







Idiomas
Páginas
Jurídico


NEN AMB HIPOGLUCÈMIES
Meritxell Torrabías
Hospital General de Vic
4/11/2013

Cas clínic
• Avisen de sala de parts: A l´hora de vida nadó hipotònic.
• NNAT (38.3sg) de PAEG (3175g) T 47 cm.
• 2a gestació, sense diabetis, TPAL 0010, triple screening baix risc. Eutòcic. Apgar 8/10/10.
• Gl: low.

• Extracció sang per estudi
• Perfussió glucosada (4 mg/kg/min) reverteix simptomatologia.
• Persisteixen hipoglucèmies– Trasllat a Hospital Granollers

• A l´exploració:
– Facies peculiar
– Orelles displàsiques
– Mans i peus amples i curts
– Hèrnia umbilical
– Hipotonia

Fotos


• Hospital Granollers:– Nou estudi ( Gl normal): normal (?)– Aport Gl (màx 5.7 mg/kg/min)– Ictericia—fototeràpia x 4 dies– Hidrocortisona– no respon– LM+dextrinomaltosa—retirada progressiva de perfussió– 1 mes: Sensor de Gl (Vall d´Hebron):
• HIPOGLUCEMIES preprandials que recuperen amb la ingesta– Diazòxid – Gl ok
– Eco abdominal: dubtosa duplicitat renal– 2 mesos: alta.
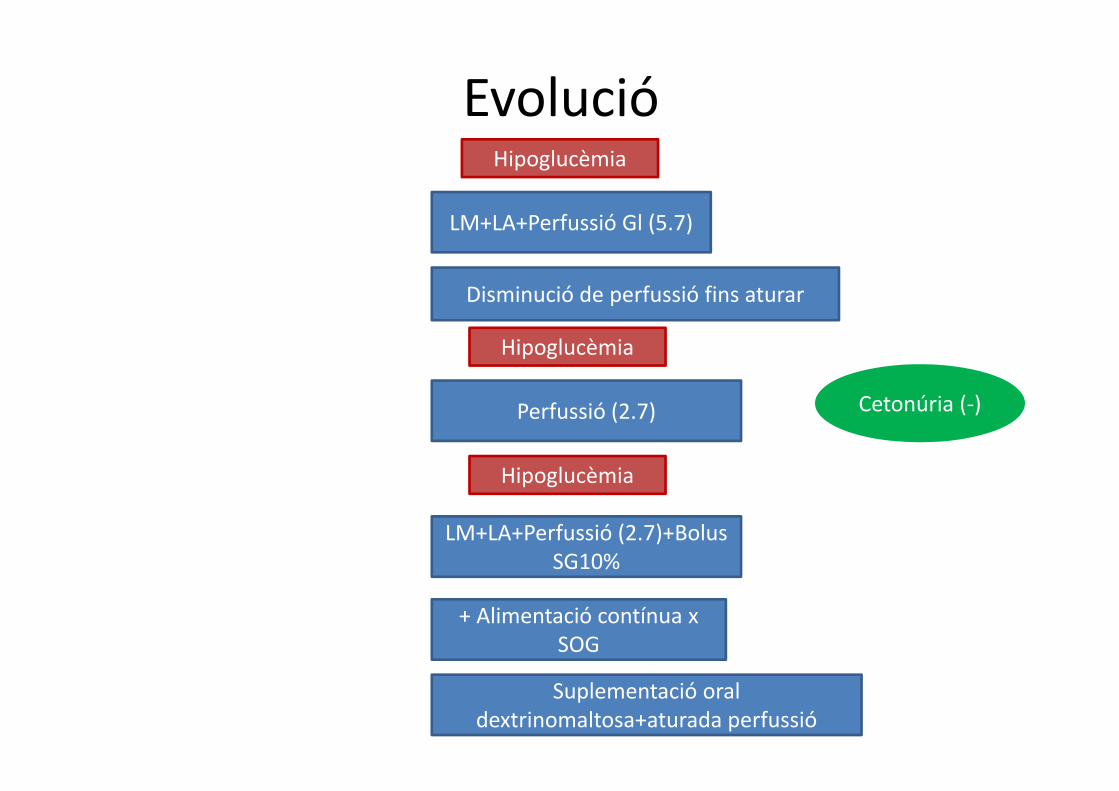
EvolucióHipoglucèmia
LM+LA+Perfussió Gl (5.7)
Disminució de perfussió fins aturar
Hipoglucèmia
Perfussió (2.7)
+ Alimentació contínua x SOG
Hipoglucèmia
LM+LA+Perfussió (2.7)+BolusSG10%
Suplementació oral dextrinomaltosa+aturada perfussió
Cetonúria (‐)
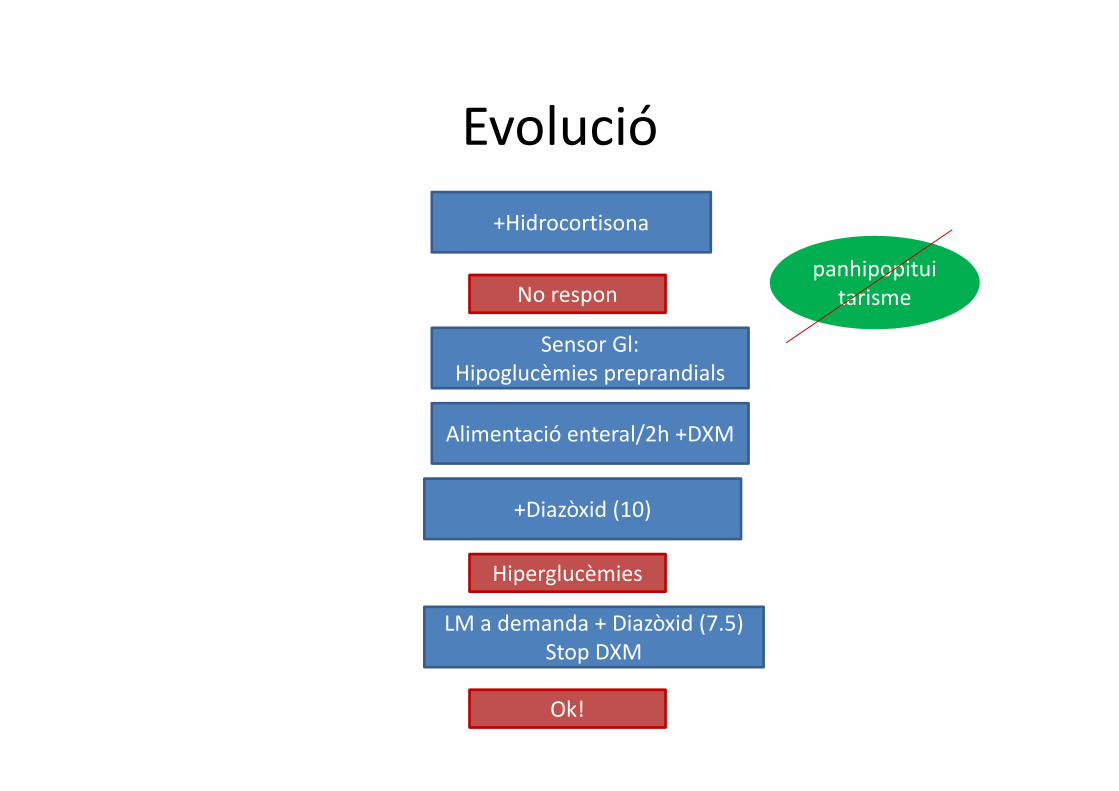
Evolució
+Hidrocortisona
No responpanhipopitui
tarisme
Sensor Gl:Hipoglucèmies preprandials
Alimentació enteral/2h +DXM
+Diazòxid (10)
LM a demanda + Diazòxid (7.5)Stop DXM
Hiperglucèmies
Ok!

Anàlisis en hipoglucèmia• Gl 37
• Insulina 18.2 mvU/ml (Gl/Ins 2, N>5)
• ACTH 51 pg/ml (9‐52), GH 17.1 ng/ml (>7.5),
• Triglicèrids 29 (30‐133), Col 67 (120‐221), Àc grassos ll 0.05 mmol/l (0.1‐0.6)
• Aminoàcids orina: normals,
• Cossos cetònics (ß‐OH‐butirat): <1 mg/dl (<3.1)
• Cariotip 46 XX
HIPERINSULINISME: • Gl/Ins <5• ÚNICA HIPOGLUCÈMIA QUE INHIBEIX LA LIPOLISIS (AGll i CC↓)

Hirsutisme per diazòxid

Sensor de glucosa 2 mesos (diazòxid 10)
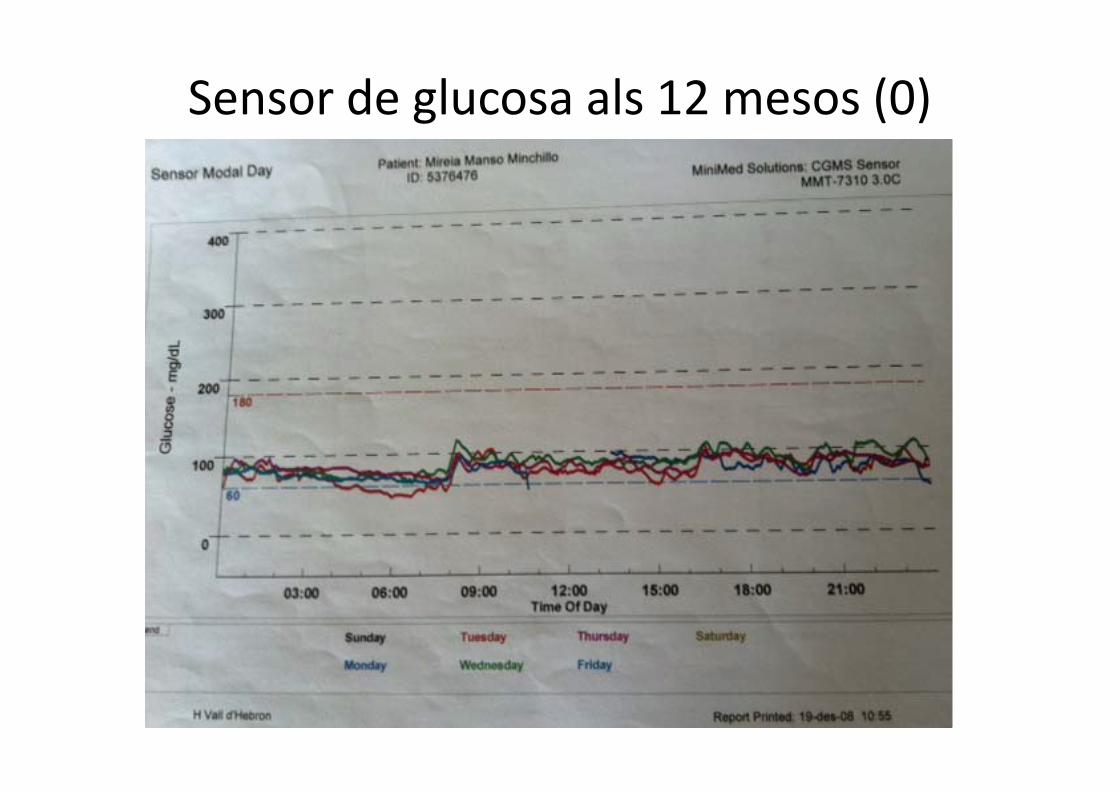
Sensor de glucosa als 12 mesos (0)

Evolució• Orelles displàsiques
– PEA normals.
• Dubtosa duplicitat renal– Gammagrafia: no duplicitat, pol inf dret hipocaptant
– Eco: ronyó dret horitzontalitzat i sospita doble sistema esquerre.
– Eco (Quirón): duplicitat renal bilateral
– CUMS normal

Evolució
• Hipotonia– Estimulació precoç– Neuropediatria: Retard lleu‐moderat, – RMN:
• Gliosis periventricular i occipital (2ària a hipoglucèmies??) adenohiòfisis petita, neurohipòfisis normal.
• Malf Arnold‐Chiari 1 i Siringomielia (neurocirurgià)
• Hèrnia umbilical– Resolució espontània
• Hipermetropia i astigmatisme– Lents

Evolució
• Hipoglucèmia– Gl 37, Ins 18.2
• Talla baixa– Test GH normal, IGF1 baixa.
– Test generació IGF‐1: 57.2‐‐>187 ng/ml.

80
100
120
140
160
180
200
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Talla
979075
3102550
0
10
20
30
40
50
60
70
80
90
3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
Pes
75
90
97
31025
50

Resum
• Hiperinsulinisme neonatal transitori• Facies peculiar• Mans tosques
• Orelles displàsiques• Talla baixa
• Retard mental• Malformació renal• Malformació SNC• Hèrnia umbilical • Defecte refracció

DIAGNÒSTIC DIFERENCIAL HIPOGLUCÈMIA NEONATAL1. Increment de la utilizació perifèrica de la glucosa
– Fill de mare diabètica (transitori 3‐5 dies)
– Eritroblastosis
– Drogues maternes (clorpropamida, propanolol, Gl ev, benzotiacides, simpaticomimètics)
– Hiperinsulinisme (tumor cèl. Beta o nesidioblastosi)
– Síndrome Wiedemann‐Beckwith (hipoglucèmia+macrosoma+macroglòssia+onfalocele+visceromegàlia+malf renals)
– Iatrogènia
– Sdme Kabuki
2. Inadequat aport de glucosa endògen o exògen
– Prematuritat
– CIR
– Dejú perllongat
– Stress Perinatal: Asfixia; Hipotèrmia; Sepsis; Shock
– Policitèmia
– Exsanguinotransfusió
– Dèficit d´hormones contrarreguladores (glucagó, GH, cortisol, ACTH)
– Alteració del metabolisme dels hidratos de carboni (Glucogenosis; fructosèmia; galactosèmia)
– Alteració del metabolisme dels aminoàcids: (Acidèmias metilvalòniques; Acidèmia Glutàrica tipus
I i II; Leucinosis (MUSD); Deficiència de 3‐hidroxi‐3 metil‐glutarilCoA liasa; Deficiència
de carnitina; Deficiències de acil‐CoA‐deshidrogenasa de cadena curta, mitjana i llarga).
Estudi genètic SUR1, KIR6.2, GCK (‐), HNF4alfa (‐)
Estudi genètic B‐W (‐)

Estudis genètics
• Regions subtelomèriques: (‐)
• Beckwith‐Wiedemann (MLPA de dosis i metilació de la regió d´imprinting
del cr11p: (‐)
• Hiperinsulinisme (‐)– Gen de la subunitat Kir6.2 dels canals de K de la cèl beta cr 11p15: (‐)
– 39 exons codificants i promotor del gen ABCC8 que codifica el receptor de les sulfonilurees pancreàtiques (SUR1): (‐)
– Gen GCK cr 7p15 (MODY2): (‐)
– Estudi genètic HNF4A (MODY 1): (‐)
• Visita genetista St Joan Déu:– Sdme Kabuki??

KABUKI (japonès)
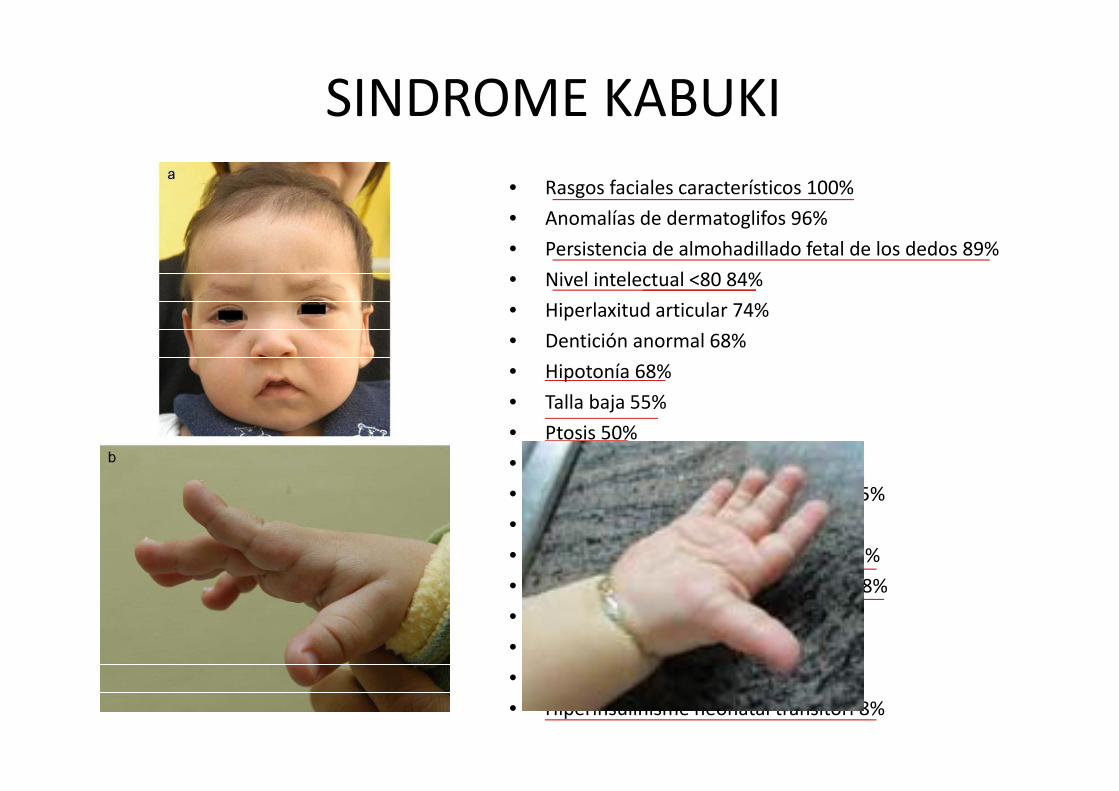
SINDROME KABUKI• Rasgos faciales característicos 100%
• Anomalías de dermatoglifos 96%
• Persistencia de almohadillado fetal de los dedos 89%
• Nivel intelectual <80 84%
• Hiperlaxitud articular 74%
• Dentición anormal 68%
• Hipotonía 68%
• Talla baja 55%
• Ptosis 50%
• Anomalías cardiovasculares 42%
• Anomalías palatinas‐labio leporino 35%
• Escoliosis 35%
• Defectos vertebrales o en costillas 32%
• Malformaciones del tracto urinario 28%
• Pérdida auditiva 27%
• Displasia de caderas 18%
• Crisis epilépticas 17%
• Hiperinsulinisme neonatal transitori 8%

SDME KABUKI
• Sdme del maquillatge Kabuki
• Descrita 1980 Japó i 1990 a Europa
• 1/32.000 Japó, 1/86.000 Oceania
• Espanya: – Any 1997 An Esp Ped: 6 casos descrits
– Any 2005 Rev Neurología: 18 casos descrits
• Casos esporàdics, però algun HAD
• Dx clínic i confirmació genètica (gen MLL2 cr 12q12‐q14)

Diagnòstic
• Sospita clínica:– Trets facials característics
– Retard creixement
– Retard mental
– Alteracions esquelètiques
– Persistència de almohadillado
• Confirmació genètica

FACIES SDME KABUKI
Top Related