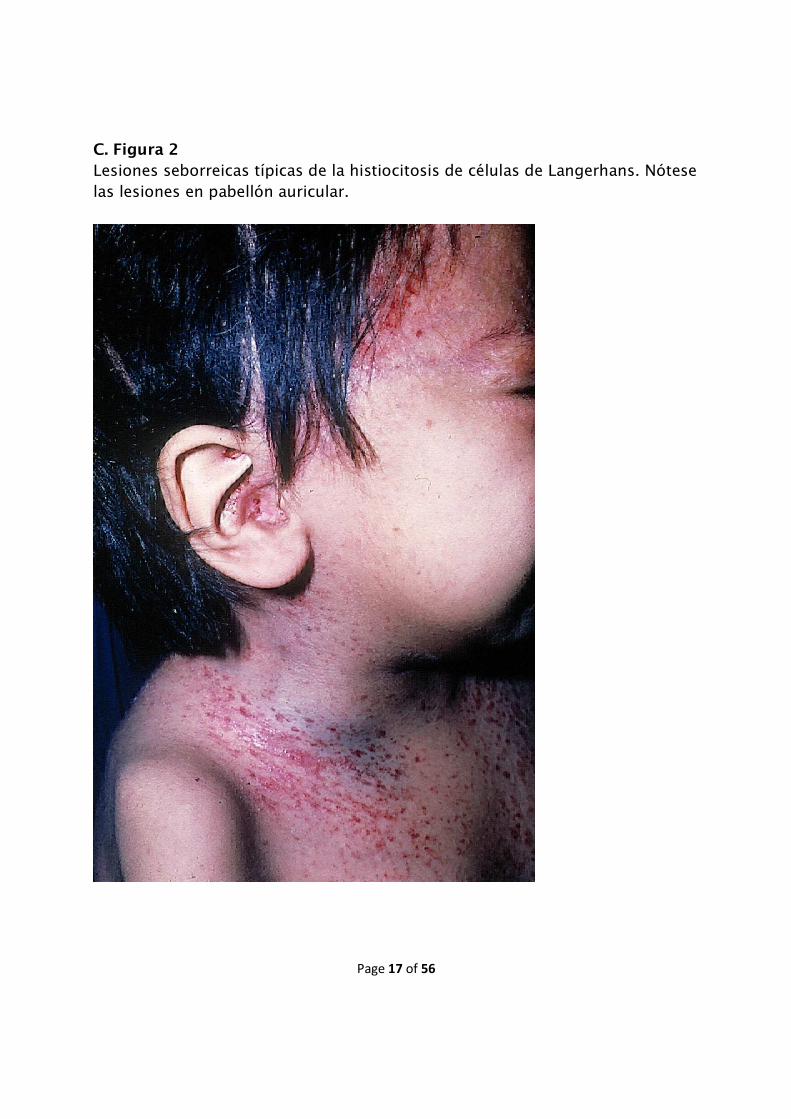
seborreica de los lactantes (C. Figuras 1 y 2).
Page 16 of 56
C. Figura 1
Lesiones cutáneas típicas de la histiocitosis de células de Langerhans.
Page 17 of 56
C. Figura 2
Lesiones seborreicas típicas de la histiocitosis de células de Langerhans. Nótese las lesiones en pabellón auricular.

Page 18 of 56
Los niños y los adultos pueden desarrollar lesiones usualmente no
pruriginosas, eritematopapulares, petequiales, en ocasiones, erosivas formando
costras serosas en el cuero cabelludo, ingles, región retroauricular, abdomen,
dorso, y tórax que se pueden parecer a las lesiones cutáneas difusas por
cándidas. En ocasiones se puede presentar alopecía.
Las lesiones ulceradas detrás de las orejas, o comprometiendo el cuero
cabelludo, genitales o región perianal pueden ser confundidas con infecciones
bacterianas o micóticas. Esta manifestación puede ser autolimitada, ya que las
lesiones en general desaparecen sin ningún tratamiento durante el primer año
de vida. Sin embargo estos pacientes deben ser monitoreados frecuentemente
por la posibilidad de evolución a una enfermedad sistémica. Los pacientes
pueden también presentar pápulas o lesiones nodulares de aspecto purpurico o
marrón en cualquier parte del cuerpo que involucionan espontáneamente.
(Enfermedad de Hashimoto-Pritzker).16
En ocasiones las lesiones cutáneas son extensas y coexisten con
visceromegalias y citopenias.
C. Figura 3
Compromiso en pliegues en un paciente con hepatoesplenomegalia.

Page 19 of 56
C.1.2 Mucosa Oral
Los síntomas que se pueden presentar incluyen: hipertrofia gingival,
sangrado, úlceras de paladar duro o blando, mucosa bucal, lengua y labios. Las
lesiones de la mucosa oral pueden preceder localizaciones de la enfermedad en
otros sitios.
C. Figura 4 Compromiso de la encía por histiocitosis de células de Langerhans.
C.1.3 Huesos Es la localización más frecuente en la HCL (alrededor del 80%).3 Las
lesiones pueden ser únicas o múltiples, asintomáticas o dolorosas. El sitio más
frecuente de compromiso óseo, es el cráneo, mostrando lesiones únicas o
múltiples típicamente osteolíticas, como en sacabocados sin reacción perióstica
circundante .

Page 20 of 56
C. Figura 5 Lesión típica en calota (Cortesía Dra Ana María Silván, Sevilla, España)
Otros sitios frecuentes son fémur, costilla, vértebras (frecuentemente
cervicales), ilíaco y húmero. Las lesiones usualmente líticas pueden causar
aplastamiento de vertebras o también masas orbitarias que pueden producir
proptosis confundiéndose con rabdomiosarcoma, leucemia, neuroblastoma,
pseudo tumor inflamatorio o tumores lipídicos de esa región. Algunas lesiones
líticas de cráneo pueden tener además compromiso de partes blandas que
comprometen a la duramadre. Las lesiones en huesos largos pueden ocasionar
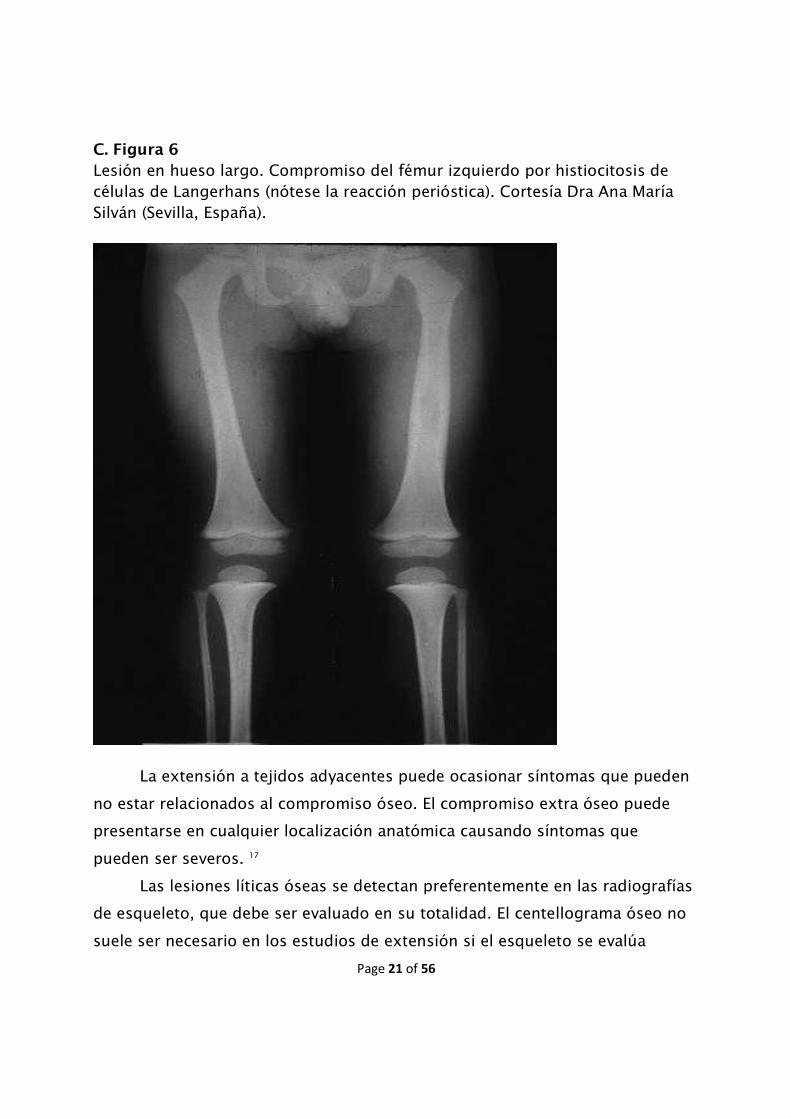
raramente fracturas y también reacción perióstica.
Page 21 of 56
C. Figura 6
Lesión en hueso largo. Compromiso del fémur izquierdo por histiocitosis de células de Langerhans (nótese la reacción perióstica). Cortesía Dra Ana María Silván (Sevilla, España).
La extensión a tejidos adyacentes puede ocasionar síntomas que pueden
no estar relacionados al compromiso óseo. El compromiso extra óseo puede
presentarse en cualquier localización anatómica causando síntomas que
pueden ser severos. 17
Las lesiones líticas óseas se detectan preferentemente en las radiografías
de esqueleto, que debe ser evaluado en su totalidad. El centellograma óseo no
suele ser necesario en los estudios de extensión si el esqueleto se evalúa

Page 22 of 56
cuidadosamente con radiología. Las lesiones de los huesos de la cara, fosa
craneal anterior o media (hueso temporal, etmoides, esfenoides, región
zigomática) con extensión tumoral intracraneana forman parte del grupo de
riesgo de compromiso del SNC. Estos pacientes tienen tres veces más riesgo de
desarrollar DI y también con más riesgo de otras lesiones del SNC que serán
descriptas más adelante. Las lesiones que se localizan en el oído medio (hueso
temporal) son frecuentes y ocasionan habitualmente supuración ótica
persistente que puede ser difícil de distinguir de procesos infecciosos.
C. Figura 7
Compromiso por histiocitosis de células de Langerhans del hueso temporal (flecha) produciendo lesión osteolítica.
Las lesiones vertebrales con compromiso de partes blandas a menudo
presentan dolor y compromiso neurológico. Las lesiones gingivales y en huesos
maxilares pueden causar caída de piezas dentarias.

Page 23 of 56
C.2 Ganglios Linfáticos y Timo
Los ganglios cervicales son los más frecuentemente comprometidos,
pueden ser de consistencia blanda o dura y tener edema linfático asociado. En
ocasiones, la HCL puede presentarse como una hiperplasia tímica o
compromiso ganglionar mediastinal que puede confundirse con un linfoma o
un proceso infeccioso pudiendo causar síntomas de tipo bronquial obstructivo
(tos, disnea, cianosis).4
C.3 Hígado y Bazo
En la HCL, el hígado y el bazo son considerados órganos de riesgo ya
que el compromiso de estos órganos influencia negativamente el pronóstico. La
edad mediana de los niños con compromiso hepático es de alrededor de 2 años
y ellos se pueden presentar con hepatomegalia o hepatoesplenomegalia,
fosfatasa alcalina elevada, aumento de transaminasas y gama-glutamil
transpeptidasa. La hepatomegalia puede estar acompañada por disfunción que
puede asociarse con hipoalbumiemia, ascitis, hiperbilirrubinemia y déficit de
factores de coagulación. La ecografía de hígado puede mostrar imágenes
hipoecoicas o señales de baja intensidad a lo largo de las venas portales o
tractos biliares. Una de las más severas complicaciones del compromiso
hepático es la colestasis progresiva que evoluciona a una colangitis
esclerosante, las biopsias usualmente no revelan la presencia de CL, pero si de
un infiltrado linfocitario periportal.5 Se cree que las citoquinas elaboradas por
los linfocitos periportales puede dañar el tracto biliar. La gran mayoría de los
pacientes con colangitis esclerosante no responderán a la medicación y
requerirán transplante hepático. La presencia de hipoalbuminemia al
diagnóstico en los pacientes con compromiso hepático mostró una significativa
asociación con un peor pronóstico, según un estudio reportado.6
Page 24 of 56
La esplenomegalia masiva puede causar citopenias por el
hiperesplenismo y compromiso respiratorio mecánico, sin embargo, la
esplenectomía está raramente indicada.
C.4 Sistema Gastrointestinal
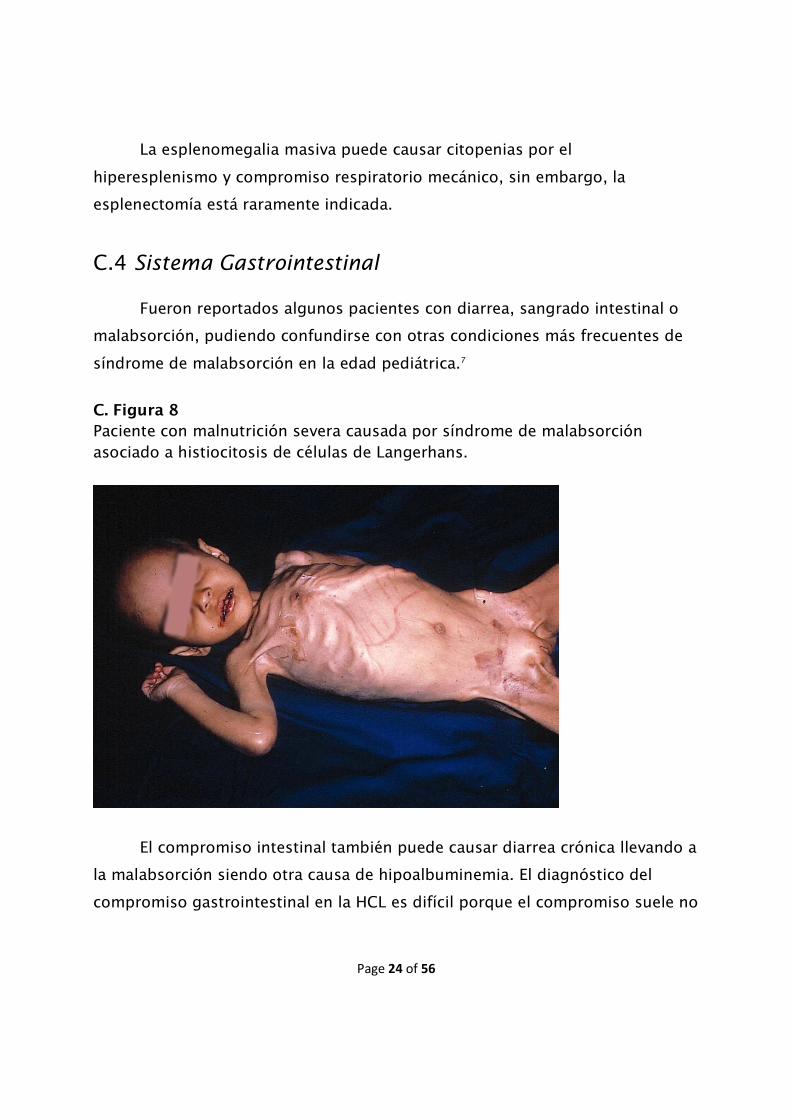
Fueron reportados algunos pacientes con diarrea, sangrado intestinal o
malabsorción, pudiendo confundirse con otras condiciones más frecuentes de
síndrome de malabsorción en la edad pediátrica.7
C. Figura 8
Paciente con malnutrición severa causada por síndrome de malabsorción asociado a histiocitosis de células de Langerhans.
El compromiso intestinal también puede causar diarrea crónica llevando a
la malabsorción siendo otra causa de hipoalbuminemia. El diagnóstico del
compromiso gastrointestinal en la HCL es difícil porque el compromiso suele no
Page 25 of 56
afectar la mucosa en forma uniforme. Es necesaria una evaluación endoscópica
cuidadosa con múltiples biopsias, para confirmar la HCL en esa localización.
C.6 Pulmones
El compromiso pulmonar puede ser asintomático o presentar taquipnea,
disnea, tos, o dolor torácico. Los pulmones están afectados menos
frecuentemente en los niños que en los adultos, en los que el cigarrillo juega
un rol etiológico clave. La radiografía suele mostrar un infiltrado intersticial
inespecífico. La tomografía computada de tórax de alta resolución es necesaria
para visualizar el patrón quístico /nodular con destrucción de tejido
pulmonar,que se puede presentar en estos pacientes.
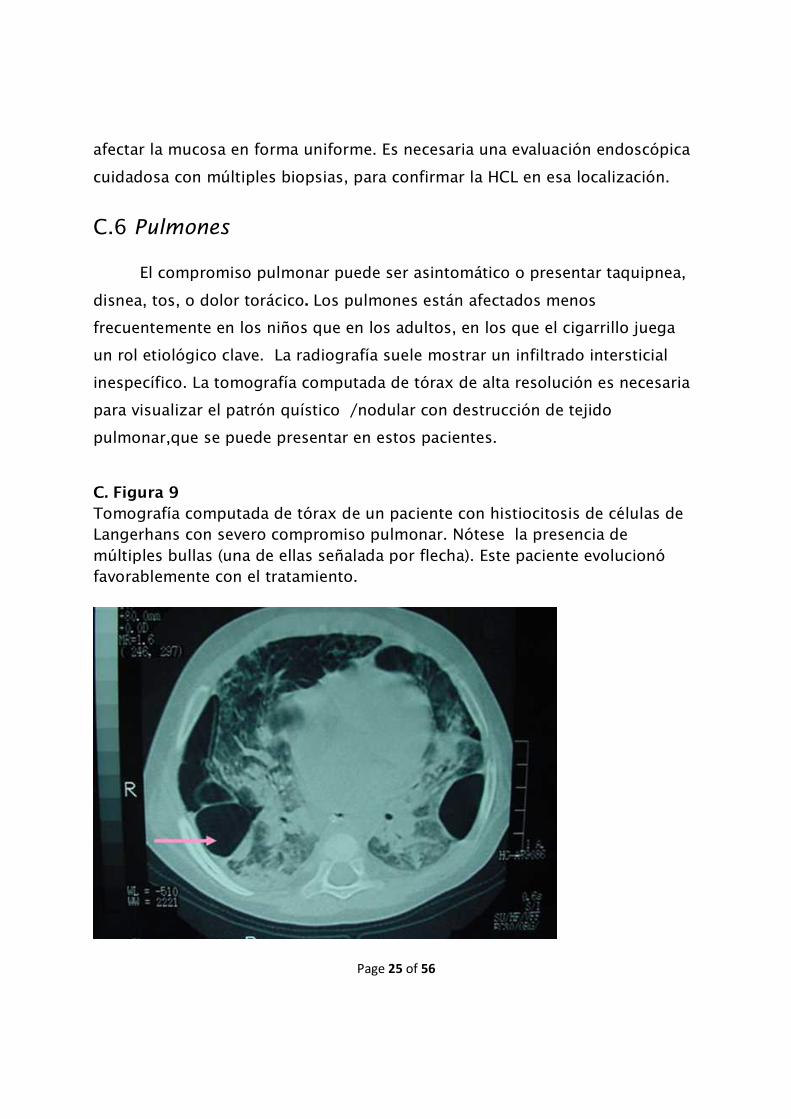
C. Figura 9
Tomografía computada de tórax de un paciente con histiocitosis de células de Langerhans con severo compromiso pulmonar. Nótese la presencia de múltiples bullas (una de ellas señalada por flecha). Este paciente evolucionó favorablemente con el tratamiento.

Page 26 of 56
Los niños con compromiso pulmonar y de órganos de bajo riesgo tienen
una sobrevida a 5 años sin diferencias significativas cuando se las compara con
aquellos pacientes que tienen solo compromiso de órganos de bajo riesgo.1
Las pruebas funcionales pulmonares pueden ser anormales, con un
compromiso restrictivo y disminución del volumen pulmonar. La disminución
de la capacidad de difusión pulmonar puede también preceder el comienzo de
la hipertensión pulmonar. En los niños pequeños con enfermedad pulmonar
extensa el tratamiento puede detener la destrucción tisular y mecanismos de
reparación pueden restaurar parcialmente el funcionamiento pulmonar.
C.7 Sistema Hematopoyético El compromiso del sistema hematopoyético se presenta más
frecuentemente en niños pequeños (anemia, trombocitopenia y/o neutropenia)
que tienen una enfermedad diseminada que suele comprometer hígado, bazo,
ganglios linfáticos y/o piel.6, 8 Otros pacientes tienen solo citopenias leves. En
un estudio realizado en niños con este sistema afectado se les encontró
compromiso de médula ósea con presencia de células de Langerhans en
estudios por análisis de citometría de flujo o inmunohistoquímicos de la
médula ósea. Los pacientes con una enfermedad de alto riesgo pueden
presentar hemofagocitosis médular. En estos pacientes puede ser difícil decir
cuál de las dos entidades es primaria: la HCL o la Linfohistiocitosis
Hemofagocítica.18
La presencia de anemia y trombocitopenia con o sin leucopenia al
diagnóstico en los pacientes con compromiso del sistema hematopoyético
mostró una significativa asociación con un peor pronóstico, según un estudio
reportado.6
En un estudio realizado en biopsias de médula de médula ósea se
encontraron CL en un bajo porcentaje de casos (14%). Los hallazgos más
frecuentes fueron: mielofibrosis, hemofagocitosis (especialmente en las

Page 27 of 56
citopenias severas), mielodisplasia y displasia megacariocítica con
emperipolesis.
C.8 Sistema Nervioso Central
C.8.1 Compromiso Neuroendócrino La DI, causada por el daño de la parte posterior de la hipófisis, es la
manifestación endocrinológica más frecuente de la HCL.10 Algunos pacientes
presentan la DI previamente a las otras lesiones de la enfermedad. 11 Un estudio
sobre estos pacientes encontró que el 51 % de ellos tendrán otras lesiones
diagnósticas de HCL durante el año de detectada la DI.12 Un estudio realizado
sobre 589 pacientes, mostró que el riesgo a 10 años de tener una DI es del 24
%.13 Estos investigadores no constataron una disminución de la incidencia de la
DI en pacientes tratados con quimioterapia. Sin embargo otro estudio reportó
una disminución en la incidencia de DI del 40 % al 20 % con 6 meses de
tratamiento con prednisona y vinblastina, en los pacientes con riesgo de
compromiso del SNC.14
Las biopsias de la hipófisis son raramente necesarias. Más a menudo, el
diagnóstico se hace con una biopsia de piel, hueso o ganglio linfático en un
paciente que ya tiene la anormalidad de la hipófisis. La frecuencia de la DI está
aumentada en los pacientes con lesiones óseas en “sitios especiales” ya que el
75% de los pacientes con DI tuvieron estos compromisos previamente. Un
estudio mostró que la frecuencia de la DI en pacientes tratados con
poliquimioterapia fue del 11% y más del 50% en aquellos que no fueron
tratados agresivamente.14 Aproximadamente la mitad de los pacientes con HCL
y DI, desarrollaran compromiso de la parte anterior de la hipófisis con
deficiencia de las hormonas de crecimiento, tiroidea y gonadotróficas durante
los 10 años que siguen al diagnóstico de la DI.

Page 28 of 56
C.8.2 Otros compromisos crónicos del Sistema Nervioso Central Los pacientes con HCL pueden desarrollar lesiones tumorales en los
plexos coroideos o substancia blanca.15 Estas lesiones contienen células de
Langerhans CD1a positivas y linfocitos CD 8 positivos que en ocasiones forman
lesiones pseudotumorales (C. Figura 5). Los pacientes con HCL pueden tener
un síndrome neurodegenerativo crónico (1 a 4 %) que se manifiesta con
disartria, ataxia, dismetría y en ocasiones cambios de conducta. La resonancia
magnética nuclear muestra hiperintensidad del núcleo dentado y substancia
blanca del cerebelo (en T 2) (C. Figura 6) o lesiones hiperintensas en los
núcleos de la base (en T1) y atrofia del cerebelo. Los hallazgos diagnósticos
en las imágenes pueden preceder por muchos años el comienzo de los
síntomas o ser coincidentes. Un estudio mostró que 14 de 25 pacientes con
HCL presentaron déficit neuro psicológicos con una mediana de seguimiento de
10 años, 7 presentaban DI y 5 tenían evidencias radiológicas de cambios
neurodegenerativos en sistema nervioso.

Page 29 of 56
C. Figura 10
Lesiones pseudotumorales en cerebelo causadas por histiocitosis de céluals de Langerhans.
C Figura 11 Lesiones desmielinizantes en fosa posterior
Page 30 of 56
C. References 1Braier J, Latella A, Balancini B et al. Outcome in children with pulmonary Langerhans cell Histiocytosis. Pediatr Blood Cancer. 2004; 43: 765-769. 2Grois N, Potschger U, Prosch H et al. Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatr Blood Cancer. 2006; 46: 228-233. 3Wang J, Wu X, Xi ZJ. Langerhans cell histiocytosis of bone in children: a clinicopathologic study of 108 cases. World J Pediatr. 2010; 6: 255-259. 4Yagci B, Varan A, Uner A et al. Thymic Langerhans cell histiocytosis mimicking lymphoma. Pediatr Blood Cancer. 2008; 51: 833-835. 5Lee RJ, Leung C, Lim EJ et al. Liver transplantation in an adult with sclerosing cholangitis due to multisystem Langerhans cell histiocytosis. Am J Transplant. 2011; 11: 1755-1756. 6Braier JL, Rosso D, Latella A et al. Importance of multi-lineage hematologic involvement and hypoalbuminemia at diagnosis in patients with "risk-organ" multi-system Langerhans cell histiocytosis. J Pediatr Hematol. Oncol 2010; 32: e122-125. 7Singhi AD, Montgomery EA. Gastrointestinal tract langerhans cell histiocytosis: A clinicopathologic study of 12 patients. Am J Surg Pathol. 2011; 35: 305-310. 8Nezelof C, Frileux-Herbet F, Cronier-Sachot J. Disseminated histiocytosis X: analysis of prognostic factors based on a retrospective study of 50 cases. Cancer. 1979; 44: 1824-1838. 9Galluzzo ML, Braier J, Rosenzweig SD. et al. Bone marrow findings at diagnosis in patients with multisystem langerhans cell histiocytosis. Pediatr Dev Pathol. 2010; 13: 101-106.

Page 31 of 56
10Makras P, Alexandraki KI, Chrousos GP et al. Endocrine manifestations in Langerhans cell histiocytosis. Trends Endocrinol Metab. 2007; 18: 252-257. 11Marchand I, Barkaoui MA, Garel C et al. Central Diabetes Insipidus as the Inaugural Manifestation of Langerhans Cell Histiocytosis: Natural History and Medical Evaluation of 26 Children and Adolescents. J Clin Endocrinol Metab. 2011; 96: E1352-E1360. 12Prosch H, Grois N, Prayer D et al. Central diabetes insipidus as presenting symptom of Langerhans cell histiocytosis. Pediatr Blood Cancer. 2004; 43: 594-599. 13Donadieu J, Rolon MA, Thomas C et al. Endocrine involvement in pediatric-onset Langerhans' cell histiocytosis: a population-based study. J Pediatr. 2004; 144: 344-350. 14Grois N, Flucher-Wolfram B, Heitger A, et al. Diabetes insipidus in Langerhans cell histiocytosis: results from the DAL-HX 83 study. Med Pediatr Oncol. 1995; 24: 248-256. 15Grois N, Fahrner B, Arceci RJ, et al. Central nervous system disease in Langerhans cell histiocytosis. J Pediatr. 2010; 156: 873-881, 881 e871. 16 Soto V. Langerhans Cell Histiocytosis with Isolated Cutaneous Involvement in an Infant, Oncopedia#390. Released on Oncopedia: 06/14/2010. URL: https://www.cure4kids.org/ums.oncopedia/case_detail/?id390 17 Chantada G. Tumor-like Multiple Flat Bone Involvement in Langerhans Cell Histiocytosis, Oncopedia #210. Released on Oncopedia: 09/29/2008. URL: http://www.cure4kids.org/ums/oncopedia/case_detail/?id=210 18 Soto V. Langerhans Cell Histiocytosis Occuring After Hemophatocytic Syndrome in a 4-Year-Old Girl, Oncopedia #150. Released on Oncopedia 08/03/2009. URL: http://www.cure4kids.org/ums/oncopedia/case_detail/?id=150
D. Evaluacion Diagnostica (NCI)
D.1 Historia Clínica Se debe realizar una historia clínica completa haciendo referencia a:
naturaleza y duración de los síntomas siguientes: dolor, tumefacción, erupción
de piel, otorrea, irritabilidad, fiebre, pérdida de apetito, diarrea, pérdida o
pobre progreso de peso, retardo de crecimiento, polidipsia, poliuria, disnea,
exposición al tabaco, decaimiento, cambios de conducta o perturbaciones
neurológicas.

Page 32 of 56
D.2 Examen Físico En todos los pacientes, es necesario incluir talla, peso y evaluar el
desarrollo puberal (estadificación de Tanner), extensa evaluación de
compromiso cutáneo, presencia de ictericia, palidez, edema, adenopatías,
supuración de oído, anormalidades orbitarias, lesiones en paladar y encías,
estado de dentición, tumefacción de partes blandas, lesiones de mucosas anal y
genital, taquipnea, tiraje intercostal, ascitis, tamaños de hígado y bazo. En el
examen neurológico, evaluar anormalidades de nervios craneanos, hiporreflexia
tendinosa, déficits visuales y disfunción cerebelosa. Esta evaluación clínica
deberá ser realizada en cada visita de control.
D. Tabla 1
Estudios de evaluación al diagnóstico o reactivación o progresión de la HCL, de acuerdo a los criterios de la Sociedad de Histiocitosis
Analisis de laboratorio: • Hemograma completo. Eritrosedimentación
• Proteinograma electroforético, bilirrubina, transaminasas, fosfatasa alcalina, gama glutamiltranspeptidasa
• Urea, creatinina, electrolitos • Ferritina • Coagulograma: TP, TTPK, fibrinógeno
Analisis de orina:
• Densidad y osmolaridad urinaria Ecografia abdominal:
• Tamaño y eco estructura del hígado y bazo Radiografia de tórax Radiografia de todo el esqueleto

Page 33 of 56
D.2.1 Biopsia Diagnóstica Más frecuentemente efectuada en piel, hueso (incluyendo oído medio) y
ganglios linfáticos.
El diagnóstico de la HCL se basa en el examen histológico e
inmunofenotípico de la lesión. El objetivo principal es la identificación
morfológica de la típica CL teñida con hematoxilina-eosina. Además Las
tinciones con anti CD1a y/o anti CD207 (Langerina) son requeridas para el
diagnóstico definitivo de HCL. Se ha demostrado que la expresión de Langerina
confirma la presencia de gránulos de Birbeck por lo que ya no es indispensable
la detección de gránulos de Birbeck para establecer el diagnóstico definitivo de
la HCL.
Los preparados teñidos con Hematoxilina-eosina y S100 no son
considerados suficientes para el diagnóstico de la HCL, ya que otros histiocitos
y macrófagos pueden dar positivos esas tinciones. En el caso de vértebra plana
sin compromiso de partes blandas, en el que el riesgo del procedimiento de la
biopsia es mayor que el beneficio de obtener el tejido para el diagnóstico, se
justifica una actitud expectante.
D. Tabla 2 Otros estudios a utilizarse de acuerdo a la indicación clínica (Sociedad de Histiocitosis) Indicación Estudio Compromiso de órganos de riesgo
• Estudio HLA
Bicitopenia, pancitopenia. Mono-citopenia persistente o prolongada
• Aspirado y biopsia de la médula ósea para descartar otras causas. Detección de CL. Otras alteraciones: mielofibrosis, mielodisplasia, hemofagocitosis.(x)
Disfunción hepática
• La biopsia es recomendada si existe un compromiso hepático significatico (ej. en casos en los que se sospecha colangitis esclerosante)
Compromiso pulmonar (Rx de tórax anormal o signos o síntomas sugestivos de
• Tomografía computada (TC) de alta resolución • Estudios de función pulmonar si la edad lo
permite

Page 34 of 56
compromiso pulmonar) TC anormal o sospecha de otras causas como infecciones atípicas
• Lavado bronco-alveolar (BAL), >5% de células CD1a-positivas en el BAL son diagnósticas en no fumadores
• Biopsia pulmonar si el BAL no es diagnóstico
Sospecha de lesiones óseas en el área cráneo-facial, incluyendo el maxilar inferior o superior Sospecha de lesiones vertebrales Tomografía de Emisión de Positrones (PET). Centellograma óseo
• Resonancia Magnética Nuclear (RMN) de cerebro
• Se puede considerar además la TC para mejor visualización de la lesión ósea
• RMN espinal para excluir compresión medular y evaluar masas en partes blandas
• Pueden ser útiles para evaluar la respuesta al
tratamiento (lesiones óseas u otras).
Anormalidades visuales o neurológicas
• Evaluación neurológica clínica • Evaluación europsicométrica • RMN de cerebro
Sospecha de compromiso endocrinológico (baja talla, retardo de crecimiento, poliuria, polidipsia, síndromes hipotalámicos, pubertad precoz o demorada) y/o alteraciones en las imágenes hipotalámicas o pituitarias
• Evaluación endocrinológica (incluyendo test de la deprivación hídrica y pruebas para evaluar la hipófisis anterior)
• MRI de cerebro
Otorrea o sospecha de compromiso auditivo
• Evaluación fonoaudiológica • RMN de cerebro • TC del hueso temporal
Diarrea crónica inexplicada, retraso de crecimiento o evidencia de mala absorción intestinal
• Endoscopia y biopsia
(x) Aspirado y biopsia de médula ósea: especialmente en pacientes con compromiso
multisistémico con presencia de citopenias o dolores óseos difusos o síndrome febril
prolongado al diagnóstico, para descartar otras etiologías.

Page 35 of 56
E. Tratamiento
Para planear el tratamiento de la HCL, es necesario considerar si el
paciente presenta enfermedad unisistémica, multisitémica y si presenta
compromiso de órganos de riesgo.
E.1 Enfermedad Unisistémica El tratamiento adecuado debe basarse en la distinta afectación de los
órganos involucrados.
Los casos con compromiso unisitémico en el hueso pueden ser tratados
de múltiples formas. En casos que solo se evidencie una sola lesión que
comprometa al hueso frontal, parietal, occipital u otra lesión única localizada
en cualquier otro hueso pueden plantearse solamente tratamiento con el
curetaje con el que se obtiene la biopsia, ya que algunos de estos pacientes
pueden presentar una regresión de la lesión. Las lesiones localizadas
sintomáticas también pueden requerir tratamiento para mejorar los síntomas,
utilizando esteroides como la prednisona por via oral o en otros casos, se
puede requerir administración intra-lesional de esteroides.1, 2 Otra alternativa
incluye la indometacina (anti inflamatorio no esteroídeo), la cual puede ser útil
y tener un rol en el tratamiento de primera línea en pacientes con compromiso
óseo unisistémico.3
En pacientes que presentan lesiones vertebrales o femorales en riesgo de
colapso puede ser necesario tomar una conducta más agresiva con corticoides
o quimioterapia, con miras a lograr una rápida resolución de los síntomas. En
estos casos, también puede estar indicada la radioterapia localizada a baja
dosis.4 Cuando existe inestabilidad de las vértebras cervicales y síntomas
neurológicos, pueden ser necesarios tratamientos ortopédicos
complementarios.

Page 36 of 56
Las lesiones localizadas en el hueso temporal (mastoides, oído medio) u
órbita son considerados sitios especiales para los lineamientos de la HS y se
recomienda el uso de prednisona y vinblastina, durante 6 a 12 meses, lo cual
pretende disminuir el riesgo de desarrollar diabetes insípida a la vez de mejorar
las tasas de respuesta.5
En los pacientes con HCL limitada a la piel, se puede observar una actitud
expectante, ya que se puede producir en muchos casos la regresión de la lesión
de manera espontánea. En casos que no resuelven espontáneamente, se
pueden utilizar una variedad de tratamientos incluyendo esteroides tópicos,
metotrexate oral,6 talidomida oral,7 aplicación tópica de mostaza nitrogenada8 y
psoralenos con luz ultravioleta.9 La elección de cada uno de ellos dependerá de
la disponibildad y la experiencia local, además de la ubicación y tipo de lesión.
E.2 Enfermedad multisitémica (Filosofía de tratamiento basada en los estudio de la Sociedad de Histiocitosis) E.2.1 Etapa previa a la Sociedad de Histiocitosis En la etapa que precedió a la creación de la Sociedad de Histiocitosis /
Histiocyte Society (HS), la enfermedad tuvo grandes cambios en la estrategia
terapéutica que reflejó los cambios en la comprensión de la enfermedad.
Durante la década del sesenta, la HCL fue considerada una enfermedad maligna
y por este motivo fue tratada con una o más drogas citotóxicas agregadas a los
corticoesteroides con diferentes resultados. Lamentablemente la mayoría de
los estudios publicados reflejan la experiencia retrospectiva de una sola
institución, con un limitado número de casos recolectados durante un
prolongado tiempo de observación. Entre las décadas del cincuenta y la década
del setenta hubo significativas contribuciones para una mejor descripción de
las manifestaciones clínicas, recolección de la información sobre la historia

Page 37 of 56
natural de la enfermedad, elaboración de programas de tratamiento, y mejoría
del pronóstico en general, lograda por algunos estudios multi-institucionales.
Sin embargo, debido a la falta de criterios uniformes en la evaluación y
estadificación, los grupos de pacientes reportados variaban en lo que se refiere
a extensión de la enfermedad. Esto impidió una comparación precisa de la
información reportada y dificultó sacar conclusiones que favorecieran mejores
tratamientos.
Los mayores estudios clínicos prospectivos cooperativo de la década del
ochenta fueron los del grupo italiano AEIOP y el del grupo DAL-HX 83 ambos10
basados en dar quimioterapia sistémica, inmediatamente después de
confirmado el diagnóstico de HCL. La tasa de sobrevida global estuvo en el
orden del 90% en estos estudios. Quizás el hallazgo más significativo del
estudio DAL-HX 83 fue su baja tasa de reactivación (23%), lo cual sugería que
un tratamiento iniciado tempranamente puede modificar el curso natural de la
enfermedad.10 Las secuelas permanentes relacionadas con la enfermedad se
encontraron en 33% a un 48% en estos estudios. Sorprendentemente la DI
ocurrió solamente en el 20 % y 10 % de los casos respectivamente.
E.2.2 Etapa de los ensayos clínicos prospectivos internacionales (Sociedad de Histiocitosis) Clínicos e investigadores básicos interesados en los Histiocitos y sus
enfermedades, se encontraron en 1985, en Filadelfia, en una histórica reunión
internacional en la que se fundó la Sociedad de Histiocitosis. Esta Sociedad
aportó un foro para el intercambio de información y la elaboración de
actividades organizadas de importancia fundamental. Como resultado de
esfuerzos colaborativos, algunos criterios fueron publicados por la Sociedad de
Histiocitosis.11, 12 Estas publicaciones establecieron un lenguaje uniforme en
cuanto a la clasificación de las enfermedades histiocitarias, criterios de
diagnóstico y normas de evaluación y seguimiento. El objetivo principal de la

Page 38 of 56
Sociedad de Histiocitosis fue y es facilitar estudios internacionales cooperativos
a gran escala en la HCL.
Tres ensayos prospectivos de tratamiento han sido realizados hasta
ahora. Los logros y hallazgos importantes se resumen a continuación.
E.2.3 Estudio LCH-I (1991-1995) El LCH-I fue el primer estudio internacional de la sociedad de Histiocitosis
para el tratamiento de la HCL multisistémica. En este estudio, todos los
pacientes elegibles fueron randomizados al azar para recibir 24 semanas de
tratamiento con vinblastina o etopósido. Se administraron inicialmente pulsos
con metilprednisolona a altas dosis, en las 2 ramas, para prevenir síntomas
generales. Los pacientes fueron evaluados en intervalos determinados.
Quinientos veintitrés pacientes fueron registrados de los cuales 210 tenían
compromiso multisistémico y 143 fueron randomizados. La comparación de los
2 regímenes no arrojó diferencias significativas con respecto a la respuesta
inicial (57% vs. 49%), probabilidad de reactivación (61% vs. 55%), mortalidad
(24% vs. 20%) y secuelas (39% vs. 51%) incluyendo DI (22% vs. 23%).12
Este estudio contribuyó a mejorar el tratamiento de la HCL, mostrando que:
• La vinblastina y el etopósido usados como drogas únicas, son igualmente
efectivos en la HCL multisistémica
• El mal pronóstico está claramente asociado al compromiso de al menos
un órgano de riesgo (hígado, sistema hematopoyético, bazo, pulmones)
constatado al diagnostico.
• La respuesta al tratamiento en la semana 6, es un factor predictivo
independiente y muy fuerte para predecir el pronóstico. La enfermedad

Page 39 of 56
progresiva luego de 6 semanas de tratamiento estuvo asociada a mal
pronóstico.
Los resultados del LCH-I fueron también comparados con el DAL-HX
83/90 ya que en esos estudios se utilizaron criterios estrictos y uniformes para
el diagnóstico, la evaluación de la extensión de la enfermedad, el uso de una
quimioterapia estándar asignada según el grupo de riesgo y la respuesta al
tratamiento. Se utilizaron para ambos estudios los mismos criterios para
evaluar la extensión de la enfermedad y la respuesta al tratamiento. A pesar
que el LCH-I alcanzó una tasa similar de sobrevida, se comparó
desfavorablemente con el DAL en otros aspectos. El concepto del tratamiento
inicial del DAL-HX, aportó una clara discriminación entre los respondedores y
no respondedores. Esto sugirió que los pacientes con HCL multisistémica
severa, pueden beneficiarse con un tratamiento inicial más intenso. Se especuló
además que una interrupción rápida de la actividad de la enfermedad podría
reducir la incidencia de los efectos tardíos. En el estudio LCH-I se encontró una
incidencia alta de secuelas relacionadas a la enfermedad, aunque el lapso de
seguimiento fue menor que en DAL-HX. Todas estas conclusiones sugirieron
que el tratamiento debía ser intensificado.
Sin embargo, seguía sin aclarar si la potencial superioridad del DAL-HX
se debía a la administración continua de corticoesteroides, a la combinación de
vinblastina y etopósido o al tratamiento prolongado con 6-mercaptopurina por
1 año. Para aclarar estas preguntas, se diseñó el siguiente estudio en 1996
(estudio LCH-II).
E.2.4 Estudio LCH-II (1996-2001) El estudio LCH-II exploró la intensificación del tratamiento con el objeto
de comparar los resultados con los estudios DAL-HX y clarificar la pregunta

Page 40 of 56
sobre el valor de agregar el etopósido al tratamiento estándar con prednisona y
vinblastina en los pacientes con HCL multisistémica.11
Este estudio randomizado se concentró en los pacientes con compromiso
de órganos de riesgo (OR) (hígado, sistema hematopoyético, bazo y pulmones)
y pacientes menores de 2 años de edad, comparando 2 ramas de tratamiento
con prednisona y vinblastina. En una de ellas se utilizaba el etopósido y en se lo
omitía. El tratamiento de continuación incluía 6-mercaptopurina agregado a
pulsos con prednisona y vinblastina. La duración total del tratamiento fue de 6
meses.
Este estudio aleatorizado para los pacientes de riesgo reveló pronósticos
similares para ambas ramas de tratamiento: respuesta rápida en la semana 6
(rama A vs. Rama B, 63% y 71%, respectivamente), probabilidad de sobrevida a 5
años (74% vs. 79%), frecuencia de reactivaciones (46% vs. 46%) y secuelas o
consecuencias permanentes (43% vs. 37%). Es importante señalar que los
pacientes menores de 2 años sin compromiso de órganos de riesgo, tuvieron
una alta tasa de respuesta (>80%) y 100% de sobrevida. Sin embargo los
pacientes con compromiso de órganos de riesgo y falta de respuesta luego de 6
semanas de tratamiento tuvieron la mayor mortalidad.11
Los resultados de este ensayo clínico mostraron una tasa de sobrevida a
5 años del 74% para los pacientes tratados con prednisona, vinblastina y 6-
mercaptopurina. La sobrevida fue del 79% para aquellos pacientes tratados con
las mismas drogas con el agregado del etopósido, no existiendo diferencias
estadísticamente significativas entre las 2 ramas.
Las conclusiones más importantes del estudio LCH II fueron:
• La edad menor a 2 años al diagnóstico no tiene significado pronóstico,
por lo que el grupo de riesgo puede ser definido solo por el compromiso
de órganos de riesgo al diagnóstico

Page 41 of 56
• Debido a la alta mortalidad por enfermedad, la intensificación del
tratamiento es razonable para este grupo de riesgo
• Usando un tratamiento inicial similar, el estudio LCH-II tuvo una duración
más corta (6 meses) que el estudio DAL-HX (12 meses) y una tasa de
reactivación más alta.
Basado en estas conclusiones, se diseñó el siguiente estudio (LCH-III), cuyos
resultados no han sido publicados aún.
E.3.5 Estudio LCH III
La Sociedad de Histiocitosis estudió en este protocolo, regímenes para
pacientes de riesgo, bajo riesgo, compromiso multióseo o lesiones con
riesgo de causar un compromiso del sistema nervioso central.
• Los pacientes con compromiso de órganos de riesgo fueron asignados al
azar para recibir prednisona, vinblastina, 6-mercaptopurina con o sin el
agregado de metotrexate (endovenoso en la inducción y oral durante el
mantenimiento). La duración del tratamiento fue de 1 año. Se repitió el
tratamiento de inducción por otras 6 semanas Intensificación) para
aquellos que no respondieron completamente al tratamiento inicial.
• Los pacientes con compromiso de más de 1 órgano de bajo riesgo (piel,
huesos, ganglios linfáticos, hipófisis) fueron asignados al azar para
recibir prednisona y vinblastina por 6 o 12 meses.
• Los pacientes con compromiso multióseo, recibieron prednisona y
vinblastina durante 6 meses para disminuir la incidencia de

Page 42 of 56
reactivaciones y secuelas. Los pacientes con lesiones de cráneo, con
riesgo de causar un compromiso del SNC, recibieron un tratamiento
similar, para prevenir ésas lesiones, especialmente la DI. Los pacientes
con lesiones vertebrales con invasión del canal raquídeo recibieron el
mismo tratamiento con el fin de prevenir la compresión medular y sus
secuelas.
Los resultados de este estudio no han sido publicados hasta el momento.
E.3 Evaluacion de La Respuesta al Tratamiento
De acuerdo con la naturaleza del HCL, las siguientes definiciones deben
ser aplicadas para evaluar el efecto del tratamiento.
E. Tabla 5 Definición del estado de la enfermedad ENFERMEDAD NO ACTIVA no evidencia de
enfermedad
Resolución de todos los signos
o síntomas
enfermedad en
regresión
Regresión de signos o
síntomas, no lesiones nuevas
ENFERMEDAD ACTIVA enfermedad estable Persistencia de signos o
síntomas, no lesiones nuevas
Enfermedad progresiva progresión de signos o
síntomas
y/o aparición de nuevas
lesiones

Page 43 of 56
E. Tabla 6 Definición de criterios de respuesta
MEJOR
Respuesta completa No enfermedad activa
regresión Enfermedad activa mejor
INTERMEDIA
mixta Nuevas lesiones en un sitio, y
regresión en otros sitios
estable sin cambios
PEOR progresión
E.3.1 Tratamiento de Salvataje
En la etapa anterior a 1994, el tratamiento de salvataje no se administró
en forma sistemática, En ese entonces, la Sociedad de Histiocitosis inició un
ensayo clínico (LCH-I-S) con el fin de mejorar la sobrevida, y eventualmente
curar, a los pacientes multisistémicos que, a pesar de recibir el tratamiento
estándar, desarrollaron enfermedad progresiva.13 Este protocolo contempló la
posibilidad de dar transplante alogénico de médula ósea con donante familiar
compatible o tratamiento inmunosupresor (globulina antitimocito,prednisona y
ciclosporina) para los que no cumplieran ese requisito. Este estudio fue cerrado
debido al bajo ingreso de pacientes. Luego de analizar la información del
estudio LCH-I, se detectó que un pequeño grupo de pacientes de alto riesgo,
con mal pronóstico pueden ser identificados muy tempranamente durante su
evolución. Para estos pacientes, la Sociedad de Histiocitosis estableció un
estudio en fase II (LCH-II-S) no randomizado con 2-clorodesoxiadenosina,
incluyendo aquellos pacientes cuya enfermedad progresaba después de recibir
un tratamiento combinado con prednisona, vinblastina y etopósido. La 2-
clorodesoxiadenosina resultó una droga activa en la HCL, aunque la tasa de

Page 44 of 56
respuesta fue mayor en los pacientes con enfermedad multifocal ósea y en los
pacientes de bajo riesgo que en aquellos con compromiso de órganos de
riesgo. Muchos de estos pacientes que no respondieron al tratamiento tuvieron
una alta mortalidad, en especial, aquellos menores de 2 años o a los que se les
inició tardíamente el tratamiento.14
E.4 Sintesis E.4.1 Conceptos extráidos de la guía de evaluación y tratamiento de la Sociedad de Histiocitosis La HCL puede tratarse ingresando pacientes, con previa aprobación del
consejo de ética de un hospital a los ensayos clínicos de la Sociedad de
Histiocitosis, con el objeto de mejorar el conocimiento sobre la biología de la
HCL y optimizar su tratamiento. El uso del protocolo de investigación para
tratar pacientes que no tienen la oportunidad de ser incluidos en esos estudios,
es fuertemente desaconsejado. Las recomendaciones que siguen intentan
proveer a los médicos una guía para el manejo de estos pacientes que no
pueden o no quieren ser incluidos en los protocolos de investigación.
• Está aceptado que las HCL con compromiso de más de un órgano o
sistema deben recibir tratamiento sistémico. Los pacientes con
compromiso hepático, esplénico, hematopoyético y/o pulmonar pueden
tener un riesgo de evolución fatal (Compromiso de órganos de riesgo), en
especial aquellos que no respondan al tratamiento inicial. Pero aún
cuando los pacientes respondan al tratamiento, pueden presentar
reactivaciones de la enfermedad que pueden tener consecuencias
permanentes como problemas ortopédicos o déficit hormonales o
neurológicos que incidan en la calidad de vida. Por lo tanto el paciente
podría necesitar diferentes estrategias terapéuticas para contrarrestar

Page 45 of 56
cuadros clínicos progresivos potencialmente mortales o reactivaciones
durante la evolución de la enfermedad. La evolución del tratamiento de la
HCL se relacionó de cerca con los cambios en la comprensión de la
biología de la enfermedad. Las dificultades en el desarrollo de
tratamientos óptimos se deben a dificultades para comprender aspectos
de la patogénesis de esta enfermedad.
• Evidencias recientes (información preliminar no publicada del ensayo
clínico LCH-III de la Sociedad de Histiocitosis), sugieren que la duración
total de 12 meses de tratamiento, reduciría la tasa de reactivación cuando
se la compara con 6 meses de similar medicación.
• Los pacientes con HCL multisistémica pueden tener un curso clínico
variable. Aquellos que no tienen compromiso de órganos de riesgo asi
como aquellos que si lo tienen pero que han respondido al tratamiento
inicial estándar tienen una excelente posibilidad de sobrevida a largo
plazo. La combinación con prednisona y vinblastina es un tratamiento
efectivo con un mínimo de toxicidad y es, por lo tanto, el tratamiento
inicial estándar para todos los pacientes en los que el tratamiento
sistémico está indicado. Para aquellos pacientes con compromiso de
órganos de riesgo que no respondan, especialmente aquellos con
progresión clínica evidente durante las 6 primeras semanas de
tratamiento, tienen un pronóstico desfavorable. Para estos pacientes, se
justifica un tratamiento de intensificación iniciado tempranamente.
• Los pacientes con enfermedad ósea multifocal tienen un excelente
pronóstico (sobrevida del 100%), pero una alta tendencia a la reactivación
de la enfermedad (30 a 50%) y sus secuelas. Esto es similar para los
pacientes con lesiones en “sitios especiales” con riesgo de compromiso
del SNC

Page 46 of 56
• El objetivo del tratamiento en estos grupos es el de prevenir
reactivaciones de la enfermedad y sus consecuencias permanentes o
secuelas. La evidencia para recomendar diferentes regímenes es sin
embargo limitada y mayormente basada en análisis retrospectivos u
opiniones de expertos.
E.6 Tratamiento de Primera Línea
E.6.1 Enfermedad Multisístémica
Se sugiere un tratamiento inicial con prednisona y vinblastina para todos
los pacientes con HCL multisistémica cualquiera sea el compromiso de órganos.
El uso de un tratamiento diferente o más intenso podrá plantearse en casos de
mala respuesta inicial. Es, de este modo, obligatoria una evaluación al terminar
las 6 semanas de tratamiento para plantear el tratamiento a seguir.
Aquellos pacientes que tuvieron compromiso de órganos de riesgo al
diagnóstico o durante el tratamiento y su enfermedad progresó, son candidatos
a opciones de tratamientos de salvataje.
Sin embargo pacientes con colangitis esclerosante o lesiones
bullosas/quísticas pulmonares sin compromiso de otros órganos de riesgo,
presentan un pronóstico y evolución diferentes necesitando diferentes
estrategias terapeúticas multidisciplinarias.15, 16
Para aquellos pacientes sin compromiso de órganos de riesgo que no
mejoraron o que mejoraron parcialmente con el tratamiento inicial, se
recomienda un segundo curso de tratamiento con las misma drogas que las
utilizadas en la inducción.

Page 47 of 56
Se recomienda que todos los pacientes que tuvieron respuesta completa
luego de 6-12 semanas de tratamiento inicial continuen con el tratamiento de
mantenimiento que consiste en pulsos de vinblastina y prednisona cada 3
semanas y 6 mercaptopurina diaria de manera continua con una duración total
de 12 meses.
Aquellos pacientes que después del segundo ciclo de tratamiento inicial
(12 semanas) todavía presentan compromiso de órganos de riesgo, se
recomienda iniciar el tratamiento de salvataje. Para aquellos pacientes sin
compromiso de órganos de riesgo, pero que no responden después del
segundo ciclo inicial, la recomendación es pasar a otro tipo de tratamiento con
drogas alternativas (ver más adelante “tratamiento de segunda línea para la HCL
de bajo riesgo”)
Enfermedad ósea multifocal, “sitios especiales”y lesiones de riesgo para el
sistema nervioso central. No existe todavía una evidencia firme sobre cuál es el
mejor tratamiento para este subgrupo de pacientes. La ventaja de un
tratamiento sistémico se basa en la potencial reducción del riesgo considerable
de secuelas, que afectarán su calidad de vida.
Información proveniente de los estudios prospectivos no randomizados
DAL-HX 83 y DAL-HX 90 sugieren que 1 año de tratamiento sistémico con
vinblastina, prednisona y 6-mercaptopurina pueden reducir tanto la reactivación
como la frecuencia de secuelas cuando se la compara con estudios previos. Por
esta razón la sociedad de Histiocitosis recomienda un tratamiento sístémico
que consista en 1 ciclo +- un segundo ciclo y un tratamiento de continuación
con pulsos de prednisona/vinblastina cada 3 semanas, con una duración total
de tratamiento de 12 meses (como en la F. Figura 1 pero sin la 6-
mercaptopurina).

Page 48 of 56
F. Figura 1 Célula de Langerhans (flecha)
E.7 Tratamiento de Segunda Línea
E.7.1 Opciones de Tratamiento de Salvataje para Pacientes
de Riesgo
No existen todavía evidencias suficientes de un tratamiento óptimo para
los pacientes con HCL multisistémica progresiva severa que no respondan al
tratamiento estándar. Han sido reportados recientemente, resultados
promisorios con un régimen que usa una combinación con 2-
clorodesoxiadenosina (2-CdA) y citarabina (Ara-C) y también con el trasplante
de células madre luego de un tratamiento con un régimen condicionante de de
baja intensidad (RIC-SCT). Sin embargo, ambas publicaciones se basan en un
limitado número de casos y necesitan confirmación con estudios clínicos

Page 49 of 56
prospectivos. La Sociedad de Histiocitosis ha abierto ensayos para ambas
opciones. Para aquellos raros casos que necesitan un tratamiento de salvataje,
se recomienda contactar a un experto en HCL o los investigadores principales
de los ensayos clínicos en curso.
E.7.2 Tratamiento de Segunda Línea para Los Pacientes de Bajo Riesgo Todavía no fueron halladas las mejores opciones de secunda línea para
pacientes con HCL, persistente o recurrente, en órganos de bajo riesgo. Este
tema será estudiado en un futuro ensayo clínico de la Sociedad de Histiocitosis.
Han sido usados con cierto éxito diferentes tratamientos como la combinación
de vincristina, prednisona y citarabina y 2-clorodesoxiadenosina como única
medicación.
E.7.3 Enfermedad del Sistema Nervioso Central
El tratamiento para los pacientes con compromiso del SNC dependerá del
tipo, extensión de la lesión y el tratamiento previo. La terapeútica deberá ser
elegida de manera individual.

Page 50 of 56
E.Tabla 7 Evaluaciónde seguimiento post tratamiento. Año 1* Años 2 – 5*
EXAMEN CLINICO CADA 6 SEMANAS CADA 6 MESES PESO, TALLA, ESTADIO PUBERAL
CADA 6 MESES CADA 6 MESES
EXAMENES DE LABORATORIO EN PACIENTES QUE HAN TENIDO COMPROMISO DE ORGANOS: HEMOGRAMA. ERITROSEDIMENTACION, PRUEBAS DE FUNCION HEPATICA Y RENAL, DENSIDAD/OSMOLARIDAD URINARIA
CADA 3 MESES CADA AÑO
RADIOGRAFIAS DE LESIONES OSEAS
SOLO SI SE SOSPECHAN NUEVAS LESIONES O REACTIVACIÓN
SOLO SI SE SOSPECHAN NUEVAS LESIONES O REACTIVACIÓN
AUDIOMETRIA EN PACIENTES CON HISTORIA DE COMPROMISO DE OIDO/MASTOIDES
CADA AÑO CADA 5 AÑOS
SI COMPROMISO PULMONAR: TC DE ALTA RESOLUCION Y PRUEBAS FUNCIONALES PULMONARES
CADA 6 MESES SOLO SI SE SOSPECHA PROGRESION
ECOGRAFIA EN PACIENTES CON COMPROMISO HEPATICO
CADA 6 MESES CADA AÑO
RMN DE CEREBRO EN PACIENTES CON DI U OTRAS ENDOCRINOPATIAS O LESIONES DE RIESGO PARA EL SNC
CADA AÑO CADA 2 AÑOS
EVALUACION NEUROPSICOMETRICA EN PACIENTES CON COMPROMISO DEL SNC
CADA AÑO CADA 2 AÑOS
*Estos criterios se deben adaptar o modificar según los órganos comprometidos y las indicaciones clínicas.

Page 51 of 56
Los autores aconsejan controles clínicos (según necesidad: estudios de
laboratorio y radiológicos) en el segundo año, mensual para los pacientes de
riesgo, bimestral para los multisitémicos de alto riesgo y sitios especiales y
cada 4 meses para el resto, por la alta posibilidad de reactivaciones en este
periodo con secuelas ulteriores.
E. References
1Egeler RM, Thompson RC, Jr., Voute PA, Nesbit ME, Jr. Intralesional infiltration of corticosteroids in localized Langerhans' cell histiocytosis. J Pediatr Orthop. 1992; 12: 811-814. 2Bernstrand C, Bjork O, Ahstrom L, Henter JI. Intralesional steroids in Langerhans cell histiocytosis of bone. Acta Paediatr. 1996; 85: 502-504. 3Munn SE, Olliver L, Broadbent V, Pritchard J. Use of indomethacin in Langerhans cell histiocytosis. Med Pediatr Oncol. 1999; 32: 247-249. 4Olschewski T, Seegenschmiedt MH. Radiotherapy for bony manifestations of Langerhans cell histiocytosis. Review and proposal for an international registry. Strahlenther Onkol. 2006; 182: 72-79. 5Grois N, Potschger U, Prosch H et al. Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatr Blood Cancer. 2006; 46: 228-233. 6Steen AE, Steen KH, Bauer R, Bieber T. Successful treatment of cutaneous Langerhans cell histiocytosis with low-dose methotrexate. Br J Dermatol. 2001; 145: 137-140. 7Sander CS, Kaatz M, Elsner P. Successful treatment of cutaneous langerhans cell histiocytosis with thalidomide. Dermatology. 2004; 208: 149-152. 8Sheehan MP, Atherton DJ, Broadbent V, Pritchard J. Topical nitrogen mustard: an effective treatment for cutaneous Langerhans cell histiocytosis. J Pediatr. 1991; 119: 317-321. 9von Stebut E, Schadmand-Fischer S, Brauninger W et al. Successful treatment of adult multisystemic Langerhans cell histiocytosis with psoralen-UV-A, prednisolone, mercaptopurine, and vinblastine. Arch Dermatol. 2008; 144: 649-653. 10Gadner H, Heitger A, Grois N et al. Treatment strategy for disseminated Langerhans cell histiocytosis. DAL HX-83 Study Group. Med Pediatr Oncol. 1994; 23: 72-80. 11Gadner H, Grois N, Potschger U et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification. Blood. 2008; 111: 2556-2562. 12Gadner H, Grois N, Arico M et al. A randomized trial of treatment for multisystem Langerhans' cell histiocytosis. J Pediatr. 2001; 138: 728-734. 13Minkov M, Grois N, Braier J et al. Immunosuppressive treatment for chemotherapy-resistant multisystem Langerhans cell histiocytosis. Med Pediatr Oncol. 2003; 40: 253-256. 14Weitzman S, Braier J, Donadieu J et al. 2'-Chlorodeoxyadenosine (2-CdA) as salvage therapy for Langerhans cell histiocytosis (LCH). results of the LCH-S-98 protocol of the Histiocyte Society. Pediatr Blood Cancer. 2009; 53: 1271-1276.

Page 52 of 56
15Braier J, Ciocca M, Latella A, et al. Cholestasis, sclerosing cholangitis, and liver transplantation in Langerhans cell Histiocytosis. Med Pediatr Oncol. 2002; 38: 178-182. 16Braier J, Latella A, Balancini B et al. Isolated pulmonary Langerhans cell histiocytosis presenting with recurrent pneumothorax. Pediatr Blood Cancer. 2007; 48: 241-244.
F. Reactivaciones y Efectos Tardíos
F.1 Reactivaciones
La reactivación se define en la HCL por la reaparición de la enfermedad
luego de haber logrado una respuesta completa a un tratamiento previo. La
reactivación es un evento relativamente frecuente y, cuando ocurre, a menudo
se produce temprano durante la evolución de la enfermedad (88% de los
episodios de reactivación se producen en los primeros dos años de evolución
de la enfermedad). La incidencia de las reactivaciones se correlaciona con la
extensión de la enfermedad al diagnóstico, siendo más frecuente en las formas
multisistémicas. Los sitios en donde las reactivaciones se manifiestan más
frecuentemente son: huesos (incluyendo oído medio) y región hipotálamo-
hipofisaria (bajo la forma de DI), siendo poco frecuentes las localizaciones en
órganos de riesgo. La mortalidad de los pacientes con reactivaciones es muy
baja. Sin embargo, la presencia de secuelas se correlaciona con la incidencia de
las reactivaciones.1, 2
F.2 Efectos Tardios
Durante el curso de la HCL, es frecuentemente difícil establecer, en
alguno de los órganos comprometidos, cuando está cursando una enfermedad
activa o por el contrario cuando ya está establecida la secuela (por ejemplo.
hígado, pulmones, huesos, DI, sistema nervioso central). Se puede también
constatar al diagnóstico y durante la evolución de la enfermedad la coexistencia

Page 53 of 56
de hallazgos en determinados órganos sugerentes de enfermedad activa con
otros que ya presentan compromiso secuelar
Las consecuencias permanentes o efectos tardíos en la HCL están
relacionados con la enfermedad y menos frecuentemente con el tratamiento,
siguen siendo un desafío para el equipo tratante. Los niños con compromiso de
órganos de bajo riesgo (piel, huesos, ganglios con compromiso de órganos de
bajo riesgo tienen un 24% de incidencia de secuelas a largo plazo mientras
que esta tasa es mayor en los que tienen compromiso multisistémico, llegando
hasta casi 71% de secuelas a largo plazo.3
F.2.1 Secuelas Óseas Las secuelas óseas más temidas se atribuyen al compromiso vertebral,
que puede traer cuadros de inestabilidad por colapso vertebral o en ocasiones
por escoliosis. La vertebra plana se reconstituye en la mayoría de los casos de
manera completa durante los cinco años que siguen al compromiso inicial. En
casos con compromiso femoral, es posible observar a largo plazo inestabilidad
en la articulación de la cadera por lesiones de la columna, fémur, tibia, húmero
o cadera, pueden presentarse en un 20 % de los pacientes. Estos problemas
pueden causar escoliosis, asimetría facial o de miembros. Las lesiones del oído
medio pueden dejar como secuelas: hipoacusia que ha sido reportada hasta en
un 13 % de los casos y colesteatoma.4
F.2.2 Secuelas Dentarias En algunos pacientes con compromiso oseo mandibular, pueden ocurrir
secuelas dentarias a largo plazo, incluyendo la pérdida de piezas dentarias.

Page 54 of 56
F.2.3 Secuelas Endocrinológicas Los pacientes con DI, tienen un riesgo mayor de presentar
panhipopituitarismo y deben ser monitoreados cuidadosamente para controlar
su crecimiento. A los 10 años del diagnóstico, se estima que hasta un 54 % de
los pacientes con DI pueden desarrollar déficit de hormona de crecimiento.5 Un
tema que causa preocupación es la probabilidad de que el suplemento con
hormona de crecimiento (HC) pudiera causar un aumento en la tasa de
reactivación aunque eso no se ha podido comprobar. Además del déficit de HC,
los problemas de crecimiento y desarrollo pueden ser frecuentes durante la
enfermedad, debe vigilarse los efectos del uso de corticoides, los cuales
también pueden traer efectos no deseados a largo plazo.
F.2.4 Secuelas Neurológicas Las lesiones del Sistema Nervioso Central relacionadas a la HCL ocurren
más a menudo en pacientes con DI o lesiones de riesgo de compromiso de
sistema nervioso central en cráneo (mastoides, órbita, hueso temporal)[6]. El
compromiso neurológico puede manifestarse de múltiples formas, desde
hallazgos asintomáticos en los estudios de imágenes hasta cuadros
infrecuentes de severa discapacidad. Menos frecuentemente, se evidencian
lesiones de desmielinización los estudios de imágenes, asociados a trastornos
de conducta o en la memoria. En menores ocasiones, se evidencian lesiones
pseudotumorales en las imágenes.
F.2.5 Secuelas Pulmonares
En ocasiones, puede ocurrir una enfermedad restrictiva pulmonar crónica.
Los pacientes con compromiso pulmonar deben ser seguidos con pruebas de
función respiratoria.

Page 55 of 56
F.2.6 Secuelas Hematológicas
La insuficiencia medular secundaria al tratamiento de la HCL, es rara y
está asociada con un mayor riesgo de enfermedad maligna. Los pacientes con
HCL tienen un riesgo incrementado de desarrollar cáncer. La Leucemia
mieloide aguda ha sido típicamente descrita , usualmente luego del tratamiento
en pacientes con HCL. Se especuló en el rol del etopósido en la inducción de
esta complicación, aunque no está claro todavía la asociación entre estos
factores.8 Ha sido descripta la leucemia linfoide aguda en niños con HCL, tanto
con fenotipo T o B.9, 10 La asociación entre HCL y otras enfermedades malignas
(retinoblastoma, tumores cerebrales, carcinoma hepatocelular, tumor de Ewing)
ha sido también reportada en algunos pacientes y en algunos de ellos la
neoplasia precedió a la HCL.7
F.2.7 Secuelas Hepáticas
De acuerdo a lo previamente descripto en la sección Compromiso
Específico de Organos, el compromiso hepático puede evolucionar a una
colangitis esclerosante que raramente responde a algún tratamiento que no sea
el trasplante de hígado.11
F. References 1Minkov M, Steiner M, Potschger U, et al. Reactivations in multisystem Langerhans cell histiocytosis: data of the international LCH registry. J Pediatr. 2008;153:700-5,705.e1-2. 2Pollono D, Rey G, Latella A, et al. Reactivation and risk of sequelae in Langerhans cell histiocytosis. Pediatr Blood Cancer. 2007;48:696-699. 3Haupt R, Nanduri V, Calevo MG, et al. Permanent consequences in Langerhans cell histiocytosis patients: a pilot study from the Histiocyte Society-Late Effects Study Group. Pediatr Blood Cancer. 2004;42:438-444. 4Roger G, Dupre M, Leboulanger N, et al. Cholesteatoma secondary to temporal bone involvement by Langerhans cell histiocytosis: a complication amenable to curative surgery. Otol Neurotol. 2009;30:190-193.

Page 56 of 56
5Donadieu J, Rolon MA, Pion I, et al. Incidence of growth hormone deficiency in pediatric-onset Langerhans cell histiocytosis: efficacy and safety of growth hormone treatment. J Clin Endocrinol Metab. 2004;89:604-609. 6Grois N, Fahrner B, Arceci RJ, et al. Central nervous system disease in Langerhans cell histiocytosis. J Pediatr. 2010;156:873-81,881.e.1. 7Egeler RM, Neglia JP, Puccetti DM, et al. Association of Langerhans cell histiocytosis with malignant neoplasms. Cancer. 1993;71:865-873. 8Ghosn MG, Haddad AC, Nassar MN, et al. Acute myeloid leukemia and Langerhans' cell histiocytosis: multiple theories for an unusual presentation. Leuk Res. 2010;34:406-408. 9Trebo MM, Attarbaschi A, Mann G, et al. Histiocytosis following T-acute lymphoblastic leukemia: a BFM study. Leuk Lymphoma. 2005;46:1735-1741. 10Pastor-Jane L, Escoda-Teigell L, Martinez-Gonzalez S, et al. Multiorgan histiocytosis after B-cell acute lymphoblastic leukemia. Am J Dermatopathol. 2011;33:516-520. 11Braier J, Ciocca M, Latella A, et al. Cholestasis, sclerosing cholangitis, and liver transplantation in Langerhans cell Histiocytosis. Med Pediatr Oncol. 2002;38:178-182.
Top Related