Dinámica clásica y cuántica disipativa de adsorbatos en...
218
UNIVERSIDAD COMPLUTENSE DE MADRID Facultad de Ciencias Físicas Departamento de Física Aplicada I (Termología) Dinámica clásica y cuántica disipativa de adsorbatos en superficies metálicas Memoria para optar al grado de DOCTOR EN CIENCIAS FÍSICAS presentada por JOSÉ LUIS VEGA GONZÁLEZ Director de tesis: Prof. Salvador Mirét Artés Departamento de Física Atómica, Molecular y de Agregados Instituto de Matemáticas y Física Fundamental, C.S.I.C. Madrid, 2006
Transcript of Dinámica clásica y cuántica disipativa de adsorbatos en...

UNIVERSIDAD COMPLUTENSE DE MADRID
Facultad de Ciencias Físicas
Departamento de Física Aplicada I (Termología)
Dinámica clásica y cuánticadisipativa de adsorbatos en
superficies metálicas
Memoria para optar al grado de
DOCTOR EN CIENCIAS FÍSICAS
presentada por
JOSÉ LUIS VEGA GONZÁLEZ
Director de tesis:
Prof. Salvador Mirét Artés
Departamento de Física Atómica, Molecular y de Agregados
Instituto de Matemáticas y Física Fundamental, C.S.I.C.
Madrid, 2006

UNIVERSIDAD COMPLUTENSE DE MADRID
Facultad de Ciencias Físicas
Departamento de Física Aplicada I (Termología)
Dinámica clásica y cuánticadisipativa de adsorbatos en
superficies metálicas
Memoria para optar al grado de
DOCTOR EN CIENCIAS FÍSICAS
presentada por
JOSÉ LUIS VEGA GONZÁLEZ
Director de tesis:
Prof. Salvador Mirét Artés
Departamento de Física Atómica, Molecular y de Agregados
Instituto de Matemáticas y Física Fundamental, C.S.I.C.
Madrid, 2006

El ruido, pieza fundamental de este mundo loco y entretenido,
sin él todo sería más predecible y aburrido.

A mis Padres y Hermanos/a, a la Loma, y
por supuesto...a mi ruido.

Quiero comenzar esta memoria de Investigación agradeciendo al Profesor de
Investigación Salvador Mirét Artés, por toda su dedicación, paciencia, entusiasmo
y esfuerzo que ha tenido en la dirección de mi tesis Doctoral. Además de director,
siempre ha sido un padre, maestro y amigo.
¡Gracias Salva!.
También quiero agraceder esta memoria al Doctor Raúl Guantes Navacerrada,
una persona con una calidad humana excepcional, que ha sido un referente y pieza
fundamental en ésta, mi tesis.
¡Gracias Raúl!
Quiero extender mi agradecimiento, a los Profesores de Investigación Gerardo
Delgado Barrio, Pablo Villarreal Herrán y Carmela Valdemoro López, tres
personas inteligentes y entrañables donde las haya, que me han aportado muchas
cosas buenas, y que me llena de orgullo y satisfacción haber podido compartir tan
buenos momentos juntos.
Dicho esto, no puedo olvidarme del resto del grupo tan excepcional con los
que he compartido tantas y tantas horas. Desde los Doctores: Octavio Roncero,
José Campos, Alberto García, Marta Isabel Hernández, Tomás González,
Rita Prosmiti, Pilar de Lara, Estela Carmona, Juan Carlos Juanes, Manuel
Lara, Maximiliano Bartolomei, Agustín Grau, Angel Sanz...hasta el personal
administrativo del IMAFF: María Jesús Vallejo, Josefina Sánchez, y Adela
Fernández...además de los nuevos leoncitos...Ruth, Pedro y Ricardo y a mi
hermana marroquí Latifa Benjouali.
Y como no, a los leones: David (¡Que grande eres chaval!), Sergio (¡Gracias
por tantos y tan buenos consejos!), Álvaro (¡No cambies nunca!), y Cristina,
Susana y Lola (¡Sois la caña!).
¡Chavales, simplemente...os quiero!

No quiero finalizar este agradecimiento, sin las personas que se han cruzado
en mi vida en esta etapa de formación, y me han aportado todo lo bueno que había
dentro de ellas, comenzando en Badajoz (Josemari), pasando por Gainesville
(Jorge, Adriana, Juan Pedro, María Fernanda), Augsburg (Juan, Karen, Gloria,
Jessica), Oslo (Runar), Londres (Stephen), Rehovot (Sergey, Ilya, Eva), Madrid
(Eduardo, Lope y Ricardo) y terminando por los amigos de mi pueblo: Antonio,
Manolo y Juan Antonio.
¡Gracias por todo!
y sobre todo a mis Padres, Hermanos y mis tíos Emilia y Paco.
¡Gracias, Muchísimas Gracias por todo!

Índice general.
1. INTRODUCCIÓN. 4
2. EXPERIMENTO. 11
2.1. Introducción. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
2.2. Sondas de superficies. . . . . . . . . . . . . . . . . . . . . . . . . 11
2.3. Técnicas experimentales para medir el proceso de difusión y
vibración. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
2.3.1. Microscopía de campo iónico. . . . . . . . . . . . . . . . 13
2.3.2. Microscopía por efecto túnel. . . . . . . . . . . . . . . . 14
2.3.3. Espectroscopía vibracional. . . . . . . . . . . . . . . . . 15
2.4. Técnica QHAS. . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.4.1. Haz atómico. . . . . . . . . . . . . . . . . . . . . . . . . 16
2.4.2. Preparación de la muestra. . . . . . . . . . . . . . . . . . 17
2.4.3. Detectores. . . . . . . . . . . . . . . . . . . . . . . . . . 18
2.5. Resultados experimentales . . . . . . . . . . . . . . . . . . . . . 21
2.5.1. Na-Cu(001) . . . . . . . . . . . . . . . . . . . . . . . . . 22
2.5.2. CO-Cu (001) . . . . . . . . . . . . . . . . . . . . . . . . 30
2.5.3. CO-Pt (111) . . . . . . . . . . . . . . . . . . . . . . . . . 32
3. FORMALISMO TEÓRICO. 34
3.1. Procesos estocásticos. . . . . . . . . . . . . . . . . . . . . . . . . 34
3.1.1. Proceso del caminante aleatorio. . . . . . . . . . . . . . . 34
1

ÍNDICE GENERAL. 2
3.1.2. Proceso de Wiener. . . . . . . . . . . . . . . . . . . . . . 37
3.1.3. Proceso de Orstein-Uhlenbeck . . . . . . . . . . . . . . . 39
3.2. Ecuación de difusión. . . . . . . . . . . . . . . . . . . . . . . . . 47
3.3. Ecuación de Fokker-Planck. . . . . . . . . . . . . . . . . . . . . 50
3.4. Difusión de átomos bajo la presencia un potencial. . . . . . . . . 55
3.4.1. Ecuación de Kramers. . . . . . . . . . . . . . . . . . . . 55
3.4.2. Ecuación generalizada de Langevin. . . . . . . . . . . . . 57
3.4.3. Formalismo clásico teórico para medir difusión de átomos
sobre superficies por QHAS. . . . . . . . . . . . . . . . . 58
3.4.4. Aproximaciones dentro del formalismo teórico. . . . . . 63
3.4.4.1. Modelos de difusión continua. . . . . . . . . . 63
3.4.4.2. Modelo de difusión discreta. . . . . . . . . . . 66
3.4.4.3. Aproximación gaussiana. . . . . . . . . . . . . 70
3.4.4.4. Difusión de partícula aislada. . . . . . . . . . . 71
3.4.5. Difusión normal y anómala. . . . . . . . . . . . . . . . . 73
3.5. Vibración de baja frecuencia. . . . . . . . . . . . . . . . . . . . . 78
3.6. Modelo anarmónico. . . . . . . . . . . . . . . . . . . . . . . . . 80
3.7. Difusión y vibración de baja frecuencia. . . . . . . . . . . . . . . 82
3.8. Efectos cuánticos. . . . . . . . . . . . . . . . . . . . . . . . . . . 83
4. RESULTADOS. 85
4.1. Algoritmo numérico para resolver la ecuación de Langevin. . . . . 85
4.2. Superficie de Energía Potencial. . . . . . . . . . . . . . . . . . . 89
4.3. Dinámica caótica. . . . . . . . . . . . . . . . . . . . . . . . . . . 92
4.4. Espectro de potencias y difusión anómala. . . . . . . . . . . . . . 100
4.5. Influencia del ruido gaussiano blanco. . . . . . . . . . . . . . . . 113
4.6. Teoría de Kramers aplicada a la difusión de adsorbatos por
superficies. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 117
4.6.1. Teoría unidimensional. . . . . . . . . . . . . . . . . . . . 117
4.6.2. Teoría multidimensional. . . . . . . . . . . . . . . . . . . 121
4.6.3. Corrección de barrera finita. . . . . . . . . . . . . . . . . 122

ÍNDICE GENERAL. 3
4.6.4. Resultados Numéricos. . . . . . . . . . . . . . . . . . . . 124
4.6.4.1. Potencial separable. . . . . . . . . . . . . . . . 124
4.6.4.2. Potencial no separable. . . . . . . . . . . . . . 129
4.6.4.3. Potencial separable y no separable. . . . . . . . 134
4.7. Teoría hamiltoniana de línea vibracional de átomos adsorbidos
sobre superficies. . . . . . . . . . . . . . . . . . . . . . . . . . . 140
4.7.1. Formalismo hamiltoniano. . . . . . . . . . . . . . . . . . 140
4.7.2. Forma de línea para el modo vibracional. . . . . . . . . . 143
4.7.2.1. Oscilador armónico. . . . . . . . . . . . . . . . 143
4.7.2.2. Correcciones anarmónicas. . . . . . . . . . . . 145
4.7.2.3. Correcciones cuánticas. . . . . . . . . . . . . . 151
4.7.2.4. Desplazamiento y anchura del modo T depen-
diente de la temperatura. . . . . . . . . . . . . . 155
4.8. Tratamiento cuántico para explicar la forma de línea vibracional. . 162
4.8.1. Modelo de un oscilador armónico amortiguado excitado. . 162
4.8.2. Forma de línea para el modo T. . . . . . . . . . . . . . . . 166
4.8.3. Relajación vibracional en los sistemas Na/Cu(001), CO/Cu(001)
y CO/Pt(111). . . . . . . . . . . . . . . . . . . . . . . . . 170
4.9. Formas de línea para el pico cuasielástico y el modo T. . . . . . . 177
4.9.1. Forma de línea de difusión. . . . . . . . . . . . . . . . . . 177
4.9.2. Forma de línea de vibración. . . . . . . . . . . . . . . . . 183
4.10. Difusión y vibración a baja frecuencia. . . . . . . . . . . . . . . . 186
5. CONCLUSIONES. 192
A. Procesos estocásticos. 199
A.1. Procesos estacionarios. . . . . . . . . . . . . . . . . . . . . . . . 199
A.2. Proceso Gaussiano. . . . . . . . . . . . . . . . . . . . . . . . . . 200
A.3. Proceso markoviano. . . . . . . . . . . . . . . . . . . . . . . . . 200
Bibliografía. 202

Capítulo 1
INTRODUCCIÓN.
La difusión de adsorbatos (átomos, moléculas) es uno de los procesos más
elementales que tiene lugar en la superficie de un cristal. Este estudio es impor-
tante por razones prácticas y conceptuales. Por un lado, la difusión de adsorbatos
es una etapa preliminar de fenómenos de superficie más complejos tales como,
por ejemplo, la desorción asociativa y catálisis heterogénea. La necesidad de un
conocimiento cuantitativo y preciso de los coeficientes y velocidades de difusión,
así como una detallada caracterización de los mecanismos de difusión y de las
interacciones adsorbato-sustrato, han contribuido de forma decisiva a una mejora
de las correspondientes técnicas experimentales. Existen numerosas técnicas con
muy buena precisión para medir coeficientes de difusión así como frecuencias de
vibración de adsorbatos, cada una con sus correspondientes rangos de aplicabili-
dad y ventajas [1, 2, 3, 4]. Por otro lado, la teoría básica de la difusión atómica
y molecular es conceptualmente muy rica, con importantes implicaciones en las
diferentes ramas de la física, química y matemáticas. En general, existen diferen-
tes puntos de vista para analizar un determinado proceso. Y, dependiendo del caso
específico, todos pueden ser igualmente válidos, o unos pueden ser más apropia-
dos que otros. Por ejemplo, un proceso de difusión puede ser considerado como un
problema de movimiento browniano, cuya teoría se remonta a los trabajos de Ray-
leigh, Einstein, Smoluchowsky, Langevin, Fokker, Planck, y otros en el comienzo
4

CAPÍTULO 1. INTRODUCCIÓN. 5
del siglo pasado. Desde este punto de vista, las partículas se difunden siguiendo
trayectorias estocásticas bajo la acción de una fuerza aleatoria proveniente de un
gran número de átomos que constituyen la superficie cristalina (sustrato) a una
determinada temperatura, comportándose de la misma forma que las partículas
brownianas [5, 6]. La teoría de procesos estocásticos fue generalizada y sus ba-
ses matemáticas fueron asentadas por Stratonovich e Itô. Si a este tratamiento
estocástico que describe el movimiento browniano se le añade una fuerza adiabá-
tica ejercida sobre la partícula, se puede describir la difusión de adsorbatos sobre
superficies; donde por fuerza adiabática interpretamos la fuerza que deriva del
potencial de interacción adsorbato-sustrato. Pero la difusión atómica y molecular
puede también ser vista como un problema de una partícula encerrada en un pozo
de potencial y que puede escapar mediante activación térmica difundiéndose por
la superficie del cristal. Esta línea de investigación fue iniciada con el famoso tra-
bajo de Kramers en 1940 [7], estando todavía activa [8, 9]. Mientras que el primer
enfoque está íntimamente relacionado con la mecánica estadística fuera del equi-
librio, el segundo se encuentra mucho más relacionado con la teoría del estado
de transición de las reacciones químicas. Existe además otro punto de vista que
conviene resaltar, en el que se supone despreciable el acoplamiento no adiabático
(fuerza de fricción y aleatoria) entre el adsorbato y la superficie, dando lugar a
una dinámica determinista del adsorbato regida únicamente por las leyes de New-
ton. Aunque conceptualmente fue una idea propuesta por Boltzmann, deducida
termodinámicamente a partir de las ecuaciones microscópicas de movimiento pa-
ra pequeños sistemas con solo unos grados de libertad, en las últimas décadas se
ha encontrado una justificación rigurosa gracias al desarrollo de la teoría ergódica
de sistemas dinámicos no lineales [10]. Existen numerosos trabajos y libros pu-
blicados acerca de esta teoría del transporte caótico [11, 12, 13, 14]. La mayoría
de estos trabajos que combinan teoría y experimento sobre los procesos de difu-
sión y vibración adsorbato-superficie se han basado en los dos primeros y más
tradicionales puntos de vista, mientras que los desarrollos teóricos de la teoría de
transporte no lineal han sido aplicados principalmente a modelos simples tratados

CAPÍTULO 1. INTRODUCCIÓN. 6
de forma analítica.
Por otro lado, es bien sabido que los movimientos de difusión y vibración
de adsorbatos en superficies son dos procesos elementales los cuales son de
fundamental importancia en la física de superficies. La técnica experimental de
la dispersión cuasielástica de átomos de He (QHAS) se ha revelado asimisma
como una alternativa y muy conveniente herramienta para el análisis de ambos
movimientos, difusión [15, 16, 17, 18, 19] y vibración de baja frecuencia
[20, 21, 22, 23] de adsorbatos en superficies. De las medidas de tiempos de vuelo
[24], convertidos a una escala de transferencia energética, se puede obtener una
amplia información de los diferentes procesos que tienen lugar en la superficie,
manifestándose en este rango de energía por medio de picos de diferente
intensidad y formas de línea. En un típico espectro de transferencia energética,
principalmente se observa un pico de alta intensidad cuyo máximo corresponde
a una transferencia energética casi nula (el denominado pico cuasielástico o
pico Q), que nos da información del movimiento difusivo del adsorbato sobre
la superficie. Segundo, se observan diferentes picos, debido al intercambio
energético del átomo de He con los fonones de la superficie tal como la onda
Rayleigh (RW) y la resonancia longitudinal (LR), proporcionando información
sobre la dispersión inelástica que tiene lugar en la superficie (correspondiente a
los procesos de creación y aniquilación de los fonones del cristal). Y tercero,
picos adicionales débiles a baja transferencia energética atribuidos al movimiento
de traslación y/o rotación frustrado paralelo a la superficie de los adsorbatos,
modos T y R respectivamente. Teóricamente, la correspondiente forma de
línea de todos estos picos están representados por la magnitud que se mide
experimentalmente denominado factor de estructura dinámico S(∆K, ω), donde
∆K es la transferencia de momento paralelo a la superficie y ω = ∆E/~ es la
frecuencia, siendo ∆E la energía transferida a la sonda atómica. Esta magnitud
dinámica se define como la trasformada de Fourier espacio-temporal de la función
de correlación espacio-temporal de van Hove G(R, t) [25, 3]. Esta función nos
da idea de la probabilidad que dada una partícula inicialmente (t = 0) en el

CAPÍTULO 1. INTRODUCCIÓN. 7
origen R = (0, 0), cualquier partícula (incluyendo ella misma) se encuentre en
una posición de la superficie R = (x, y) en un tiempo posterior t 6= 0.
Con motivo de extraer información valiosa sobre la dinámica del adsorbato,
así como información de la interacción adsorbato-superficie, se han desarrollado
algunas aproximaciones teóricas para explicar dicho fenómeno de superficie,
tratando de relacionar la cantidad observable S(∆K, ω) con magnitudes físicas
tales como coeficientes de difusión y fricción y frecuencias vibracionales.
Tradicionalmente, se han empleado dos modelos físicos para obtener información
acerca de la dinámica de difusión:
(i) a pequeñas transferencias de momento paralelo, ∆K → 0 (sondeo a largas
distancias) la estructura de la superficie de energía potencial (SEP) adiabática que
simula la superficie no interviene y la difusión del adsorbato sobre la superficie
se considera como una partícula browniana sometida a una fricción y a un ruido
térmico como consecuencia de la interacción no adiabática adsorbato-sustrato, y
en la que se desprecia la interacción con otros adsorbatos; por tanto, este proceso
de difusión puede ser analizado bajo un formalismo estocástico. A partir de este
modelo de difusión libre, se puede obtener el coeficiente de difusión de una
partícula aislada que se difunde por la superficie, supuestamente plana (modelo
continuo de difusión);
(ii) en el caso contrario, a grandes transferencias de momento paralelo, la SEP
adiabática juega un papel muy importante en este proceso de difusión, por tanto
se emplea un modelo discreto de saltos instantáneos activados térmicamente entre
diferentes posiciones del cristal para explicar la difusión (modelo de Chudley-
Elliot [26]). En este régimen, el movimiento de vibración del adsorbato puede
asistir al movimiento de difusión.
Por otro lado, el pico correspondiente al modo-T presenta dos efectos, uno
de desplazamiento en energía y otro de anchura energética cuando aumenta la
temperatura en la superficie. Dichos efectos han sido explicados hasta la fecha
mediante un tratamiento anarmónico en el que se supone transiciones entre
los niveles vibracionales de un oscilador anarmónico [20], teniendo en cuenta

CAPÍTULO 1. INTRODUCCIÓN. 8
además que se parte de una población inicial que obedece una distribución de
Boltzman. Estos efectos nos proporcionan información acerca de la curvatura de la
interacción adiabática y del coeficiente de fricción cuando se extrapola la anchura
energética [anchura a la mitad de la altura (FWHM)] a temperatura cero.
En cualquier caso, un análisis detallado para las diferentes formas de línea de
los picos Q y T está todavía por desarrollarse y, por tanto, el principal objetivo
en esta memoria de investigación ha sido contribuir a esta tarea del desarrollo de
un buen modelo teórico que tenga en cuenta ambos movimientos de difusión y
vibración, y así poder reproducir teóricamente las formas de línea de los picos Q
y T medidos experimentalmente. Básicamente, nos hemos centrado en sistemas
físicos del que se tiene mucha información experimental, pero para los que no se
ha desarrollado una teoría completa que explique estas formas de línea obtenidas
en los espectros de transferencia energética medidos por QHAS. En esta memoria
de investigación se van a estudiar sistemas con baja concentración de adsorbatos,
para poder así comprender mucho mejor la difusión y la vibración del adsorbato
sobre la superficie, no teniendo en cuenta la interacción con otros adsorbatos.
Este primer análisis nos va a aportar mucha información sobre la dinámica del
sistema adsorbato-superficie, para así en un futuro poder estudiar y comprender
mejor la dinámica completa adsorbatos-superficie teniendo en cuenta ahora sí la
interacción entre adsorbatos.
Otro de nuestros objetivos ha sido el empleo de la teoría de Kramers
desarrollada para superficies monodimensionales que, por primera vez, se ha
aplicado a procesos de difusión de adsorbatos sobre superficies observadas
mediante la técnica QHAS.
Se ha realizado también un tratamiento clásico bajo una teoría hamiltoniana
además de un tratamiento cuántico considerando un oscilador armónico (adsorba-
to) acoplado a un gran número de osciladores armónicos no acoplados entre sí que
representan el baño térmico (superficie a una temperatura T ), e intentar reproducir
la forma de línea del modo T medida experimentalmente, así como dar una expli-
cación de los efectos que presenta este pico T cuando se aumenta la temperatura

CAPÍTULO 1. INTRODUCCIÓN. 9
de la superficie.
Y, por último, se ha estudiado bajo un formalismo determinista regido por
las ecuaciones de Newton el comportamiento caótico (dinámica no lineal) que
aparece en estos sistemas como consecuencia de la PES adiabática empleada
para simular esta interacción potencial adsorbato-sustrato, dando como resultado
difusión anómala (comportamiento no browniano).
Por otro lado, sabemos que los movimientos de difusión y vibración a baja
frecuencia están gobernados por diferentes escalas de longitud y de tiempo y es
por eso que muchas veces se pueden tratar de forma independiente. Además, se
sabe que pueden estar acoplados cuando la energía térmica kBT (donde kB es la
constante de Boltzman) es similar o superior a la altura de la barrera energética
de difusión siempre y cuando nos encontremos en la zona de Brillouin de ∆K
grandes [27, 28, 29, 30]. Hasta donde se sabe, el primer intento de considerar las
dos clases de movimientos conjuntamente se encuentra recogido en el trabajo de
Chen y Ying [27, 28] en el que se utilizó una teoría microscópica basada en el
formalismo de los operadores de proyección de Mori [31]. Posteriormente, se han
aplicado teorías estocásticas alternativas dentro del formalismo de Fokker-Planck
[32], o el formalismo equivalente de Langevin [33, 34] que han reproducido
satisfactoriamente los experimentos mediante interpretaciones clásicas [16, 17,
22, 27, 28, 29, 30, 14, 35, 36] o mecanocuánticas [37].
Esta memoria de investigación se ha organizado de la siguiente forma. En el
siguiente capítulo, se ha realizado una breve introducción a las principales técnicas
experimentales empleadas para medir los procesos de difusión y vibración de
adsorbatos sobre superficies, presentando una descripción más detallada de la
técnica QHAS. Esta técnica permite medir los procesos de vibración y difusión de
forma conjunta. Se mostrarán los resultados experimentales obtenidos por QHAS
para los diferentes sistemas estudiados: Na/Cu(001), CO/Cu(001), y CO/Pt(111).
En el capítulo tres se expone brevemente el formalismo estocástico empleado para
analizar el fenómeno de la difusión de adsorbatos sobre superficies y se describe
la teoría básica que se encuentra detrás de la técnica QHAS y su conexión con el

CAPÍTULO 1. INTRODUCCIÓN. 10
punto de vista estocástico. La correspondiente teoría es una extensión de aquélla
desarrollada para la colisión de neutrones térmicos en superficies. Por otro lado,
se introduce el formalismo desde el punto de vista desarrollado por Kramers en el
que se considera la partícula encerrada y vibrando dentro de un pozo de potencial,
pudiendo escapar por activación térmica y dando lugar al fenómeno de difusión.
Con estos formalismos teóricos se obtiene una información muy valiosa acerca
de las propiedades de transporte de los adsorbatos sobre la superficie, así como
una mayor comprensión de la dinámica de adsorción. En el capítulo cuatro, se
muestran todos los resultados obtenidos en esta memoria de investigación. Desde
el cálculo de magnitudes como coeficientes de difusión, frecuencias vibracionales,
frecuencias y distribuciones de salto, hasta el desarollo de nuevas teorías para la
obtención de formas de línea que se ajusten bien a los picos medidos por QHAS
para la difusión y la vibración. También se mostrará el efecto de estrechamiento
observado por primera vez en estos sistemas en los picos Q y T [38]. Los
experimentales ajustan usualmente los picos Q y T a una lorenciana. Como se
verá en esta memoria de investigación, este ajuste no está siempre justificado.
Por último, como objetivo final se muestra el tratamiento teórico propuesto en
esta memoria para el que se ha tenido en cuenta la contribución de cada proceso
de difusión y vibración medidos con la técnica QHAS. Ya que, como se ha
dicho anteriormente, cuando se dan las condiciones idóneas de transferencia de
momento paralelo ∆K, barrera de difusión y temperatura estos procesos se dan
simultáneamente, contribuyendo ambos a las formas de líneas medidas en el
experimento.
Por último, se presentan las conclusiones obtenidas en este trabajo de
investigación sobre los procesos de difusión y vibración que tienen lugar en el
fenómeno de adsorción de átomos y moléculas sobre superficies.

Capítulo 2
EXPERIMENTO.
2.1. Introducción.
En este capítulo se van a describir las principales técnicas experimentales que
existen actualmente para medir los procesos de difusión y vibración que tienen
lugar en la superficie de un cristal. Primeramente, se va a comenzar hablando
de las sondas de superficies que se emplean para observar tales fenómenos.
Seguidamente, se procederá a analizar brevemente dichas técnicas experimentales
y, por último, se describirá de una forma más detallada la técnica QHAS cuya
sonda son los átomos de He. Finalmente, se mostrarán y discutirán los resultados
experimentales obtenidos por esta técnica para los diferentes sistemas adsorbato-
sustrato analizados en esta memoria de investigación.
2.2. Sondas de superficies.
En la actualidad, una gran parte de la información sobre los procesos
dinámicos que ocurren en la interacción átomo-superficie se obtiene a partir
de experimentos de dispersión de haces atómicos/moleculares por superficies.
Cuando un haz de átomos incide sobre la superficie de un cristal, la colisión
induce cambios tanto en las propiedades del haz como en las del cristal. Todos
11

CAPÍTULO 2. EXPERIMENTO. 12
estos experimentos están diseñados para medir solamente los cambios ocurridos
en las propiedades del haz incidente. Cuando se está interesado en el análisis de
superficies, se deben utilizar técnicas experimentales que no penetren mucho en
el cristal, de ahí la importancia de buscar la técnica adecuada para tal análisis.
La experiencia nos dice que los dispositivos de difracción de rayos X y de
neutrones térmicos no son muy convenientes para el análisis de superficies ya
que son haces que penetran mucho en el cristal; en cambio, los electrones a baja
energía (10-300 eV) y los átomos de He sondean las capas más externas de la
superficie. En particular, con el haz atómico, se tiene que a grandes distancias
el átomo está débilmente atraído por interacciones de van der Waals, mientras
que a cortas distancias el solapamiento de la distribución electrónica del He y la
correspondiente distribución de la superficie produce una repulsión muy fuerte.
Esto ocasiona que el He sea sensible exclusivamente a la capa cristalina más
externa y, en particular, a la densidad electrónica sobre la superficie. Además,
su baja energía y su naturaleza inerte aseguran que sea una sonda no destructiva.
También debido a su masa, el átomo de He tiene una longitud de onda comparable
con las distancias interatómicas de la superficie, presentando así fenómenos de
difracción que nos permite conocer su estructura[39] y densidad electrónica[40].
Los átomos de He presentan una relación entre energía y momento muy favorable
para que se creen fonones específicos en la superficie y poder también así medir
dispersión inelástica. Por último, destacar que es muy sensible a la existencia
de impurezas y defectos en la superficie, que nos permite analizar el desorden,
distribución de adsorbatos, así como la difusión de átomos sobre superficies. Estas
características convierten al He en la sonda atómica por excelencia para el análisis
de superficies. A continuación se muestra una tabla con las diferentes sondas de
superficies:

CAPÍTULO 2. EXPERIMENTO. 13
Helio Electrones Neutrones Rayos X
Energía (meV) 5-100 (0.1-3)105 1-100 (0.4-3)107
Penetración no de capas 1 3-5 1 1Resolución en E (meV) 0.2 3 0.001 >8Resolución en k (Å−1) 10−2 10−2 (0.1-1)10−3 10−4
Máxima presión ambiental (Pa) 10−3 10−3 >105 >105
2.3. Técnicas experimentales para medir el proceso
de difusión y vibración.
Los coeficientes de difusión pueden variar en ordenes de magnitud, depen-
diendo de la temperatura a la que se encuentra la superficie. Para medir difusión
lenta, donde el tiempo entre salto y salto es del orden de segundos se utiliza la
microscopía por efecto túnel (STM) y la microscopía de campo iónico (FIM). En
cambio, si el tiempo de saltos es del orden de microsegundos, la técnica que es
sensible para medir difusión rápida es la dispersión cuasielástica de átomos de
He. A diferencia de la STM y FIM, QHAS nos proporciona información de la
dinámica de difusión de forma indirecta.
2.3.1. Microscopía de campo iónico.
La microscopía de campo iónico [41] proporcionó la primera medida directa
de difusión de átomos adsorbidos. Esta técnica evolucionó a partir de la
microscopía por campo de emisión (FEM)[1] con la que se observó directamente
el proceso dinámico de difusión. Un metal sirve como sustrato para los átomos
adsorbidos. A temperaturas suficientemente bajas, la difusión es tan lenta que
puede ser ignorada. Seguidamente, un gas inerte es ionizado por un fuerte
campo eléctrico y los iones emitidos son detectados sobre una pantalla. La
ionización depende sensiblemente del entorno local, de ahí que se pueda observar
directamente la localización de los átomos adsorbidos en la superficie. El método

CAPÍTULO 2. EXPERIMENTO. 14
tiene una resolución de 1-2 Å proporcionando una imagen detallada de dicha
localización. Después de la primera medida, el metal se calienta rápidamente
teniendo que enfriarlo para poder realizar una nueva medida. Para obtener
información sobre los saltos individuales de los átomos adsorbidos, el periodo
de altas temperaturas debe ser lo suficientemente corto para que las partículas
adsorbidas no experimenten muchos saltos. Ya que, de ser así, se obtendría
una distribución gaussiana con una anchura que daría un coeficiente de difusión
global.
2.3.2. Microscopía por efecto túnel.
La Microscopía por efecto túnel [42] consiste en generar un cambio
de corriente por efecto túnel en un filamento metálico, obteniendo así una
observación directa la difusión de las partículas adsorbidas a la superficie. Este
efecto hace que la superficie se caliente rápidamente, originando un proceso de
difusión. Una vez que el filamento se enfría, se procede a una nueva medida.
Finalmente, la difusión atómica es recogida en forma de película en la que se
puede ver en detalle el mecanismo de difusión tipo “caminante aleatorio” [43]
donde la partícula adsorbida salta hacia delante y hacia atrás a través de una
superficie bien caracterizada. Estas medidas han mostrado también que estos
átomos pueden, en un simple salto , viajar sobre más de una celdilla unidad de
la red cristalina. El aspecto más delicado de esta técnica es el fuerte campo que
produce el filamento, pues puede provocar un cambio en el entorno electrónico de
las partículas localmente adsorbidas afectando así al proceso natural de difusión
[44, 45, 46].
Ambas técnicas FIM y STM se encuentran limitadas a los procesos de difusión
lenta, siendo el tiempo medio entre saltos de las partículas adsorbidas del orden de
10−2 s. Por tanto, para procesos de difusión más rápida, se deben utilizar métodos
indirectos, tal y como la QHAS. A continuación se muestra una tabla con las
técnicas experimentales mencionadas para el estudio del movimiento de difusión:

CAPÍTULO 2. EXPERIMENTO. 15
Técnica Rango de D (cm2s−1)
STM 10−19 − 10−16
FIM 10−19 − 10−16
FEM 10−14 − 10−9
QHAS 5,10−6
Por último, tenemos que decir que existen más técnicas de difusión sobre
superficies que se pueden encontrar en la literatura [47, 48, 4, 1].
2.3.3. Espectroscopía vibracional.
La espectroscopía vibracional de especies adsorbidas (átomos o moléculas)
sobre superficies es una técnica poderosa para extraer información de la
interacción adsorbato-adsorbato y adsorbato-sustrato que gobiernan procesos tales
como captura, desorción, disociación, así como procesos complejos de difusión y
reacciones en superficies. Los modos vibracionales de los adsorbatos se clasifican
en modos internos (altas frecuencias) y modos externos (bajas frecuencias). Los
primeros están localizados dentro del propio adsorbato, y sus amortiguamientos
o relajaciones sobre la superficie se producen a través de mecanismos de
excitación originando la creación de pares electrones-huecos [49, 50], los cuales
son independientes de la temperatura [51]. En cambio, los modos externos
corresponden a movimientos frustrados sobre la superficie (por ejemplo, para una
molécula estos podrían ser un movimiento frustrado de rotación, de vibración
o de traslación), pudiendo intercambiar energía con los fonones de la superficie
mediante un mecanismo de amortiguamiento dependiente de la temperatura o
un mecanismo de excitación par electrón-hueco . Las correspondientes energías
vibracionales han sido determinadas por diferentes técnicas tales como dispersión
inelástica de neutrones [52, 53] y electrones inelástica [54], espectroscopía
infrarroja [55] y más recientemente, mediante QHAS.
A continuación, describimos en una tabla las técnicas más comunes que se
emplean para medir la vibración:

CAPÍTULO 2. EXPERIMENTO. 16
Técnica Rango Espectral (meV) Resolución (meV) (monocapas)
INS .100 >10−2 ≥1IRS &22 0.1 >10−3
EELS &5 >1 >10−4
QHAS 0.2− ≈50 >0.08 >10−3
Cuadro 2.4: INS (Dispersión Inelástica de Neutrones), IRS (EspectroscopíaInfrarroja), EELS (Dispersión Inelástica Electrónica).
2.4. Técnica QHAS.
El aparato instrumental de la técnica QHAS [21] está constituido básicamente
por los siguientes dispositivos: una fuente de haces moleculares, una muestra que
se sitúa dentro de una cámara de vacío, detectores para registrar los estados finales
del haz y aparatos auxiliares que controlan el estado de la muestra durante el
tiempo que dura el experimento. El montaje conceptualmente es muy simple, pero
su realización en la práctica requiere el uso de técnicas sofisticadas para poder
medir este tipo de interacción microscópica.
2.4.1. Haz atómico.
Para llevar a cabo el experimento, el haz tiene que ser intenso, monocromático
y con una divergencia angular mínima. Esto se consigue mediante una expansión
adiabática en la que el haz de gas de He se genera a alta presión (30-200
bar) y se hace fluir a través de una pequeña “boquilla” (con un diámetro de
aproximadamente 10 µm)[56, 57]. Debido a la expansión en el vacío, el gas se
enfría, aumentando así su monocromaticidad. Con este procedimiento se consigue
un haz intenso con una distribución de velocidades tal que ∆v/v . 1 %, siendo v
la velocidad de los átomos de He. Las velocidades del haz de He que se utilizan en
los experimentos estan comprendidas entre 3000 m/s ( ki =16 Å−1, λdB =0.3 Å,
donde λdB es la longitud de onda de De Broglie) y 700 m/s (ki =4 Å−1, λdB =1.5

CAPÍTULO 2. EXPERIMENTO. 17
Å). Estas velocidades se obtienen simplemente cambiando la temperatura de la
fuente desde 300 K a 40 K. Seguidamente, el haz es transformado en pulsos
mediante un disco giratorio de rendijas triangulares denominado “chopper”. Este
haz puede interseptar las rendijas triangulares a diferentes alturas ocasionando
así pulsos de diferente duración y, variando la frecuencia de rotación del disco, se
puede adaptar el pulso del haz a los requerimientos del experimento. Por último, el
haz alcanza la cámara de vacío donde se encuentra la muestra que se va a estudiar,
situándose ésta en un manipulador que permite optimizar las posiciones x-y-z del
cristal.
2.4.2. Preparación de la muestra.
Las muestras han de ser monocristales con superficies limpias y bien
caracterizadas. Una tarea difícil en el caso de superficies metálicas. Tal proceso
de preparación consta de dos etapas:
(i) La primera consiste en la preparación del monocristal. Éste tiene que estar
limpio de impurezas y con la superficie a estudiar bien caracterizada. Para ello
se realizan diferentes tratamientos químicos y mecánicos. Mediante difracción de
rayos X se llega a conocer la ordenación del cristal.
(ii) La segunda etapa se realiza una vez que la muestra se sitúa dentro de la
cámara de vacío, y continúa durante todo el experimento, controlando en todo
momento la limpieza y la ordenación del cristal. Esta etapa de preparación de la
muestra es muy delicada y requiere la ayuda de diversas técnicas de análisis de
superficies.
Por otro lado, la cámara de dispersión se rellena por partículas, las cuales
pueden ser adsorbidas por la superficie cristalina. La concentración de partículas
adsorbidas a la superficie viene determinado por una densidad superficial relativa
simbolizada por ϑ.

CAPÍTULO 2. EXPERIMENTO. 18
2.4.3. Detectores.
Los detectores de los haces dispersados por la muestra deben tener una alta
sensibilidad selectiva al haz molecular, de esta forma se minimiza la relación
señal-ruido y el tiempo de medida. Esto es importante para superficies metálicas
como las del Cu, ya que debido a que se contaminan fácilmente, el tiempo de
realización del experimento tiene que ser limitado. Además, estos detectores
deben medir el flujo saliente en todas las direcciones posibles, por lo que se
requiere que puedan rotar y que posean una pequeña apertura, pudiendo así
conocer la estructura periódica de la superficie. En el experimento, el detector que
se utiliza es un espectrómetro de masas asociado a un ionizador por bombardeo de
electrones optimizado para alcanzar una alta sensibilidad. Para reducir el fondo de
He que resulta en el detector proveniente del flujo de haz de la cámara de vacío,
se sitúan algunas cámaras de bombeo entre la cámara de dispersión y el detector.
También se utilizan cámaras de bombeo adicionales para reducir la presión parcial
a 10−15 mbar (≈10 átomos de He/cm3), obteniéndose en el detector un fondo de
helio del 10−5 % respecto del flujo original que sale de la cámara de dispersión.
Un aspecto muy importante en este tipo de estudios es la colocación geométrica
del detector. En la mayoría de los aparatos experimentales se mantiene un ángulo
constante θSD entre el ángulo incidente θi del haz de He proveniente de la fuente,
y el ángulo θf dispersado por la muestra . En la siguiente figura se muestran los
dispositivos básicos que constituyen el aparato QHAS, así como su disposición
geométrica, tan importante en este tipo de procesos.

CAPÍTULO 2. EXPERIMENTO. 19
Figura 1. En la Fig. 1a podemos ver la fuente de gas He que es transformadoen pulsos por el disco giratorio ("chopper"), la superficie cristalina donde seencuentran los átomos adsorbidos y, por último el detector para recoger el hazde helio dispersado por la muestra. Además, se observa la distribución típica develocidad del haz incidente y del haz dispersado en el experimento por QHAS.Esta distribución angular de intensidad dispersada por la muestra y recogida enel detector se obtiene haciendo girar la muestra, recogiéndose en el espectrodiferentes intensidades del haz para cada ángulo de rotación. En la Fig. 1b seobserva la cinemática de dispersión que se encuentra completamente definida porlos vectores de ondas inicial y final, ki y kf respectivamente, así como el ánguloincidente θi . Los componentes del plano de la superficie de los vectores de ondasson Ki y Kf .

CAPÍTULO 2. EXPERIMENTO. 20
Cuando el haz de He es dispersado por los átomos adsorbidos que se
difunden en la superficie, se detecta una pérdida de coherencia en el haz
incidente; este efecto origina una distribución de energía un poco más ancha
que la correspondiente si estuviese limpia. La pequeña anchura energética que
se produce por átomos de He dispersados elásticamente por los adsorbatos
que se difunden en la superficie recibe el nombre de dispersión cuasielástica
[58, 59, 60, 19]. Esta distribución de energía dispersada se mide mediante el
análisis de tiempos de vuelo: “Tiempo que tarda un pulso de haz de He desde
que sale del “chopper” hasta que alcanza el detector”. Los átomos de He que
pierden energía al interaccionar con la superficie son más lentos, y requieren más
tiempo en llegar al detector; en cambio, los átomos que al interaccionar con ésta
ganan energía llegan antes. Esta distribución de tiempos de vuelo que se recoge
en el detector es transformada a un espectro por transferencia energética, cuya
correspondiente forma de línea se conoce como pico cuasielástico o pico Q, (véase
la Fig. 2).
Una vez que se mide la anchura energética del pico cuasielástico, se relaciona
con la transferencia de momento paralelo a las partículas del haz, obteniéndose
así toda la información correspondiente al mecanismo de difusión. También
hay que mencionar que esta técnica experimental está limitada a sistemas con
rápida difusión con coeficientes del orden de 10−6cm2s−1. La técnica posee una
interpretación teórica más compleja que las anteriormente mencionadas FIM o
STM, pero presenta la ventaja que no es una técnica destructiva como las otras.
Por otro lado, la QHAS ha sido la primera técnica con la que se obtuvieron
los primeros resultados experimentales fiables sobre las vibraciones a baja
energía, difíciles de detectar por las técnicas anteriores. La primera medida del
pico inelástico correspondiente al movimiento vibracional translacional frustrado
paralelo a la superficie (modo T) fue realizada en 1986 por Lahee, Toennies, and
Wöll [61]. No siempre estos dos picos, Q y T, aparecen en el espectro tan bien
separados por sus escalas temporales, ya que la temperatura de superficie juega
un papel clave en el acoplamiento o desacoplamiento de estos dos mecanismos,

CAPÍTULO 2. EXPERIMENTO. 21
Figura 2. Patrón de tiempo de vuelo y espectro de transferencia energética ∆E.Un ejemplo de espectro de tiempos de vuelo está dado en a). El mismo espectro,convertido a transferencia energética se muestra en b).
observándose un solapamiento de estos picos debido a la contribución de la
vibración (modo T) sobre la difusión (pico Q) en el caso de estar acoplados.
De ahí la importancia de esta técnica a la hora de estudiar ambos mecanismos
de forma detallada y precisa. Decir que, aunque esta técnica no depende de la
conductividad que posea el cristal, puede ser igualmente aplicable a los aislantes,
semiconductores y metales, además de poder observar tanto los procesos de
difusión, como los procesos de vibración a baja energía.
2.5. Resultados experimentales
En esta sección vamos a mostrar los resultados experimentales recogidos
en los trabajos realizados por Toennies y col.[15, 16, 17, 20, 22], empleando

CAPÍTULO 2. EXPERIMENTO. 22
la técnica QHAS para los sistemas Na-Cu(001), CO-Cu(001) y CO-Pt(111).
Mostraremos de forma separada los resultados experimentales que se obtuvieron
para tales sistemas, para más adelante en los sucesivos capítulos analizarlos e
interpretarlos detenidamente.
2.5.1. Na-Cu(001)
Estos resultados experimentales se han llevado a cabo por HUGO II[62, 63],
un aparato de QHAS de alta resolución que incorpora una fuente supersónica
de haz de He de ∆v/v . 1 %, una cámara de vacío donde se situó la muestra a
2,10−11 mbar, y un brazo de tiempo de vuelo de 1.4 m a un ángulo fijo θSD = 95,8
de en la dirección del haz incidente. Se varía el ángulo del haz incidente (θi) con
respecto a la normal a la superficie. La transferencia de momento paralelo viene
dada por la siguiente relación cinemática:
∆K = Kf − Ki = kfsin(θSD − θi) − kisin(θi) (2.1)
siendo ki y kf las magnitudes de los vectores de ondas inicial y final. En un
experimento típico de, por ejemplo, Na/Cu(001) la resolución angular es de
δθ = 0,3, y la resolución de la transferencia de momento paralelo δ(∆K)
>0.03 Å−1. Se consigue una resolución en la transferencia energética de δE=
0.3 meV. Para obtener una superficie cristalina limpia, la superficie de Cu (001)
es sometida a ciclos repetitivos de “sputtering” hasta que la espectroscopía
Auger detecta una impureza inferior al 0.005 %. Se puede comprobar por
dispersión elástica e inelástica una superficie suave con un alto ordenamiento.
La concentración ϑNa=0.028, da una reflectividad de I/I0=0.5, donde I0 es la
intensidad especular. Dicho parámetro ϑ se mide con respecto a la densidad
atómica de la superficie Cu(001), donde se toma como referencia ϑNa=1, la
densidad superficial correspondiente a 1.53×1019 átomos de Na/cm2. Por último
indicar que la temperatura en cada espectro de tiempo de vuelo fue estable con
una ligera fluctuación de ± 1K.

CAPÍTULO 2. EXPERIMENTO. 23
En futuras referencias hablaremos de las direcciones diagonal [100] y paralela
[110] en la superficie de Cu(001). En las Figs. 3a) y 3b) se indican la estructura de
la superficie de Cu (001) en el espacio real y recíproco así como las direcciones
de difusión a lo largo de la superficie, respectivamente.
Figura 3. Diagrama esquemático que muestra la superficie de Cu(001). El espacioreal (a) y la primera zona de Brillouin de la red recíproca (b). Las dos direccionesde difusión son la diagonal [100] y la paralela [110].
En la Fig. 4 se muestra una serie de distribuciones de tiempos de vuelo
convertidas a escala energética, que fue obtenida por QHAS a varias temperaturas
para una concentración de Na de ϑNa = 0,028 sobre Cu(001) y ∆K=0.86 Å−1,
orientado en la dirección [100].
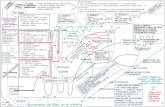
CAPÍTULO 2. EXPERIMENTO. 24
Figura 4. Espectro de transferencia de energía (pico Q) a diferentes T de lasuperficie. Los círculos abiertos muestran los puntos experimentales, mientrasque las líneas discontinuas muestran la función respuesta del instrumento. Lalínea sólida a través de los datos experimentales refleja un perfil de Voight(convolución de la función respuesta del instrumento (función gaussiana) con unperfil lorenciano optimizado[64]). Se puede ver en el recuadro de la parte superiorde la figura, el espectro de tiempo de vuelo a 180 K ampliado para poder observarel modo T.
Vemos cómo la anchura energética del pico Q medido experimentalmente
ajusta bien con la correspondiente al perfil de Voight, (línea continua) obtenido
como convolución de una lorenciana y la función respuesta del instrumento
(gaussiana). La anchura del pico cuasielástico se encuentra enmascarada por la
anchura intrínseca del instrumento de medida. Esto es debido a que las anchuras
energéticas son comparables. Para extraer la anchura cuasielástica del perfil de

CAPÍTULO 2. EXPERIMENTO. 25
Voight, los experimentalistas suponen que el pico Q obedece a una lorenciana , y la
convolucionan con la gaussiana del instrumento mediante la siguiente expresión:
FT (Iexp) = FT (Iresp) × FT (L) (2.2)
siendo Iexp(~ω) la distribución energética medida experimentalmente, Iresp(~ω)
la función respuesta del instrumento (gaussiana), y L(~ω) es la función lorenciana
que representa el pico Q correspondiente a la difusión, donde FT () simboliza la
transformada de Fourier de las funciones. Por tanto, como la anchura gaussiana
del instrumento es conocida, el único parámetro ajustable es la anchura de
la lorenciana. De este modo, vamos variando la anchura de la lorenciana,
quedándonos con la anchura que convolucionada con la gaussiana produzca un
perfil de Voight que concuerde con el pico experimental. Decir que, según los
experimentalistas, la función que mejor se ajusta a las formas de línea de los picos
Q y T es una lorenciana, pero como ya comentamos en la introducción, uno de los
objetivos de nuestra memoria de invesigación ha sido analizar las formas de línea
de estos picos con mucho más detenimiento, ya que no está claro que obedezcan
a simples lorencianas.
A continuación se muestra en la Fig. 5 distintos espectros de tiempos de vuelo
convertidos a transferencia energética para una concentración de Na ϑNa = 0,047
para un rango de diferentes ángulos incidentes comprendidos entre 37,9 y 46,9
a una temperatura de superficie T = 50 K. A esta temperatura tan baja la difusión
es suficientemente lenta que no afecta a la anchura de los picos cuasielástico
e inelásticos; debido a ésto los picos medidos son particularmente estrechos.
En adición al pico cuasielástico, se observan distintos picos inelásticos con
transferencia de energía positiva correspondiente a los eventos de aniquilación
de fonones y con transferencia de energía negativa correspondiente a los eventos
de creación.

CAPÍTULO 2. EXPERIMENTO. 26
Figura 5. La energía del haz incidente empleada fue de 20 meV orientado a lolargo de la dirección [100]. En dicha figura puede observarse cómo, además delpico cuasielástico (∆E =0) y los picos inelásticos T (∆E = ±5,8meV), aparecendos picos inelásticos, el de dispersión Rayleigh (RW) y la resonancia longitudinalfonónica del sustrato (LR), dos picos de bastante relevancia en los experimentosde dispersión inelástica. En esta figura también se puede apreciar cómo la posicióndel modo T no cambia cuando se varía el ángulo incidente, dicho de otro modo,no varía su posición para los diferentes valores de ∆K, por lo que podemos decirque el modo T no presenta dispersión.
Seguidamente, la no dispersión del modo T puede observarse claramente en
la Fig. 6. En esta gráfica los picos inelásticos obtenidos de los espectros de
tiempos de vuelo convertidos a transferencia de energía se representan frente a la
transferencia de momento paralelo, ∆K. Vemos cómo al cambiar la transferencia
de momento paralelo o lo que es lo mismo el ángulo incidente, la curva de
dispersión correspondiente al modo T se mantiene constante, al contrario de las
curvas RW y LR. Estas curvas de dispersión del sustrato de Cu(001) RW y LR
son observadas previamente considerando la superficie de Cu(001) limpia, que

CAPÍTULO 2. EXPERIMENTO. 27
una vez introducido el adsorbato de Na, aparecen los correspondientes armónicos
del modo T.
Figura 6. Curva de dispersión a través de la dirección [100] a una temperatura desuperficie T = 50 K para un ϑNa = 0,047 adsorbido sobre Cu(001). Las curvasde dispersión de RW y LR del sustrato limpio Cu(001) están indicadas con líneassólidas. El modo T de los átomos de Na se muestran con la línea con puntos. Loscírculos abiertos muestran los datos experimentales. Vemos aquí cómo el modo Tno presenta dispersión.
Por otro lado, también se ha podido medir la dependencia de la anchura del
pico cuasielástico con la transferencia de momento paralelo. Esta dependencia
[65, 3] es de gran interés a la hora de interpretar el comportamiento difusivo de
las partículas adsorbidas a la superficie. En la Fig. 7 se recoge esta dependencia
para las diferentes temperaturas de superficie 200, 250 y 300 K respectivamente a

CAPÍTULO 2. EXPERIMENTO. 28
lo largo de las direcciones [100] y [110].
Figura 7. Dependencia de la anchura energética a mitad de la altura (FWHM)del pico cuasielástico Γ con la transferencia de momento paralelo ∆K. Loscírculos abiertos muestran los datos experimentales , y la línea continua muestralos resultados teóricos obtenidos por A.P. Graham y col.[16]. La energía del hazincidente fue de 11.2 meV con una concentración ϑNa=0.028 para la dirección[100] y ϑNa=0.047 para la dirección [110].
Por último, también se ha medido experimentalmente la dependencia de la
anchura energética y la posición en energía (~ω) del modo T con la temperatura
de superficie T , esta dependencia se muestra en la Fig. 8.

CAPÍTULO 2. EXPERIMENTO. 29
Figura 8. Dependencia de la posición y FWHM del pico T frente a la temperatura.Los círculos abiertos muestran los datos experimentales, y la línea continuamuestra el ajuste teórico realizado. La energía del haz incidente fue tomada a20 meV con un ángulo incidente de 57.9 a través de la dirección [100] con unaconcentración ϑNa=0.028. La ventana nos muestra un típico espectro de tiempode vuelo para una temperatura de 125 K transformado a la escala de transferenciaenergética. Apareciendo los eventos inelásticos de creación (TC) y aniquilación(TA) del modo T, además de los picos RW y LR.
Puede observarse en la gráfica superior cómo la posición del modo T
experimenta un desplazamiento lineal hacia el rojo a medida que aumenta la
temperatura, mientras que en la gráfica inferior se observa cómo su anchura
energética crece linealmente con ésta. Por último, señalar que si se extrapola la
posición y anchura energética del modo T a 0 K, se obtienen valores de 6 meV

CAPÍTULO 2. EXPERIMENTO. 30
y 0.6 meV, correspondiente a la frecuencia natural de vibración del adsorbato y
coeficiente de fricción.
2.5.2. CO-Cu (001)
En la sección anterior presentamos los datos experimentales obtenidos por
QHAS para átomos de Na adsorbidos sobre la superficie de Cu (001). A
continuación, vamos a presentar los datos experimentales obtenidos con QHAS
para moléculas de CO adsorbidas sobre la superficie de Cu(001) [66, 20]. El haz
de He tiene una energía Ei= 11.2 meV con una resolución energética de 0.33 meV,
y la concentración de moléculas de CO fue inferior al 3 %. En la Fig. 9 se muestra
el pico T medido a diferentes temperaturas.
Figura 9. Comparación del modo T medido para una concentración de CO del 3 %adsorbido sobre Cu(001) con la función respuesta el instrumento (gaussiana). Loscírculos abiertos muestran los datos experimentales y la línea continua muestra elajuste de una lorenciana convolucionada con la función respuesta del instrumento(- - - -), originando el correspondiente perfil de Voight.
También se ha podido medir la dependencia de la anchura energética

CAPÍTULO 2. EXPERIMENTO. 31
deconvolucionada del modo T y su posición energética (~ω) con la temperatura,
(véase Fig. 10).
Figura 10. Dependencia con la temperatura de a) La posición en energía y, b) LaFWHM deconvolucionada del modo T. Los círculos y triángulos vacíos muestranlos datos experimentales, mientras que la línea continua representa el ajusteobtenido por Graham y col.[20] aplicando un modelo anarmónico. Los datosexperimentales fueron tomados a un ∆K= -1.27 Å−1.
Cabe destacar en la gráfica superior, el desplazamiento lineal, ahora al azul, de
la posición del modo T experimentada por las moléculas de CO con el aumento de

CAPÍTULO 2. EXPERIMENTO. 32
la temperatura, al contrario del desplazamiento al rojo que presentaba la posición
el modo T correspondiente a los átomos de Na. Por último, en la gráfica inferior
se puede observar un crecimiento lineal de la anchura energética de dicho modo
T con la temperatura. El desplazamiento de energía medido experimentalmente
puede ser ajustado por una dependencia lineal ∆E = a+b.T con a = 3,88±0,005
meV y b = (1,04±0,05)µeV/K, indicado por la línea continua. La anchura
energética γ [FWHM del modo T] muestra también un incremento distinto con
la temperatura, el cual puede ser ajustado nuevamente por una dependencia lineal
γ = α + β.T con α = 55±5 µeV y β = (1,04 ± 0,08)µeV/K. Estos
parámetros a y α se obtuvieron extrapolando la posición y anchura energética
a 0K, respectivamente. A T = 60 K la transferencia energética concuerda
perfectamente con el valor de ∆E = 3,94 ± 0,07 µeV obtenidos por Ellis, Witte
y Toennies [67] a la misma temperatura.
2.5.3. CO-Pt (111)
Por último, se van a presentar los datos experimentales correspondientes
al modo T que se obtienen por QHAS para las moléculas de CO adsorbidas
sobre la superficie de Pt(111) . Debido a su baja frecuencia vibracional , este
modo se encuentra térmicamente poblado a una temperatura por debajo de la
temperatura de desorción T ∼ 400K [68] y, por tanto, determina las propiedades
termodinámicas del adsorbato. Estos experimentos han sido obtenidos usando la
QHAS de alta resolución en un rango de temperaturas comprendido entre 50-200
K para cuatro isómeros diferentes. La concentración fue estimado a ϑCO=0.03
[69] . En Fig. 11, se midió la dependencia con la temperatura del desplazamiento
energético (~ω) del modo T para cuatro isómeros. Como novedad, se observa la
no dependencia de la anchura energética Γ con la temperatura, efecto no aparecido
en los sistemas anteriores.
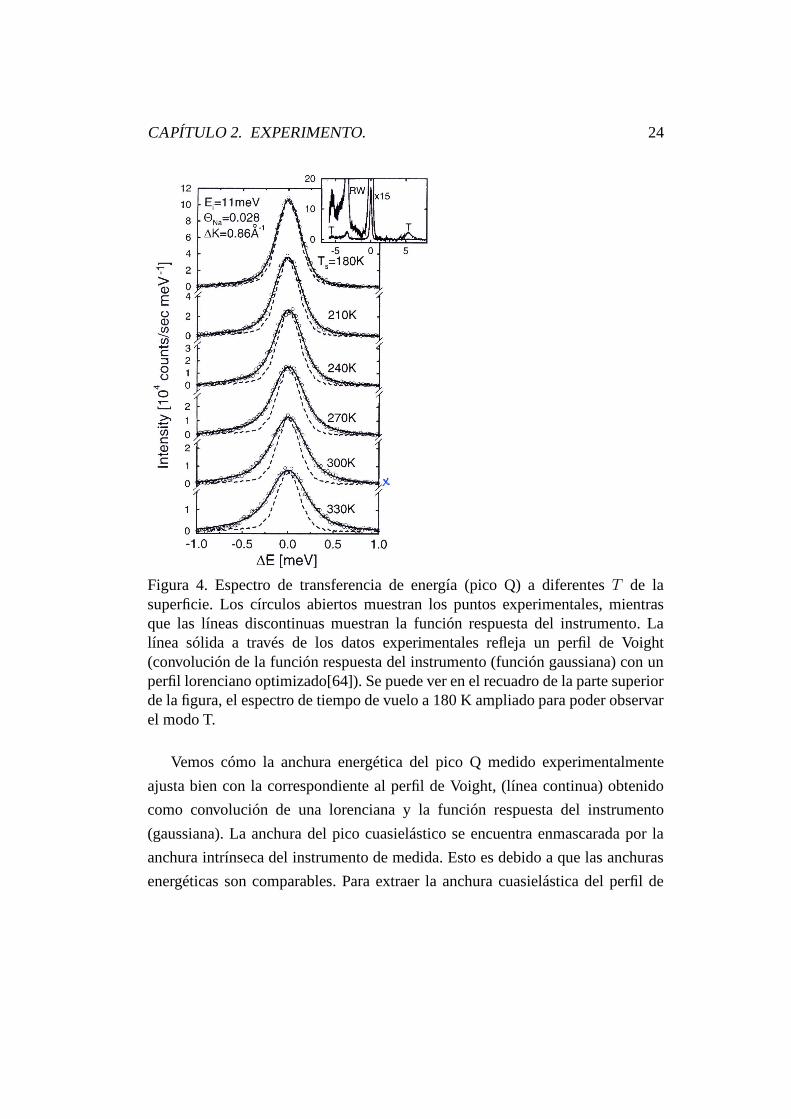
CAPÍTULO 2. EXPERIMENTO. 33
Figura 11. Posición en energía y FWHM del pico de translación frustrada paralela(modo T) de CO sobre Pt(111) frente a la temperatura de superficie T paralos cuatro isómeros diferentes. Se puede ver claramente el desplazamiento enfrecuencias debido al cambio de isótopos, así como el incremento en frecuenciacon la temperatura. En cambio, se observa que la anchura energética de los picosson independientes de los isómeros y de la temperatura, dentro de la resoluciónexperimental.

Capítulo 3
FORMALISMO TEÓRICO.
En este capítulo se describe la teoría estocástica clásica empleada para
comprender los procesos de difusión y vibración a baja frecuencia de adsorbatos
no interactuantes sobre superficies, medidos de forma satisfactoria por la técnica
de QHAS.
3.1. Procesos estocásticos.
3.1.1. Proceso del caminante aleatorio.
El fenómeno natural conocido hoy en día como movimiento browniano tiene
una larga e interesante historia. El primer registro, aunque no así la primera
observación del fenómeno, data de 1828 [5] cuando el botánico Robert Brown
recogió en una revista científica que: “Granos de polen suspendidos en un cierto
líquido y vistos a través de un microscopio, realizaban un movimiento regular
e inexplicable”. Este extraño movimiento fue objeto de muchas discusiones,
formulándose todo tipo de hipótesis con la intención de dar una explicación al
fenómeno observado. Hoy en día este movimiento es entendido y explicado por
las múltiples colisiones aleatorias de las moléculas del líquido circundante con
los granos de polen. Llegar a tal conclusión llevó muchos años, pues previamente
34

CAPÍTULO 3. FORMALISMO TEÓRICO. 35
tuvo que aceptarse la teoría cinética molecular de la materia, y finalmente el
certero trabajo de Einstein en 1905 [6] sobre el movimiento browniano contribuyó
decididamente a tal fin. Las observaciones reales y directas del movimiento de los
granos de polen sugieren que el fenómeno satisface las siguientes propiedades: a)
el movimiento es continuo, b) tiene desplazamientos independientes en intervalos
de tiempos disjuntos, c) el desplazamiento de un grano de polen es la suma de un
largo número de desplazamientos independientes e infinitamente pequeños debido
a las colisiones con las moléculas circundantes. Aplicando el teorema del límite
central nos conduce a que el desplazamiento de dicho grano sigue una distribución
gaussiana.
Por otro lado, la estructura matemática de un proceso estocástico ( proceso
que viene descrito por un conjunto de variables aleatorias Bt : t > 0), resulta
exitosa para simular este tipo de fenómenos. Por tanto, la variable estocástica Bt
se puede interpretar como la posición de una partícula browniana a un tiempo t.
Esta variable estocástica cumple las siguientes propiedades matemáticas para el
caso unidimensional:
1). B0 = 0 con probabilidad 1.
2). Las trayectorias Bt son continuas.
3). Los desplazamientos Bt − Bs (t > s) son independientes y estacionarios
(Apéndice 1).
4). Los desplazamientos obedecen a una distribución normal N(0,t-s) para
0<s<t.
Estas propiedades se dedujeron como consecuencia directa de las observacio-
nes del fenómeno físico, no garantizando para ello que tal modelo matemático
existiese.
En 1905 apareció el primer modelo matemático que explicaba este fenómeno,
fue el denominado “caminante aleatorio”, un proceso discreto en espacio y
tiempo, en el sentido de que los valores que toma la variable aleatoria Sn son
enteros para cualquier paso de tiempo discreto n, esto se puede ver en la Fig. 1.

CAPÍTULO 3. FORMALISMO TEÓRICO. 36
Figura 1. Un caminante aleatorio Sn. Vemos cómo la partícula ha visitado lospuntos 0,1,0,-1,0,1,2 (eje y) sucesivamente a cada paso de tiempo unidad.
La partícula se mueve con probabilidad p si da un paso de una unidad hacia la
derecha, y con probabilidad q = 1−p si ésta da un paso unidad hacia la izquierda.
En general, Sn describe la posición de la partícula después de n pasos de tiempo,
Entonces:
Sn =n
∑
i
Xi (3.1)
dondeX1,X2, ... es una secuencia de variables aleatorias independientes que toma
las valores +1 y −1 con probabilidades p y q. Este caminante aleatorio Sn
presenta dos propiedades básicas:
1) Tiempo homogéneo, en el que para cada tiempo discreto tn y tm no
negativos, Sn y Sn+m−Sn obedecen a la misma distribución. (Se supone S0 = 0).
2) Los desplazamientos Sn − Sm son independientes en intervalos de tiempo
disjuntos.
Como puede verse, el modelo del caminante aleatorio describe el movimiento

CAPÍTULO 3. FORMALISMO TEÓRICO. 37
de una partícula dando saltos, los cuales se encuentran idénticamente distribuidos,
y son independientes entre sí. El inconveniente que presentó este modelo discreto
para explicar el movimiento browniano es que este último es un movimiento
continuo en espacio y tiempo, por lo que se tuvo que buscar un modelo continuo
análogo al del caminante aleatorio.
3.1.2. Proceso de Wiener.
Fue en 1923 cuando el matemático Norbert Wiener demostró la existencia de
un modelo continuo para explicar el movimiento browniano. Por ese motivo dicho
proceso estocástico recibe también el nombre de proceso de Wiener, simbolizado
como: Wt : t > 0. Por tanto, se puede decir de forma aproximada que el
movimiento browniano representa el fenómeno físico, mientras que su modelo
matemático se conoce como proceso de Wiener. Matemáticamente, se comprueba
que la irregularidad en las trayectorias se debe a que su varianza varía linealmente
con el tiempo, y por tanto diverge a t → ∞. Por otro lado, se obtiene que las
trayectorias coinciden bastante bien con el movimiento errático de las partículas
brownianas. Véase en Fig. 2 una trayectoria de Wiener proyectada sobre una de
sus coordenadas.

CAPÍTULO 3. FORMALISMO TEÓRICO. 38
Figura 2. Podemos ver cómo el proceso de Wiener describe la trayectoria erráticade la posición de una partícula browniana.
El proceso de Wiener, además de gaussiano, también es markoviano (Apén-
dice 1), donde el estado futuro de la partícula únicamente depende de su estado
actual, no de su pasado. Además, decir que el valor medio es nulo, < Wt >= 0, y
su función de correlación es constante: < W (t).W (s) >= min(t, s). Por último,
destacar que sus trayectorias son continuas pero no diferenciables en ningún pun-
to, ya que si definimos Bht = [W(t+h) −Wt]/h como una variable gaussiana con
media cero y con varianza 1/h, cuando h → 0 esta distribución diverge o, lo que
es lo mismo, el cociente no converge en probabilidad a ninguna variable aleatoria
finita en ningún instante de tiempo. Esta no diferenciabilidad en sus trayectorias
provoca que tratar el movimiento browniano como un proceso de Wiener es sólo
una idealización, ya que si Wt representa la posición de la partícula browniana,
ésto significa que su velocidad es infinita, cosa que no es posible. Por este motivo,
se propusieron otros modelos alternativos, tales como el de Orstein-Uhlenbeck
para explicar el movimiento browniano.

CAPÍTULO 3. FORMALISMO TEÓRICO. 39
3.1.3. Proceso de Orstein-Uhlenbeck
El proceso de Wiener se usa para modelizar el movimiento browniano, siendo
el elegido como proceso estocástico para representar la posición de la partícula.
El problema que surge es que este modelo no es del todo realista, pues como se ha
visto la velocidad instantánea de la partícula no queda definida debido a que sus
trayectorias no son diferenciables en ningún punto.
Orstein y Uhlenbeck [70] establecieron en 1930 un nuevo modelo para salvar
esta dificultad. Estos autores consideran un proceso donde la variable estocástica
es ahora la velocidad, y en el que la posición se obtiene por integración de ésta,
no teniendo que recurrir al modelo de Wiener. El punto de partida de este modelo
se basa en la ecuación estocástica fenomenológica que propuso Langevin en 1908
[71] :
mv(t) = −mγv(t) + F (t) (3.2)
Esta expresión es la ecuación de Langevin en su forma más simple, que explica
el comportamiento de una partícula browniana en una dimensión. Esta ecuación
fenomenológica supone que la fuerza que actúa sobre la partícula se puede
descomponer en una parte determinista, denominada fuerza de fricción, que según
la ley de Stokes, es proporcional a la velocidad y se opone al movimiento de ésta,
−mγv(t), siendo γ el coeficiente de fricción que depende de la viscosidad del
fluido, y otra aleatoria F (t) que representa la parte errática del movimiento debido
al choque con las moléculas del fluido. Se supone que esta fuerza estocástica
satisface dos condiciones [72]:
1. Que el proceso F (t) es un proceso gaussiano.
2. Su tiempo de correlación es infinitamente corto, es decir,
< F (t0)F (t0 + t) >= Kδ(t) (3.3)
donde K es una constante que da una estimación de la intensidad de la colisión.
La suposición gaussiana es muy razonable debido a que la masa de la partícula

CAPÍTULO 3. FORMALISMO TEÓRICO. 40
browniana es muchísimo mayor que las masas de las moléculas de su entorno,
y por tanto su movimiento viene determinado por el gran número de golpes
sucesivos con las moléculas del medio. Este hecho permite aplicar el teorema del
límite central a F (t) que obedecerá a una distribución gaussiana. Esta situación
también justifica la segunda suposición, ya que la correlación entre sucesivos
impactos se considera apreciable sólo para el movimiento molecular, siendo
extremadamente pequeña (δ−correlada) comparada con la escala de tiempo de la
partícula browniana, esto hace que el movimiento que experimenta la partícula
en un instante determinado no viene influido por su pasado (aproximación
markoviana). Así pues, debido a que la masa de las moléculas del fluido
es mucho mas pequeña que la masa de la partícula browniana, el efecto de
una simple colisión sobre esta última no es apreciable, y como consecuencia,
las posiciones y las velocidades de dicha partícula browniana permanecen
prácticamente inalteradas. Es sólo después de un largo número de colisiones
en el que su acción acumulativa produce un efecto perceptible en la dinámica
de la partícula browniana. Esto significa que si se hacen observaciones de la
partícula browniana en una escala de tiempo del orden de los segundos, la
fuerza debido a las colisiones variará mucho mientras que la fuerza de fricción
lo hará muy poco. De esta manera se reconocen dos escalas de tiempos muy
diferentes. Una escala de tiempo microscópica que viene determinada por el
tiempo τc que dura una colisión entre la partícula browniana y las moléculas del
medio (10−21 s en un líquido) y otra escala de tiempo macroscópica τr = 1/γ
(10−7 − 10−9 s) denominado tiempo de relajación, debido a la fricción que
experimenta la partícula al moverse en el fluido. Estas fluctuaciones a escala
microscópica que están ocurriendo en el fluido que se encuentra en equilibrio
térmico son las que producen que el sistema (partícula browniana) inicialmente
en un estado de no-equilibrio se relaje al equilibrio. El teorema que relaciona esta
magnitud fenomenológica (macroscópica) γ con las fluctuaciones microscópicas
a las que se encuentra sometida la partícula browniana se conoce como teorema
de fluctuación-disipación. Tenemos que decir que este teorema es válido dentro de

CAPÍTULO 3. FORMALISMO TEÓRICO. 41
un régimen lineal, es decir las desviaciones que se producen fuera del equilibrio,
no se encuentran muy alejadas de él, pudiendo estudiarse dentro de una teoría de
respuesta lineal.
Esta relajación de sistemas en estados de no-equilibrio para alcanzar el estado
de equilibrio está regida por la “hipótesis de regresión” establecida en 1930 por
Lars Onsager [73], que viene a decirnos que el amortiguamiento o relajación
que sufre la partícula browniana como consecuencia de la interacción con las
moléculas del líquido y la fuerza fluctuante que experimentan las moléculas
de éste no son independientes, ya que ambas provienen de dicho acoplamiento
sistema (no equilibrio) - entorno (equilibrio). Fue Kubo a través del denominado
“teorema de fluctuación-disipación” quien estableció una relación cuantitativa
entre las magnitudes macroscópicas y microscópicas [74], que en una dimensión
adopta la forma:
mγ(ω) =1
kBT
∫ ∞
−∞< δF (t0)δF (t0 + t) > e−iωtdt (3.4)
Las fluctuaciones de la fuerza que experimenta la partícula debido a los golpes con
las moléculas son las que hacen que ésta se relaje al equilibrio. Estas fluctuaciones
de la fuerza vienen definidas por [75]:
δF (t) = F (t)− < F (t) > (3.5)
y por tanto la función de correlación de las fluctuaciones se puede reescribir como:
< δF (t0)δF (t0 + t) >=< F (t0)F (t0 + t) > − < F (t) >2 (3.6)
Para el caso en que nos encontramos, entre estos dos tiempos característicos, mi-
croscópico τc y macroscópico τr, la fuerza F (t) fluctuará mucho, manteniéndose
la posición y velocidad de la partícula prácticamente constantes. Por tanto, es ra-
zonable suponer que en este intervalo de tiempo sobre la partícula actúan fuerzas
en un sentido y en el sentido opuesto, de tal forma que en valor medio la fuerza
resultante es cero, es decir, < F (t) >= 0. Entonces:

CAPÍTULO 3. FORMALISMO TEÓRICO. 42
< δF (t0)δF (t0 + t) >=< F (t0)F (t0 + t) > (3.7)
y la relación de Kubo adopta la forma:
mγ(ω) =1
kBT
∫ ∞
−∞< F (t0)F (t0 + t) > e−iωtdt (3.8)
Para el caso que nos ocupa, en el que tenemos dos escalas de tiempos bien
diferenciadas (aproximación markoviana), se cumple la siguiente condición:
τr τc (3.9)
y por tanto podemos decir que:
γ(ω) = γ(0) = 1/τr = cte (3.10)
entonces el teorema de fluctuación disipación queda como:
γ =1
mkBT
∫ ∞
0
< F (t0)F (t0 + t) > dt (3.11)
Si ahora sustituimos (3.3) en (3.4) tenemos:
K = γmkBT (3.12)
una estimación de la intensidad con que golpean las moléculas del fluido a la
partícula.
Si ahora se divide la ecuación de Langevin (3.2) por la masa de la partícula m,
se obtiene:
v(t) = −γv(t) + ξ(t) (3.13)
A una ecuación diferencial como ésta donde uno de los términos varía de forma
aleatoria, se le denomina ecuación diferencial estocástica. Con estas suposiciones,
la fuerza gaussiana por unidad de masa ξ(t) queda perfectamente definida

CAPÍTULO 3. FORMALISMO TEÓRICO. 43
conocida su media y su función de correlación:
< ξ(t) >= 0 (3.14)
< ξ(t0)ξ(t0 + t) >=γkBT
mδ(t) (3.15)
La fluctuación δξ(t) = ξ(t) con media nula y δ−correlada en el tiempo, recibe el
nombre de ruido blanco, ya que su densidad espectral ξ(ω) = cte o, dicho de otro
modo, presenta un espectro plano cuya magnitud es común a todas las frecuencias.
Por otro lado, si se calcula las propiedades de la integral del ruido ξ(t):
Y (t) =
∫ t
0
ξ(s).ds (3.16)
dado que ξ(t) obedece a un proceso gaussiano, Y(t) también lo es y por tanto su
media y varianza vienen dadas por:
< Y (t) >= 0 (3.17)
σ2[Y (t) − Y (s)] = |t− s| (3.18)
Por tanto, se puede ver que Y(t) no es más que un proceso de Wiener. Esta
conclusión nos llevaría a interpretar el ruido blanco como la derivada de un
proceso de Wiener. Sin embargo, esta definición nos entraña una doble dificultad:
1) las trayectorias de W(t) no son diferenciables en ningún punto, y 2) la integral
en (3.16) no se encuentra bien definida, pues no es una integral de Riemann. Pese
a estos inconvenientes, la definición de ruido dada por la identidad:
dW (t)
dt= ξ(t) (3.19)
se puede establecer de manera coherente en el contexto de los denominados
procesos estocásticos generalizados.

CAPÍTULO 3. FORMALISMO TEÓRICO. 44
La solución de la ecuación diferencial estocástica lineal de primer orden
(3.13), viene dada por:
v(t) = v0e−γt +
∫ t
0
ξ(s)eγ(s−t)ds (3.20)
Como la integral es un operador lineal, al aplicarla sobre un proceso gaussiano
(ruido) da como resultado otro proceso gaussiano. Por tanto, la solución v(t)
también es gaussiana, siempre y cuando la condición inicial v0 sea una variable
aleatoria gaussiana, independiente de ξ(t). Sabemos que un proceso gaussiano
viene caracterizado completamente por su media y su función de correlación y,
por tanto, para la variable estocástica gaussiana v(t) se tiene que [76, 77]:
< v(t) >=< v0 > e−γt (3.21)
ya que < ξ(t) >= 0. Además,
< v(t)v(t′) >=< v20 > e−γ(t+t
′)+
γkBT
me−γ(t+t
′)
∫ t
0
ds
∫ t′
0
eγ(s+s′)δ(s− s′)ds′ (3.22)
Entonces, cuando la partícula browniana se relaja alcanzando el equilibrio térmico
con el fluido o, dicho de otro modo, cuando alcanza el estado estacionario (t →∞), el proceso de Orstein-Uhlenbeck (O-U) presenta una media < v(t) >= 0 y
una función de correlación que decae exponencialmente con el tiempo dada por:
< v(t)v(t′) >=< v(t)v(t+ τ) >=< v(0)v(τ) >= C(τ) = (3.23)
< v20 > e−γ|τ | =
kBT
me−γ|τ |

CAPÍTULO 3. FORMALISMO TEÓRICO. 45
Este tipo de decaimiento exponencial se da siempre que el proceso sea
gaussiano, markoviano y estacionario [78], y el único proceso estocástico que
cumple estas características es el de O-U. Este proceso presenta trayectorias
continuas con probabilidad uno. Además, viendo su función de correlación C(τ)
se sabe que su densidad espectral no es constante:
C(ω) =2kBTγ
m(ω2 + γ2)(3.24)
pudiendo ver que no todas las frecuencias contribuyen por igual al proceso.
Por otro lado, se define un tiempo de correlación τcorr como el tiempo que
permanece correlacionada la variable estocástica, en este caso v(t). Pasado este
tiempo, la correlación entre la variable a diferentes tiempos es nulo. Formalmente,
si el proceso es estacionario, el tiempo de correlación viene definido por:
τcorr =1
C(0)
∫ ∞
0
C(τ)dτ (3.25)
Es fácil demostrar por (3.25), que el tiempo de correlación en un proceso de O-U
viene dado por:
τcorr =1
γ(3.26)
Como vemos aquí, este tiempo de correlación no es más que el tiempo de
relajación del sistema al equilibrio, es decir, tiempo a partir del cual la partícula
comienza a sentir el efecto macroscópico de las moléculas circundantes. Por tanto,
γ nos da una estimación de la velocidad con que la partícula browniana alcanza
el estado de equilibrio o, lo que es lo mismo, la rapidez con que la partícula
browniana pierde la correlación en su velocidad.
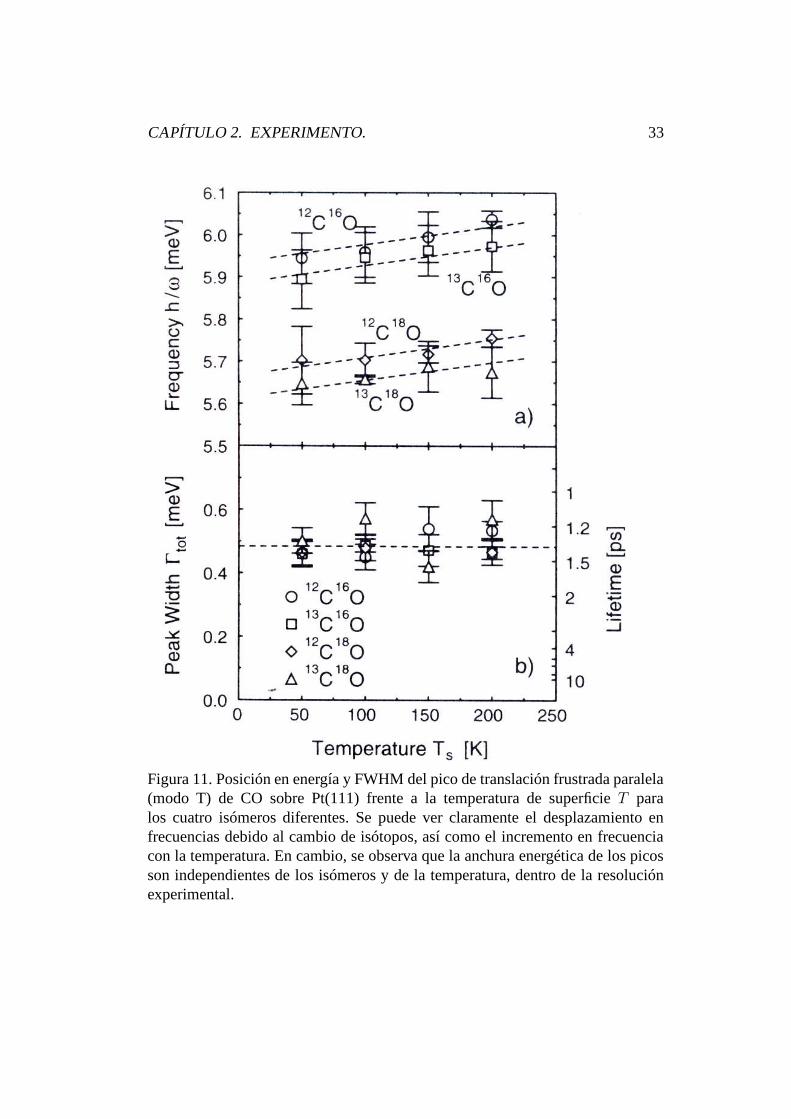
Por último, si se integra el proceso de O-U :
x(t) =
∫ t
0
v(s)ds (3.27)
se obtiene la posición de la partícula browniana, donde a t = 0, suponemos
CAPÍTULO 3. FORMALISMO TEÓRICO. 46
que x(0) = 0. Como ya se mencionó anteriormente, integrando un proceso
gaussiano, el resultado es nuevamente otro proceso gaussiano. Por tanto, x(t)
vendrá caracterizada completamente por su media y su varianza:
< x(t) >= 0 (3.28)
σ2(t) =< x2(t) >=2kBT
mγ[t+ (
e−2γt − 2
2γ)]. (3.29)
Si tomamos γ → ∞, x(t) converge a un proceso de Wiener. Este resultado es
interesante, ya que se obtiene una justificación de la identidad dWdt
= ξ(t) cuando
γ → ∞ en la relación:
dx(t)
dt= v(t) (3.30)
En la Fig. 3 vemos cómo varía el proceso de O-U, y como tiende al proceso de
Wiener cuando τcorr → 0 .
Figura 3. Proceso de O-U estacionario (X(t)=v(t)) para diferentes valores detiempo de correlación. a) τcorr = 0,1, b)τcorr = 0,05 y c) τcorr = 0,01.
Para terminar esta sección acerca de las ecuaciones estocásticas, diremos que
si en una ecuación diferencial estocástica el término estocástico aparece de forma
lineal, se la denomina ecuación de Langevin (se va a trabajar en una dimensión
para simplificar las ecuaciones). De este modo, la forma general de la ecuación de
Langevin viene dada por:

CAPÍTULO 3. FORMALISMO TEÓRICO. 47
dx
dt= q(x, t) + g(x, t)ξ(t) (3.31)
Si esta función g(x, t) es constante , se habla de ruido aditivo, en otro caso se
habla de ruido multiplicativo. Finalmente q(x, t) obedece al término de arrastre,
mientras que g(x, t) corresponde al término difusivo. Cuando en dicha ecuación
este ruido, que consideraremos blanco, aparece como multiplicativo, el proceso
resultante x(t) será en general una función no continua del tiempo. Cuando esto
ocurre, hay dos maneras de resolver esta Ecuación Diferencial Estocástica (SDE),
sujeta a dos interpretaciones diferentes, de Itô y de Stratonovich. Como nota
importante, apuntar que para el ruido aditivo, ruido que se ha utilizado en nuestro
estudio, ambas interpretaciones coinciden.
3.2. Ecuación de difusión.
El primer sistema estadístico que fue estudiado con ecuaciones diferenciales
estocásticas fue el movimiento browniano. Este procedimiento de estudiar las
propiedades de sistemas estadísticos dentro y fuera del equilibrio de forma
aproximada mediante el empleo de ecuaciones estocásticas, es lo que se conoce
como formalismo de la ecuación de Langevin. Fue Einstein, el primero que dio
una explicación al movimiento browniano. En lugar de fijarse en las trayectorias
de una sola partícula, formuló una descripción probabilística válida para un
conjunto de ellas. En este trabajo, se introdujo el concepto de densidad de
partículas para referirse al colectivo de partículas. Si n(r, t) es la densidad de
partículas que podemos encontrar en el punto r en el instante de tiempo t, la
ecuación de movimiento para esta densidad de partículas n, verifica la ecuación
de difusión:
∂n
∂t= D∇2n (3.32)

CAPÍTULO 3. FORMALISMO TEÓRICO. 48
siendo D la constante de difusión, y ∇2 el operador laplaciano en tres
dimensiones. Si la condición inicial es suponer las partículas a r = 0 , la solución
de la ecuación anterior (3.32) viene dada por:
n(r, t) =N
(4πDt)3/2e−r2/4Dt (3.33)
con una posición media nula, y una varianza que aumenta linealmente con el
tiempo:
< r(t) >= 0 (3.34)
< r2(t) >= 6Dt (3.35)
Después de los conceptos introducidos en la sección anterior, diríamos que
r(t) no es más que un proceso de Wiener tridimensional (es decir, que cada
componente es un proceso de Wiener). Sin embargo, la aproximación anterior
al problema es fenomenológica , ya que no nos permite calcular la constante de
difusión en términos de cantidades microscópicas. Fue Langevin el que más tarde
realizó un tratamiento diferente que, de algún modo, puede considerarse como
complementario al de Einstein. En su planteamiento, Langevin quiso describir
la trayectoria de una partícula browniana como el resultado de aplicar la ley de
Newton a dicha partícula, suponiendo que dos fuerzas actuaban sobre esta: una
de fricción, y otra aleatoria ξ(t). De este modo, la ecuación de movimiento que
propuso Langevin resultó ser :
r(t) = −γr(t) + ξ(t) (3.36)
donde r es la velocidad tridimensional (derivada temporal de la posición r).
Langevin supuso que las propiedades de esta fuerza eran de media nula y
correlación con la posición de la partícula nula también:

CAPÍTULO 3. FORMALISMO TEÓRICO. 49
< ξ(t) >= 0 (3.37)
< r(t).ξ(t) >= 0 (3.38)
así, multiplicando (3.36) por r y tomando promedios respecto a las realizaciones
de ξ(t) :
1
2
d2 < r2 >
dt2=< r2 > −1
2γd < r >
dt(3.39)
Langevin supuso que a tiempos largos la partícula browniana alcanzaba el
equilibrio térmico con el fluido que la rodeaba, por tanto se cumplía el teorema de
equipartición de la energía:
1
2m < r2 >=
3kBT
2(3.40)
siendo kB la constante de Boltzmann, y T la temperatura del fluido. De esta
manera, la ec. (3.39) se pudo resolver, y en el régimen asintótico (t 1γ
) se
obtuvo que:
< r(t) >= 0 (3.41)
< r2(t) >=6kBT
mγt (3.42)
llegando a la misma ley de difusión obtenida por Einstein (3.35) pero con una
expresión explícita para el coeficiente de difusión:
D =kBT
mγ(3.43)
tenemos que decir que la partícula browniana a tiempos cortos (t 1γ
) presenta
un régimen balístico < r2(t) >= 3kBTm
t2 (movimiento de una partícula libre
sin experimentar colisiones) con una velocidad igual a√
3kBT/m, y es en el

CAPÍTULO 3. FORMALISMO TEÓRICO. 50
límite asintótico (tiempos largos) cuando la partícula alcanza un régimen difusivo
< r2(t) >= 6Dt. Como puede comprobarse, las condiciones de la fuerza supuesta
por Langevin obedecen a un ruido blanco gaussiano, de esta manera la solución
de (3.39) corresponde a un proceso de O-U. La expresión para el coeficiente de
difusión D se obtuvo empleando la relación obtenida del teorema de fluctuación-
disipación (3.11), que nos relaciona una cantidad microscópica (ruido) con una
cantidad macroscópica (fricción).
3.3. Ecuación de Fokker-Planck.
Es bien sabido que la solución completa de un sistema macroscópico
consistiría en resolver todas las ecuaciones microscópicas del sistema. Ya que
no se puede hacer ésto, se tiene que utilizar una descripción estocástica, o
dicho de otro modo, describir el sistema mediante variables macroscópicas
que fluctúan. La ecuación de Fokker-Planck es una ecuación de movimiento
que determina la distribución de estas variables macroscópicas sometidas a
tales fluctuaciones. Es una de las más simples ecuaciones de movimiento
para describir el comportamiento de variables macroscópicas. El movimiento
browniano descrito por la ecuación de Langevin, también queda bien definido
usando una ecuación de Fokker-Planck, la diferencia estriba ahora en que la
variable estocástica X(t), ya sea la posición x(t) o la velocidad v(t) de la
partícula browniana, es sustituida por una función densidad de probabilidad
P(X, t) que describe la dinámica de un colectivo de partículas brownianas que
no interaccionan entre sí . Dichas funciones de densidad de probabilidad, tanto
de posición como de velocidad para este colectivo de partículas, dependen del
tiempo y de sus densidades de probabilidad iniciales. De este modo, la ecuación de
Fokker-Plank es una ecuación de movimiento que describe la evolución temporal
de la función densidad de probabilidad P(X, t). Resolviendo dicha ecuación,
partiendo inicialmente de una distribuciónP(X, 0), se obtiene la función densidad
de probabilidad P(X, t) para todos los tiempos o, lo que es lo mismo, una vez

CAPÍTULO 3. FORMALISMO TEÓRICO. 51
que esta función es conocida, se podrá conocer la posición media, así como la
velocidad media de este colectivo de partículas para cualquier tiempo t. Decir
también que la ecuación de Fokker-Planck se aplica tanto a sistemas en estado
no estacionario, alejados del equilibrio térmico, como a su estado estacionario.
Como ya se vio anteriormente, los promedios de estas variables estocásticas
macroscópicas x(t) y v(t) se obtienen de forma inmediata en procesos que son
bien descritos por ecuaciones de Langevin lineales (procesos con ruido aditivo),
tal y como el movimiento browniano. La dificultad aparece cuando los procesos
vienen descritos por ecuaciones de Langevin no lineales (procesos con ruido
multiplicativo), ya que estos promedios son mas difíciles de obtener. Es por ello
que se recurre a este tipo de ecuaciones de movimiento. Fokker [79] y Planck [80]
fueron los primeros autores que derivaron una ecuación diferencial para la función
densidad de probabilidad que describe el movimiento browniano. Esta ecuación
expresada en su forma más general viene dada por [77, 81]:
∂P(X, t)
∂t= [−∂D1(X)
∂X+
1
2
∂D2(X)
∂X2]P(X, t) (3.44)
la ec. (3.44) es una de las más simples ecuaciones de Fokker-Planck. Si se resuelve
partiendo de una distribución inicial P(X, 0) para t = 0, sujeta a unas condiciones
de contorno apropiadas, uno es capaz de obtener un función de densidad de
probabilidad P(X, t) para cualquier tiempo t. Una vez que se conoce tal función,
se puede conocer la magnitud estocástica promedio de este colectivo de partículas:
< X(t) >=
∫ ∞
−∞X(t)P(X, t)dX (3.45)
en dicha ecuación, D1(X) es el denominado coeficiente de arrastre, y D2(X)
es el coeficiente de difusión. Matemáticamente hablando, esta ecuación es una
ecuación diferencial parcial lineal de segundo orden de tipo parabólico, o lo que
es lo mismo, una ecuación de difusión con una derivada adicional de primer
orden con respecto a X , ecuación que en la literatura se denomina ecuación de
Kolmogorov [82]. En el caso del movimiento browniano, el término de arrastre

CAPÍTULO 3. FORMALISMO TEÓRICO. 52
es lineal, y el término difusivo es constante. En estas condiciones la ecuación de
Fokker-Planck se resuelve analíticamente, obteniendo como solución funciones
de densidad de probabilidad gaussianas para el estado estacionario.
Tomando la velocidad de las partículas brownianas en una dimensión,X(t) =
v(t), los términos D1 y D2 en (3.44) vienen dados por:
D1(v) =< ∆v >
∆t(3.46)
siendo < ∆v >la velocidad promedio en un intervalo de tiempo ∆t,
y
D2(v) =< (∆v)2 >
∆t(3.47)
en analogía con la ec. de Langevin, (3.13) considerando pequeños intervalos de
tiempos y tomando promedios, suponiendo que < F (t) >= 0 , y < v >∼ v ,
tenemos que:
D1 = −γv (3.48)
y
D2 =2kBTγ
m(3.49)
si se comienza partiendo de una distribución inicial de velocidades P(v, 0) =
δ(v−v0), después de un tiempo t, la función de densidad de probabilidad resultado
de resolver la ecuación de Fokker-Planck tiene la siguiente expresión:
P (v0, v, t) =1
√
2πkBT (1 − e−2γt)/mexp[− m
2kBT
(v − v0e−γt)2
(1 − e−2γt)] (3.50)
Podemos ver que a tiempos cortos la densidad de probabilidad es una delta
centrada a v = v0, y que cuando el tiempo evoluciona, esta distribución se va
ensanchando hasta convertirse en una gaussiana:

CAPÍTULO 3. FORMALISMO TEÓRICO. 53
P (v0, v, t→ ∞) = P(v) =
√
m
2πkBTexp[− mv2
2kBT] (3.51)
independiente de la condición inicial (proceso markoviano), y del tiempo (proceso
estacionario). Esta distribución de velocidades estacionaria a una determinada
temperatura coincide con la distribución maxwelliana de velocidades.
Figura 4. Función densidad de probabilidad para un colectivo de partículasbrownianas cuyas velocidades comienzan distribuidas en torno a v0 = 2VT (siendoVT la velocidad en el equilibrio térmico) a tiempo t = 0. Para t > 0 , se observauna distribución gaussiana que con el paso del tiempo se relaja a cero, hastaalcanzar la distribución de Maxwell de velocidades (línea discontinua), (3.51),como consecuencia de haber alcanzado el estado estacionario.
El mismo proceder se realiza para los desplazamientos en una dimensión
partiendo de la misma ecuación de Fokker-Planck, ec. (3.44) , y por analogía con
la ec. de Langevin, se tiene que:
D1 =< ∆x >
∆t= 0 (3.52)
siendo ahora < ∆x > el desplazamiento promedio en un intervalo de tiempo ∆t,

CAPÍTULO 3. FORMALISMO TEÓRICO. 54
y
D2 =< (∆x)2 >
∆t=
2KBT
mγ(3.53)
asumiendo tiempos largos (t 1/γ).
Por tanto, dicha ecuación se convierte en una ecuación de difusión:
∂P(x, t)
∂t= D
∂2P(x, t)
∂x2(3.54)
cuyo coeficiente de difusión vale D = kBTmγ
. De este modo, la solución de Fokker-
Planck a tiempos largos viene dada por:
P(x0, x, t) =1√
4πDtexp[−(x − x0)
2
4Dt] (3.55)
Se puede ver cómo a tiempo cero todas la partículas brownianas están centradas
en x0, y con el tiempo esta distribución delta se va ensanchando hasta obtener
dicha gaussiana.
Figura 5. Función densidad de probabilidad para un colectivo de partículas cuyasposiciones se encuentran distribuidas en torno a x0. Vemos cómo la distribuciónde posiciones es gaussiana , ensanchándose a medida que pasa el tiempo.

CAPÍTULO 3. FORMALISMO TEÓRICO. 55
Estas partículas brownianas experimentan un movimiento de tipo difusivo,
irreversible y atribuido a las fuerzas aleatorias que las moléculas de fluido ejercen
sobre las partículas. La ecuación de Fokker-Planck al igual que ocurriera con
Langevin, establece una relación entre la constante de difusión microscópica D,
con la variable macroscópica γ.
3.4. Difusión de átomos bajo la presencia un poten-
cial.
3.4.1. Ecuación de Kramers.
Los átomos difundiéndose sobre la superficie a una determinada temperatura
se comportan como partículas brownianas moviéndose bajo un potencial periódi-
co. El problema del movimiento browniano bajo la influencia de un potencial fue
por primera vez resuelto por Kramers [7] para describir las reacciones químicas.
La ecuación de Kramers no es más que una ecuación especial de Fokker-Planck.
Para un problema unidimensional, esta es una ecuación para la función de densi-
dad de probabilidad en posiciones y velocidades. En términos de la densidad de
probabilidad en el espacio de fases, la correspondiente ecuación de Fokker-Planck
(ecuación Klein-Kramers) [83], toma la forma:
∂P(x, v, t)
∂t= [−∂v
∂x+∂[γv + f ′(x)]
∂v+ γ(kBT/m)
∂2
∂v2]P(x, v, t) (3.56)
donde f ′(x) es la fuerza por unidad de masa debido al potencial por unidad de
masa f(x). Notar que, para el caso en que la fuerza por unidad de masa es lineal
esta ecuación se resuelve exactamente. Cuando el coeficiente de fricción es alto,
la ecuación de Kramers se reduce a la ecuación de Smoluchowski [84], ecuación
especial del tipo Fokker-Planck que se encuentra definida únicamente para la
función de distribución de las posiciones.

CAPÍTULO 3. FORMALISMO TEÓRICO. 56
Por otro lado, el problema del movimiento browniano en potenciales
periódicos ha suscitado un gran interés en muy diferentes campos de la física ,
tal como la física del estado sólido, química-física y teoría de la comunicación
[76, 85]. Si uno se restringe al caso unidimensional, las partículas se encuentran
sometidas a una fuerza de fricción y otra fluctuante como consecuencia de la
interacción no adiabática con el sustrato, moviéndose a través de un potencial
periódico unidimensional adiabático. Debido a la presencia de estas fuerzas
fluctuantes, éstas pueden ganar energía y dejar el pozo de potencial donde se
encuentran inicialmente e ir a otros pozos de potencial cercanos a su derecha o
a su izquierda, o incluso con el paso del tiempo moverse a pozos de potenciales
muchos más lejanos. Por tanto, para tiempos suficientemente largos, cabría
esperar un movimiento difusivo de las partículas en ambas direcciones del eje
x, como ya Einstein propuso en su día, y cuya difusión viene descrita por
una constante de difusión D. Por tanto, dicha ecuación de Langevin, o su
equivalente Fokker-Planck (Klein-Kramers) describe el movimiento de partículas
brownianas a lo largo de una superficie corrugada que viene representada por
dicho potencial periódico, y que dependiendo de una alta o baja fricción presentan
diferentes comportamientos que requieren un tratamiento teórico adecuado para
su comprensión. Para la mayoría de los átomos y moléculas adsorbidas sobre
superficies, sus masas son tan pesadas que para el rango de temperaturas de
interés, el movimiento puede tratarse clásicamente. Por tanto, para el caso que
nos ocupa, vamos a concentrar toda nuestra atención en el movimiento clásico de
los adsorbatos. Un tratamiento de difusión sobre superficies incluyendo todos los
grados de libertad del adsorbato y el sustrato es hasta el momento limitado a un
conjunto determinado de sistemas que pueden ser bien modelizados. Un cálculo
ab initio completo se encuentra todavía fuera de alcanzar este punto, incluso el
empleo de aproximaciones con modelos de interacción potencial entre átomos nos
lleva a realizar cálculos completos de Dinámica Molecular (DM) en el que sólo se
consideran los grados de libertad vibracionales y translacionales, ya que el término
de acoplamiento no adiabático de las excitaciones electrónicas hasta el momento

CAPÍTULO 3. FORMALISMO TEÓRICO. 57
no se conoce. Además, las simulaciones de DM detalladas microscópicamente
no reflejan muy bien los principales problemas al tratar la difusión sobre
superficies. La aproximación más simple es integrar todos los grados de libertad
del sustrato en las ecuaciones de movimiento quedándonos únicamente con una
ecuación estocástica para los adsorbatos. Este es precisamente el contenido de
la aproximación de la ecuación de Langevin para el estudio de difusión sobre
superficies. En esta ecuación para los adsorbatos, los efectos del sustrato aparecen
de dos formas. Primero, el potencial adiabático, el cual es justamente la energía
libre del sistema entero, en el que las posiciones de los adsorbatos se tratan como
parámetros fijos. En adición, una fuerza de fricción y otra estocástica proveniente
del acoplamiento no adiabático del adsorbato con el sustrato. Este acoplamiento
conduce a las fluctuaciones y amortiguamientos del adsorbato, produciéndose un
cambio en su dinámica, pasando de una balística a una browniana, regido por una
ecuación estocástica.
3.4.2. Ecuación generalizada de Langevin.
El problema de difusión sobre superficies, se puede resolver en su forma
general a través de una ecuación de Langevin Generalizada (GLE) para los
adsorbatos:
mR(t) = −∫ t
0
K(t− t′)R(t′)dt′ −∇RV [R(t)] + F(t) (3.57)
aquí V (R) es un potencial general que incluye el potencial adiabático ejercido
por el sustrato sobre el adsorbato así como las interacciones entre los adsorbatos.
La ec. (3.57) sirve para ilustrar un número considerable de conceptos referente
al movimiento del adsorbato. Primero, el amortiguamiento que experimenta el
adsorbato está caracterizado por una función memoria K(t − t′) que depende de
lo complicado que haya sido el pasado de dicho adsorbato y sustrato. Segundo,
no nos debe sorprender que el amortiguamiento caracterizado en forma de dicha
función memoria y la fuerza de fluctuación no sean independientes, ya que ambas

CAPÍTULO 3. FORMALISMO TEÓRICO. 58
provienen del acoplamiento con las excitaciones del sustrato. Y como ya dijimos
anteriormente, dichas cantidades vienen relacionadas de forma explícita por el
teorema de fluctuación-disipación. La forma más simple de la función memoria
ocurre en el límite markoviano cuando la escala de tiempo para el movimiento
del sustrato es mucho más rápido que la del adsorbato. Para este caso, la función
memoria puede ser aproximada por una función delta:
K(t− t′) = γδ(t− t′) (3.58)
Entonces, considerando este caso más simple de aproximación markoviana,
la ecuación de Langevin generalizada se reduce a la ya conocida ecuación
de Langevin (3.36), pero ahora teniendo en cuenta la fuerza determinista
−∇RV [R(t)] que nos da cuenta del potencial adiabático:
dv
dt= −γv −∇RV [R(t)] + ξ(t) (3.59)
Decir además, que el problema de difusión de adsorbatos sobre superficies puede
ser resuelto por diferentes caminos, tales como el método de la matriz de fracción
continua (MCFM) [83], o el método de las integrales de camino de Feymann [27].
3.4.3. Formalismo clásico teórico para medir difusión de
átomos sobre superficies por QHAS.
La teoría que se encuentra detrás de las técnicas experimentales mediante
QHAS proviene del formalismo de van Hove desarrollado para explicar la
dispersión cuasielástica de neutrones [86] , técnica con la cual se estudió la
difusión sobre líquidos. Esta teoría se pudo generalizar a la difusión sobre
superficies con algunas particularidades. Primero, se supone un sustrato perfecto,
es decir se considera una superficie perfectamente periódica. Segundo, los
adsorbatos se comportan como partículas brownianas. Tercero, se supone que
la difusión se produce por activación térmica. Cuarto, los movimientos de
difusión y de vibración se suponen desacoplados aunque es bien conocido que

CAPÍTULO 3. FORMALISMO TEÓRICO. 59
las vibraciones pueden asisitir los procesos de difusión. Y quinto, dependiendo de
la magnitud de los vectores de ondas involucrados, la estructura de la red metálica
puede ser importante o no, usando diferentes modelos de difusión según el caso
que se esté investigando. Es decir, si se consideran únicamente pequeños números
de ondas, es decir, largas longitudes de ondas, se aplica un modelo de difusión
continuo; en cambio, si se trabaja con longitudes de ondas cortas, se aplica un
modelo de difusión de saltos dentro del formalismo de una ecuación maestra [26].
En analogía con la teoría de líquidos [87, 88], suponemos que el colectivo de
partículas clásicas que se difunden sobre la superficie viene descrita por la función
de correlación espacio-temporal de van Hove, también definida como la densidad
de probabilidad promedio sobre un colectivo de partículas G(R, t)[89]. Puede ser
separada en dos componentes, una que describe el movimiento de un adsorbato
particular sobre la superficie, denominada función de autocorrelación Gs(R, t),
y la otra parte describe la correlación entre dos adsorbatos distintos sobre la
superficie, que recibe el nombre de función de correlación distinta Gd(R, t):
G(R, t) = Gs(R, t) +Gd(R, t) (3.60)
Tenemos por tanto que:
∫
R
G(R, t)dR = N (3.61)
siendo N el número de adsorbatos en la superficie.
∫
R
Gs(R, t).dR = 1 (3.62)
La función de van Hove G, como tal función de correlación espacio-temporal
presenta dos magnitudes que la caracterizan, definidas como R0 (espacio de
correlación), y tiempo de correlación T0, y donde ~/R0 y ~/T0 corresponden
con las transferencias de momento y energía respectivamente. El valor que tomen
estas dos magnitudes determina el comportamiento que pueda presentar G, ya
sea clásico (función real) y cuántico (función compleja). Ya que nosotros estamos

CAPÍTULO 3. FORMALISMO TEÓRICO. 60
interesados en la forma clásica, tenemos que recurrir a otra magnitud, denominada
“longitud de onda térmica de de Broglie” λB = ~√2mkBT
, onda promedio asociada
a los adsorbatos a una energía térmica kBT . Las partículas en un líquido o
en un gas denso presentan una λB pequeña comparada a la distancia entre los
adsorbatos. Por tanto, bajo estas condiciones de λB R0 la función de van Hove
G puede separarse en una parte propia Gs y una distinta Gd, cumpliéndose la
condición (3.60). Bajo estas mismas condiciones en el que la longitud promedio
de de Broglie es muy pequeña comparada con las distancias interatómicas R0,
no se manifiestan efectos cuánticos en Gd, lo cual nos conduce a pares de
partículas separadas por una distancia R0, adquiriendo Gd un comportamiento
clásico. Ahora bien, no está resuelto todavía la condición clásica para la condición
temporal, ya que para tiempos cortos |t| T0, la función Gs presenta un
valor complejo, prueba evidente de que los efectos cuánticos están presentes.
En cambio, se observa que a tiempos largos [86], los efectos cuánticos son
despreciables y la forma de Gs adquiere un comportamiento clásico.
Por tanto, el tratamiento de G de forma clásica es correcto siempre y cuando
nos encontremos con un colectivo de adsorbatos en fase gas o en fase líquido
diluido (alta temperatura), siempre y cuando se cumplan las condiciones espacio-
temporales λB R0 , y |t| T0 respectivamente. Por el contrario, si tuviesemos
un líquido cuántico como el caso del helio líquido a baja temperatura la situación
sería completamente diferente, donde G tendría un valor complejo para cualquier
tiempo t.
Cuando la concentración de adsorbatos es baja, y se ignora la interacción entre
adsorbatos, G(R, t) = Gs(R, t) describe la dinámica de una partícula aislada
que se difunde sobre la superficie. Es claro que dentro de la mecánica clásica, la
función de correlación temporal Gs(, t) presenta el mismo significado físico que
la densidad de probabilidad P(R, t) resultado de resolver la ecuación de Fokker-
Planck para una partícula moviéndose en un potencial periódico (superficie) bajo
la influencia de la temperatura y la fricción. Equivalentemente, la dinámica de esta
partícula aislada que se difunde puede ser estudiada por la ecuación de Langevin.

CAPÍTULO 3. FORMALISMO TEÓRICO. 61
La cantidad que se mide en los experimentos de QHAS es la probabilidad
de reflexión diferencial d2R(∆K,ω)dΩdω
(probabilidad de que un átomo de He sea
dispersado por un colectivo de adsorbatos difundiéndose en la superficie a un
cierto ángulo sólido Ω con un intercambio de energía ~ω = k2f − k2
i , si ~/(2m) =
1 ). Esta magnitud puede separarse en una parte coherente y una parte incoherente:
(
d2R(∆K, ω)
dΩdω
)
coh
= ndF2coh
∫ ∫
G(R, t)e[i(∆K−ωt)]dRdt =
ndF2coh
∑
jj′
∫
< e−i∆K.Rj′(0)ei∆K.Rj(t) > e−iωtdt =
ndF2cohS(∆K, ω) (3.63)
(
d2R(∆K, ω)
dΩdω
)
inc
= ndF2inc
∫ ∫
Gs(R, t)e[i(∆K−ωt)]dRdt =
ndF2inc
∑
j
∫
< e−i∆K.Rj(0)ei∆K.Rj(t) > .e−iωtdt =
ndF2incSs(∆K, ω) (3.64)
donde nd representa la concentración de partículas que se difunden y F es el
factor de forma atómico que dependen de la interacción entre los átomos de He y
los adsorbatos.
Podemos ver de las ecuaciones que la dispersión coherente depende de la
correlación entre las posiciones de los mismos adsorbatos a diferentes tiempos,
y sobre las correlaciones entre las posiciones de diferentes adsorbatos a diferentes
tiempos. Esto produce un efecto de interferencia. En cambio, la dispersión
incoherente depende únicamente de la correlación entre las posiciones del mismo

CAPÍTULO 3. FORMALISMO TEÓRICO. 62
adsorbato a diferentes tiempos, por tanto no se obtienen interferencias.
Esta dispersión incoherente es la que se mide bajo las condiciones de
considerar baja concentración de adsorbatos. Por tanto, para el caso que nos
ocupa, en el que suponemos que los adsorbatos no interaccionan entre sí, el factor
de estructura dinámico S(∆K, ω) = Ss(∆K, ω), al igual que ocurría con la
función de van Hove G(R, t). Dicho factor Ss(∆K, ω) recibe el nombre de factor
de estructura dinámico propio, y se expresa como:
Ss(∆K, ω) =
∫ ∞
−∞e−iωtIs(∆K, t)dt =
∫ ∞
−∞< ei∆K.[R(t)−.R(0)] > e−iωtdt
(3.65)
donde los corchetes en la integral denotan un doble promedio, uno sobre el
colectivo y el otro temporal, siendo Is(∆K, t) la función de dispersión intermedia,
que es justamente la transformada de Fourier espacial de Gs(R, t).
Para terminar esta sección tenemos que tener en cuenta en todo momento, que
los átomos de prueba que sondean la superficie deben afectar poco la dinámica
del proceso físico que allí está ocurriendo, o dicho de otro modo, la distorsión
de Gs(R, t) debe ser minimizada por los átomos de prueba que inciden sobre la
superficie del cristal, ya que su papel consiste en actuar únicamente de sonda. Por
lo tanto, esto se consigue siempre y cuando los átomos de prueba permanezcan un
tiempo de espera del orden del tiempo de relajación de tal función de correlación
Gs(R, t). Este tiempo de relajación viene determinado por el tiempo que dura un
salto realizado por el adsorbato desde un sitio de adsorción a otro, simbolizado τj,
siendo j el desplazamiento efectuado en dicho salto, que se estima del orden de
10−13 s, para una distancia igual a una típica longitud de correlación ∼ 3,10−10 m.
Por tanto la velocidad de las partículas prueba deben cumplir la siguiente relación:
vsonda ≤ a/τj ≤ 3,103m/s (3.66)
siendo ésta la velocidad típica del haz incidente de los átomos de He a temperatura
ambiente, de ahí que sea la sonda atómica por excelencia para este tipo de

CAPÍTULO 3. FORMALISMO TEÓRICO. 63
experimentos con QHAS.
3.4.4. Aproximaciones dentro del formalismo teórico.
3.4.4.1. Modelos de difusión continua.
Una simple e intuitiva figura para entender la medida de la difusión en
superficies por dispersión atómica de helio, es considerar el caso de difusión
continua donde el haz incidente se comporta como una onda plana, dispersada
sobre la superficie por los adsorbatos.
Figura 6. Representación esquemática del origen de la anchura cuasielásticaproveniente de un pico elástico debido al movimiento difusivo de los adsorbatosen superficie. Se observa un desplazamiento al rojo a) o un desplazamiento al azul(b) que conduce a la anchura cuasielástica.
En este caso de difusión continua, se supone que la partícula se difunde sobre
una superficie plana, en la que no siente el efecto del potencial adiabático. Dentro
de esta aproximación, a tiempos cortos, la partícula se comporta como si estuviese
libre (gas perfecto), y por tanto Gs(R, t) adopta la forma:

CAPÍTULO 3. FORMALISMO TEÓRICO. 64
Gs(R, t) =1
2πσ2(t)exp[−R2/2σ2(t)] (3.67)
donde
σ2(t) =< R2(t) >=< v20 > t2 = 2
kBT
mt2 (3.68)
En el otro extremo, a tiempos largos, cuando el movimiento es gobernado por
la difusión debido a las interacciones de la partícula con el sustrato, Gs(R, t)
también presenta una forma gaussiana igual que (3.67) , pero donde ahora la
varianza varía linealmente con el tiempo:
σ2(t) =< R2(t) >= 4kBT
mt = 4Dt (3.69)
Esto se traduce que a tiempos cortos la Ss(∆K, ω) de la partícula libre tendrá una
forma gaussiana, mientras que a tiempos largos la Ss(∆K, ω) de difusión tendrá
un aspecto lorenciano, (ver Fig. 7).
Figura 7. Puede verse cómo la forma de línea del pico cuasielástico a tiemposcortos (∆K largos, 2 Å−1 y 3 Å−1) obedece a una gaussiana, mientras que cuandoalcanzamos el régimen difusivo (tiempos largos, ∆K pequeño, 1 Å−1), la formade línea del pico cuasielástico es lorenciana.

CAPÍTULO 3. FORMALISMO TEÓRICO. 65
Figura 8. Varianza σ2(t) para varios regímenes en los cuales Gs(R, t) es unafunción gaussiana en R. Obsérvese la forma parabólica (partícula libre) y la formalineal (difusión).
Realizando la doble transformada de Fourier de esta función Gs(R, t) a
tiempos largos, obtenemos la expresión para Ss(∆K, ω):
Ss(∆K, ω) =1
π
|∆K|2ω2 +D2|∆K|4 (3.70)
una lorenciana con una FWHM, Γ(∆K) = 2~D|∆K|2. Esta relación parabólica
entre la anchura energética del pico cuasielástico y ∆K es cierta en el caso límite
∆K → 0, en el que únicamente las largas distancias se tienen en cuenta, y
por tanto el adsorbato ve a la superficie como un continuo (superficie plana),
insensible al efecto del potencial adiabático.
Para terminar esta sección mostramos en la Fig. 8 los diferentes comporta-
mientos de σ2(t) para los diferentes regímenes que puede sufrir el adsorbato,
siempre y cuando Gs(R, t) presente un comportamiento gaussiano.

CAPÍTULO 3. FORMALISMO TEÓRICO. 66
3.4.4.2. Modelo de difusión discreta.
En este caso, se considera la periodicidad de la superficie asumiendo que los
átomos se encuentran en posiciones discretas de una red cristalina y experimentan
saltos instantáneos entre estos sitios. La situación se representa esquemáticamente
en la Fig. 9.
Figura 9. Representación esquemática del modelo de Chudley-Elliot: cada átomorealiza un salto instantáneo, con un intervalo promedio τ entre saltos. Si el detectorse encuentra en una posición correspondiente a ∆K = nG, siendo G = 2π/a elvector de la red recíproca. Los saltos no introducen una diferencia de fase y no seobserva ninguna anchura energética. En cambio, si ∆K = (n+ 1
2)G, la coherencia
temporal del paquete de ondas es reducida a τ y se genera una indeterminaciónque dará una anchura energética dada por ∆E = ~ω= ~
τ.
A bajas temperaturas, el movimiento del átomo adsorbido consiste en
oscilaciones localizadas alrededor del mínimo VA(r) con saltos ocasionales a

CAPÍTULO 3. FORMALISMO TEÓRICO. 67
mínimos cercanos, es decir, de saltos desde un sitio de adsorción a otro. Al igual
que ocurren con todos los procesos activados térmicamente [8], la frecuencia
de salto viene expresada por un prefactor y una exponencial que depende de la
temperatura por la forma establecida por Arrhenius:
ν = ν0exp(−EA/kBT ) (3.71)
donde ν es la frecuencia total de salto, y EA la barrera de difusión clásica que se
define como la diferencia entre el valor de VA(r) en el punto silla y el mínimo,
(ver Fig. 10).
Figura 10. Representación esquemática del potencial adiabático VA(x) a lo largodel recorrido en una dimensión conectando mínimos consecutivos. La barrera deactivación EA es la diferencia de potencial entre los puntos sillas (máximos en elcaso 1D) y el mínimo.
La descripción de un átomo aislado dando saltos térmicamente activados entre
posiciones adyacentes es conceptualmente la imagen más simple para describir la

CAPÍTULO 3. FORMALISMO TEÓRICO. 68
difusión sobre superficies.
Esto se debe al acoplamiento no adiabático del átomo con las excitaciones
del sustrato. Sin este acoplamiento, no se justifica que el átomo adquiera
bastante energía para saltar sobre la barrera y se equilibre a una nueva
posición de adsorción. En la más simple aproximación markoviana, las fuerzas
estocásticas actúan sobre el átomo a diferentes tiempos pudiéndose tratar de forma
estadísticamente independientes. Con esta aproximación, el acoplamiento no
adiabático viene caracterizado por un simple parámetro de fricción γ , siendo éste
el caso que nos interesa. No obstante, para un caso más general, las fluctuaciones
y amortiguamiento que experimenta el átomo en cualquier instante de tiempo
depende de su historia previa, por tanto los efectos de memoria llegan a ser
importantes y el acoplamiento de fricción tiene que venir caracterizado por una
función memoria K(t, t′) , la cual es esencial en la función de correlación de las
fuerzas estocásticas a tiempos t y t′. Si la temperatura es suficientemente baja, el
adsorbato se pasa la mayoría del tiempo vibrando a pequeñas amplitudes en el
mínimo localizado en los sitios de adsorción. Ocasionalmente éste recibe bastante
energía del sustrato (baño térmico) para dar un salto satisfactorio, después del
cual se termaliza otra vez, en otro sitio de adsorción. Si la termalización ocurre en
celdas unidad muy cercanas a la celda unidad inicial, a este salto se le denomina
salto simple; si no es así, se habla de saltos largos o saltos múltiples. Si se supone
que se parte del origen (sitio 0), las cantidades importantes con respecto a los
saltos por difusión son la frecuencia total de salto ν y la probabilidad pl de
termalización en el sitio de adsorción a l = (lx, ly). La aproximación más simple
para extraer información sobre los detalles microscópicos del proceso de difusión
y explicar de una forma razonable la dependencia de Γ(∆K) frente a ∆K en la
Fig. 7 (Capítulo 2) es descrito por un modelo de saltos introducido por Chudley-
Elliot [26]. En tal modelo los átomos se describen posicionándolos en los sitios de
adsorción, y difundiéndose por saltos instantáneos desde un sitio a otro. Los saltos
entre los sitios son descritos por vectores j, especificando el desplazamiento
atómico causado por dicho salto, obteniendo así una frecuencia de salto νj = 1/τj

CAPÍTULO 3. FORMALISMO TEÓRICO. 69
con periodo τj. Si el átomo se desplaza mediante saltos instantáneos j, entonces
la probabilidad de encontrar éste en R a un tiempo t incrementará si el átomo se
encuentra en R − j y salta j, y del mismo modo esta probabilidad decrecerá si el
átomo se encuentra en R y salta j. En otras palabras, la probabilidad de encontrar
el átomo en R a tiempo t cambiará como:
∂Gs(R, t)
∂t=
n∑
j
1
τj[Gs(R − j, t) −Gs(R, t)] (3.72)
con la condición de contorno Gs(R, t = 0) = δ(0), n el número de saltos y
τj = 1/νj es el periodo de salto. Resolviendo la ecuación maestra [90, 91] se
obtiene:
Ss(∆K, ω) =1
π
[Γ(∆K)/2]
[Γ(∆K)/2]2 + ω2(3.73)
obteniendo una lorenciana de anchura energética Γ(∆K), donde como antes,
Ss(∆K, ω) es la doble transformada de Fourier de la función de correlación
Gs(r, t). La anchura Γ(∆K)de la lorenciana viene dada por:
Γ(∆K) =∑
j
1
τj[1 − e−i∆K.j] (3.74)
Si además se considera una red de Bravais en la que los saltos j y -j ocurren por
igual, la ec. (3.74) puede ser escrita como:
Γ(∆K) = ~∆ω(∆K) = 4~
′∑
j
νjsin2 ∆K.j
2(3.75)
donde la sumatoria prima indica sumas únicamente de saltos positivos. Con
este modelo se obtiene una descripción del factor de estructura Ss(∆K, ω) así
como su anchura Γ(∆K). Además aplicando este modelo simple a los resultados
experimentales se puede obtener frecuencias de saltos νj que como bien sabemos
caracterizan al mecanismo de difusión.

CAPÍTULO 3. FORMALISMO TEÓRICO. 70
3.4.4.3. Aproximación gaussiana.
La función I(∆K, t) es considerada como la función característica de un
proceso estocástico R(t):
I(∆K, t) ≡< e−i∆K.[R(t)−R(0)] >=< e−i∆KR t
0v∆K(t′)dt′ > (3.76)
donde v∆K es la velocidad del adsorbato proyectada a lo largo de la dirección de
la transferencia del vector de onda paralelo ∆K. Si desarrollamos una expansión
a segundo orden en cumulantes nos queda el resultado estándar:
I(∆K, t) ∼ e−∆K
2
2
R t
0dt′
R t
0dt′′<v∆K(t′)v∆K(t′′)> = e−∆K2
R t
0(t−t′)ψ(t′)dt′ (3.77)
donde ψ(t) ≡< v∆K(0)v∆K(t) > es la función de autocorrelación de
velocidades. La primera aproximación viene en el corte de la serie de cumulantes,
y la segunda igualdad es cierta si el proceso estocástico de la velocidad v∆K(t) es
estacionario y gaussiano. De este modo, diremos que para un proceso gaussiano la
ecuación (3.77) es exacta. Por tanto, una partícula que se difunde libremente (sin
interacción potencial adiabática) o sometida a un potencial adiabático cuadrático,
sometida a la acción de un ruido blanco gaussiano obedece a un proceso
gaussiano, comportándose de la misma forma que una partícula browniana. Si
además, pasado un cierto tiempo, la función de distribución de posiciones y
velocidades es invariante bajo una traslación temporal, diremos que el sistema
ha alcanzado el estado estacionario y, por tanto, la función característica de este
proceso I(∆K, t) dada por (3.77) es exacta. También sabemos por el teorema
de Doobs, que si el proceso estacionario es gaussiano y markoviano (proceso de
O-U, único proceso estocástico que cumple estas tres condiciones), la función de
correlación decae exponencialmente:
ψ(t) =< v2∆K > e−t/τr (3.78)

CAPÍTULO 3. FORMALISMO TEÓRICO. 71
Entonces, la función de dispersión intermedia puede expresarse como :
I(∆K, t) = exp[−χ2(e−t/τr + t/τr − 1)] (3.79)
con
χ = τr
√
< v2∆K >|∆K| =
D|∆K|√
< v2∆K >
≡ l|∆K| (3.80)
donde l es el libre recorrido medio y:
D = τr < v2∆K > (3.81)
es el coeficiente de difusión. Tenemos que indicar que para una partícula
no sometida a la acción de un potencial adiabático y bajo un ruido térmico
(gaussiano), el tiempo de relajación, que como ya se comentó en una sección
anterior, es el tiempo en el que por primera vez la partícula siente la interacción
macroscópica con el baño, produciéndose así un cambio en su posición y
velocidad. A partir de este tiempo, la partícula pasa de tener un comportamiento
balístico (partícula libre), a un comportamiento difusivo, pudiéndose medir el
coeficiente de difusión D.
3.4.4.4. Difusión de partícula aislada.
Consideramos un sistema de adsorbatos en equilibrio con el sustrato. Este
es el único límite en el cual los coeficientes de difusión pueden definirse de
forma precisa. El coeficiente de difusión D, es el coeficiente de transporte más
importante para el caso más simple de difusión, en el que se puede seguir el
movimiento de cada adsorbato individual [1]. En este caso,D se define en función
del desplazamiento cuadrático medio (MSD) como:
D = lımt→∞
1
2Ntd
N∑
i=1
< |Ri(t) − Ri(0)|2 > (3.82)

CAPÍTULO 3. FORMALISMO TEÓRICO. 72
donde N es el número total de adsorbatos en el sistema (N = 1 en nuestro caso),
d es la dimensión espacial (para difusión sobre la superficie d = 2), Ri(t) denota
la posición de la ith partícula a tiempo t, y < . > es el colectivo promedio en el
equilibrio. El coeficiente de difusión D se encuentra bien definido en el caso de
que el desplazamiento cuadrático medio en el límite asintótico de cada partícula
que se difunde, aumenta linealmente con el tiempo, según la ley de difusión de
Einstein (3.35) [87, 92]. La ecuación (3.82) es válida para un sistema isotrópico.
En este caso, se puede definir el correspondiente coeficiente de difusión a lo
largo de cada eje principal con las coordenadas xi(t) reemplazándolas por Ri(t)
en dicha ecuación. Hay diferentes formas alternativas , que son útiles, y parten
de la definición de (3.82) [87]. La primera es en términos de la función de
autocorrelación de velocidades:
D =1
Nd
N∑
i=1
∫ ∞
0
dt < vi(t).vi(0) > (3.83)
La ec. (3.83) es conocida como la fórmula de Green-Kubo [87]. De la fórmula
Green-Kubo, se deriva una tercera expresión para el coeficiente de difusión :
D = π lımω,∆K→0
ω2
∆KSs(∆K, ω) (3.84)
Al margen de estas simples definiciones para el coeficiente de difusión D,
hay muchos casos donde dicho concepto debe considerarse cuidadosamente.
Para muchas partículas que se difunden, el concepto de D es muy eficaz sólo
para trayectorias de partículas individuales que pueden ser identificadas. En
mecanismos más complicados que involucran el movimiento de un grupo de
partículas, se puede extender la definicion de D, y en este caso obedecería a
un coeficiente de difusión correspondiente al centro de masas del sistema de
adsorbatos que se mueven a tiempo t [90]. Por otro lado, este coeficiente de
difusión puede ser expresado en términos de la frecuencia total de saltos ν y la
distribución de longitud de saltos pl, para el caso ideal donde lo saltos no están
correlacionados (aproximación markoviana). Para este caso concreto, se puede

CAPÍTULO 3. FORMALISMO TEÓRICO. 73
expresar D como:
D =1
2dν
∑
l
l2pl =1
2dν < l2 > (3.85)
Si sólo se consideran saltos simples, entonces l2 = a2, siendo a la constante
de red. En general, habrá correlaciones entre los saltos y cuando ésto ocurrre se
realizan correcciones a la formula simple de (3.85).
Por último decir en esta sección, que en todo momento estamos considerando
un coeficiente de difusión de adsorbatos libres, no teniendo en cuenta la
interacción con otros adsorbatos. En los casos en donde se tiene encuenta dicha
interacción, este coeficiente de difusión libre recibe el nombre de “difusión de
la traza”[1], simbolizado Dt, para diferenciarlo del coeficiente de difusión que sí
tiene en cuenta dicha interacción, denominado “difusión del colectivo”, Dc.
3.4.5. Difusión normal y anómala.
Los procesos de transporte en el que el MSD viola la ley de Einstein para el
movimiento browniano, son generalmente llamados procesos de difusión anómala
[93, 94]. Esta ley se reemplaza por la forma más general:
< |x(t) − x(0)|2 + |y(t) − y(0)|2 >∼ Dδtδ (3.86)
con δ < 1(subdifusión) o δ > 1(difusión aumentada o superdifusión) describiendo
un proceso de difusión más lento o más rápido que la difusión normal del
movimiento browniano.
Una cantidad de interés que se encuentra relacionada con el coeficiente de
difusión es el espectro de potencias de velocidades o, dicho de otro modo, la
transformada de Fourier de la función de autocorrelación de las velocidades
definida como:
Z(ω) =
∫ ∞
−∞< v(t).v(0) > e−iωtdt (3.87)

CAPÍTULO 3. FORMALISMO TEÓRICO. 74
El coeficiente de difusión es simplemente (en dos dimensiones):
D =1
2Z(ω = 0) (3.88)
Cuando la difusión es normal, el espectro de potencias de velocidades debería
converger a un valor finito cuando ω tiende a cero. Por otro lado, una divergencia
del espectro de potencias como ω−α con 0 > α ≤ 1 nos conduce a la denominada
difusión anómala. Esta difusión anómala puede ser también identificada desde
diferentes cantidades tal y como el MSD, visto anteriormente, en el cual
asintóticamente crece más rapidamente con el tiempo que si lo hiciese de
forma lineal (difusión normal). El espectro de potencias de velocidades es muy
conveniente ya que se encuentra relacionado con el factor de estructura dinámico
por la siguiente expresión:
Z(ω) = ω2 lım∆K→0
Ss(∆K, ω)
∆K2(3.89)
Para la difusión normal, en el límite ∆K → 0, la relación Ss(∆K, ω)/∆K2
debería comportarse como D/ω2 para pequeños valores de ω, de acuerdo con
(3.70). Decir también que el MSD y la función de autocorrelación de velocidades
a grandes distancias se encuentran relacionadas por:
< R2(t) >∼ 2t
∫ t
0
< v(0).v(τ) > dτ (3.90)
Por tanto, y esto es importante, para una divergencia del espectro de potencias
del tipo ω−α para pequeños valores de ω, el MSD debe diverger a largos tiempos
como < R2(t) >∼ t1+α, 0 < α < 1, o < R2(t) >∼ t2 (difusión balística) para
α ≥ 1.
La difusión anómala en sistemas hamiltonianos fue estudiada hace algunos
años en un modelo de potencial [95, 96, 97]. La existencia de distribuciones
de Lévy [98] de longitudes de salto fue un dato crucial para la explicación del
comportamiento anómalo. Los caminos de Lévy y los vuelos de Lévy están bien
establecidos y son conceptos bastante empleados en la física estadística [99]. El

CAPÍTULO 3. FORMALISMO TEÓRICO. 75
término vuelo de Lévy se usa para indicar un caminante aleatorio en un espacio
n-dimensional que experimenta una distribución de Lévy de longitudes de salto
y un promedio temporal finito entre saltos. Debido a que existe un tiempo finito
de espera entre salto y salto, el proceso es markoviano, ya que en este tiempo de
espera la partícula pierde la memoria del salto anterior, y por tanto esta perdida de
memoria, hace que no haya correlación entre un salto y otro. Una característica
general de las distribuciones de Lévy es que el MSD diverge:
< |x(t) − x(0)|2 + |y(t) − y(0)|2 >→ ∞ (3.91)
correspondiente al hecho de que los saltos largos se consideran instantáneos.
Claramente, la velocidad de las partículas que se difunden no puede ser infinita,
por tanto estos vuelos irreales físicamente son sustituidos por caminos de Lévy
[100, 101], en el que ahora sí, se toma en cuenta el tiempo necesario en cada salto
experimentado por el caminante aleatorio. Básicamente, se emplean dos modelos
en la descripción teórica de la difusión anómala basándose en la estadística de
Lévy. La teoría de Geisel y col. [98, 76] usan un formalismo de renovación,
asumiendo que la partícula que se difunde se comporta como un caminante
aleatorio que desarrolla recorridos libres independientes con velocidad constante
v0 cuya duración t ( o longitudes l) se distribuyen de acuerdo a una ley inversa de
potencias:
φ(l) ∼ l−β (3.92)
otros autores han empleado un formalismo de caminante aleatorio continuo
(CTRW) [102, 103], en el cual la partícula se mueve a velocidad constante en
un tiempo t, entonces se para y elige una nueva dirección en el que el tiempo
de vida de este nuevo recorrido es aleatorio y viene dado por una probabilidad
determinada; ψ(l, t) es la densidad de probabilidad en la que la partícula se
mueve una distancia l en un tiempo t. Por tanto, se puede ver que en este nuevo
formalismo, no sólo hay que tener en cuenta una distribución de saltos, sino

CAPÍTULO 3. FORMALISMO TEÓRICO. 76
también el tiempo que duran los saltos. Dicho esto, si las longitudes de salto
y los tiempos de vida de estos saltos son variables aleatorias independientes, la
densidad de probabilidad ψ(l, t) puede expresarse como:
ψ(l, t) = φ(l)ϕ(t) (3.93)
donde φ(l) nos da la probabilidad de que un salto tenga una longitud l, y ϕ(t) la
probabilidad para un tiempo de espera en el intervalo (t, t + dt). Por tanto, para
obtener caminos de Lévy y una duración de tiempo para los saltos, se introduce
un acoplamiento espacio-temporal [102, 104, 105] y ψ(l, t) se expresa como:
ψ(l, t) = p(l|t)φ(l) (3.94)
siendo p(l|t) una probabilidad condicionada para una transición a un tiempo t,
en un desplazamiento l, y como ya sabemos φ(l) es la probabilidad de longitud
de salto. Cuando asumimos una ley inversa de potencias para φ(l) tal como
(3.92) y cuando p(l|t) = δ(t − |l|/v0) ambas teorías dan el mismo resultado
para el comportamiento del MSD y por tanto, para el espectro de potencias. En
particular, para el MSD el comportamiento asintótico predicho es el siguiente
[95, 96, 106, 107, 108]:
< R2(t) >∼
t2 0 > β ≤ 1,
t4−β 1 < β < 2,
t ln t β = 2,
t β > 2.
(3.95)
Por tanto, se observa cómo la difusión anómala está caracterizada por desplaza-
mientos cuadráticos medios que divergen más rapidamente que en su forma lineal
y este fenómeno genérico aparece tanto en sistemas conservativos como en disi-
pativos. Estos procesos ocurren en multitud de sistemas físicos, por ejemplo en
dinámica de fluidos y microestructuras de semiconductores [94]. En estos ejem-
plos se ha demostrado que la difusión anómala juega un papel muy importante,

CAPÍTULO 3. FORMALISMO TEÓRICO. 77
y en el que se han visto cómo la interpretación del movimiento browniano bajo
su modelo de caminante aleatorio falla [109], teniendo que recurrir a otras inter-
pretaciones y modelos. El término “vuelos de Lévy” se emplea para indicar un
caminante aleatorio en un espacio continuo n-dimensional, cuya distribución de
longitudes de salto obedece a una distribución de Lévy en un tiempo promedia-
do finito entre saltos. Opuesto al movimiento browniano, el cual nos conduce al
teorema del límite central estableciendo una distribución gaussiana como la distri-
bución limitadora de la suma de un número muy elevado de variables aleatorias.
El caminante aleatorio de Weierstrass está caracterizado por una distribución de
Lévy [110], que se distingue por una propiedad en la que una ley de potencias
aparece en la cola de la distribución que nos conduce a una divergencia, incluso
en los momentos de bajo orden. Por último, terminar esta sección diciendo que la
existencia de distribuciones de Lévy del tipo (3.92) afecta al comportamiento del
factor de estructura dinámico. Para ver esto, supongamos un caminante aleatorio
con una probabilidad de longitud de salto algebraica para las etapas individuales
(caminante aleatorio de Weierstrass). Entonces, la densidad de probabilidad para
el caminate en la posición x después de N pasos viene dada por la convolución:
GN(x) = φ(l) × ...× φ(l) (3.96)
donde GN (x) es una función de correlación unidimensional. En el espacio de
Fourier, la función de dispersión intermedia puede entonces expresarse como:
IN(∆K) = φ(∆K)N (3.97)
Para la distribución de probabilidad dada por (3.92), la función característica φ en
el límite ∆K, ω → 0 presenta la forma [110]:
φ(∆K) = e−c(β)|∆Ka|β−1
(3.98)
donde a es la longitud de la celda unidad. Por deficición, el tiempo t = N∆t y el

CAPÍTULO 3. FORMALISMO TEÓRICO. 78
coeficiente de difusión generalizado viene dado por:
D = c(β) lıma,∆t→0
(aβ−1/∆t) (3.99)
usando la ec. (3.65), a pequeños valores de ∆K, ω, el factor de estructura
dinámico presenta la forma funcional:
Ss(∆K, ω) =1
π
D∆Kβ−1
ω2 +D2∆K2(β−1)(3.100)
Notar que dentro de la aproximación del caminate aleatorio de Weierstrass ,
cuando β = 3, la función característica dada por (3.98) es una gaussiana y
recuperamos la difusión normal, ec. (3.70).
3.5. Vibración de baja frecuencia.
Los movimientos vibracionales de adsorbatos moleculares y atómicos sobre
superficies han sido recientemente analizados por la técnica de QHAS. Esta
técnica experimental nos ha permitido una determinación precisa de modos
vibracionales a baja frecuencia, los cuales no son accesibles por otros tipos
de medidas. Mediante la técnica QHAS , cuando se hace incidir el haz de
helio de baja energía en la superficie, estos son dispersados por los adsorbatos
que hay en ella. Si el colectivo de adsorbatos se difunden por la superficie,
se observa un pico cuasielástico, centrado a frecuencia cero con una anchura
proporcional al coeficiente de difusión. Si además, algunos átomos o moléculas
están adsorbidos en posiciones específicas de la superficie, se observa un pico
adicional centrado alrededor de la frecuencia de vibración del adsorbato cercano
al mínimo del pozo de potencial. Este es el llamado modo translacional frustrado
o pico T, y nos proporciona información sobre la fricción por amortiguamiento
experimentado por el adsorbato debido a la interacción del par hueco-electrón o
con el acoplamiento de los fonones de la superficie cristalina. Este pico nos da
también información acerca de la curvatura y anarmonicidad de la PES alrededor

CAPÍTULO 3. FORMALISMO TEÓRICO. 79
del mínimo. Para adsorbatos atómicos a baja concentración, el análisis del modo
T puede ser una medida directa de todas estas cantidades físicas, ya que sólo
están presentes las interacciones adsorbato-sustrato y no hay modos vibracionales
externos o internos acoplados al modo T. Estos experimentos por QHAS han
reflejado algunas características del modo T. Primero, es un modo sin dispersión
(es independiente de la transferencia de momento paralelo) y de naturaleza
inelástica. Segundo, la forma de línea lorenciana se ajusta bastante bien a dicho
perfil del modo T siempre y cuando no solape con el pico cuasielástico. Tercero,
se ha observado un desplazamiento en dicho modo T con la temperatura para
diferentes sistemas atómicos y moleculares. Cuarto, se observa que la anchura
del pico varía linealmente con la temperatura para algunos sistemas adsorbato-
sustrato como Na/Cu(001) o CO/Cu(001), mientras que para otros sistemas parece
ser independiente de la temperatura.
Se han desarrollado teorías que veremos en la siguiente sección para
explicar dicho desplazamiento, así como la dependencia lineal de la anchura
a alta temperatura. Al mismo tiempo, simulaciones numéricas tipo Langevin
reproducen bastante bien los resultados experimentales, no sólo para el pico
cuasielástico sino también el desplazamiento y anchura del modo T. Persson
también usó un movimiento browniano para analizar la dependencia con la
temperatura del perfil de línea de los modos vibracionales de CO adsorbido
sobre Ni(111). El movimiento de difusión es puramente browniano y se piensa
que no está correlacionado con el movimiento vibracional de los adsorbatos.
También es conocido que la difusión es asistida por la vibración. En principio,
diferentes escalas de longitud y tiempo son involucradas en ambos movimientos;
las vibraciones son localizadas dentro de mínimos de adsorción de potencial
con alta frecuencia, y los procesos de difusión, por saltos desde un sitio de
adsorción a otros vecinos, incluso a vecinos mucho más lejanos. Obviamente,
los dos movimientos pueden llegar a estar más y más acoplados. Por ejemplo,
temperaturas superiores a la barrera de difusión hacen disminuir el tiempo de
espera de los adsorbatos en un sitio de adsorción e, incluso considerando altos

CAPÍTULO 3. FORMALISMO TEÓRICO. 80
valores en la constante de fricción, se pueden obtener una distancia de salto
del orden de la celdilla unidad. El trabajo de Chen y Ying, ya mencionado
anteriormente, puede ser considerado como el primer intento teórico para tratar
los dos movimientos de igual forma. De acuerdo con los argumentos empleados
arriba, el factor de estructura dinámico, (3.70) consistirá en diversos picos: el
pico cuasielástico alrededor de ω = 0 y diferentes picos inelásticos debido a la
vibración a baja frecuencia , para los eventos de creación (ω < 0) y aniquilación
(ω > 0) del modo T. Entonces, si no hay correlación entre el movimiento rápido
debido a las vibraciones y existe el movimiento lento de difusión, la función G
puede ser escrita como la convolución de las dos contribuciones y por tanto, por el
teorema de convolución, la función total de dispersión intermedia será el producto
de dos transformadas de Fourier espaciales, la una proveniente de la vibración y
la otra de la difusión. El factor de estructura dinámico total puede entonces ser
expresado como la convolución en frecuencia de los correspondientes factores
de estructura dinámicos. Considerando la contribución cuasielástica e inelásticas,
el factor de estructura dinámico total puede ser expresado como una suma, cada
uno proveniente de un movimiento desacoplado. En cambio, si los movimientos
están acoplados (por incremento de la temperatura de la superficie), la difusión
será asistida por las vibraciones, las cuales contribuirán al pico cuasielástico, y
la correspondiente suma no es valida. Sin embargo, si la temperatura es más alta
que la barrera de difusión, no se detectarán indicios de vibración observándose un
pico cuasielástico puro.
3.6. Modelo anarmónico.
Algunos autores justifican la dependencia lineal del desplazamiento y la
anchura energética del modo T con la temperatura asumiendo transiciones
vibracionales del adsorbato en la superficie, comportándose como un oscilador
anarmónico donde las poblaciones siguen una distribución de Boltzman. Sin
embargo, la naturaleza del acoplamiento anarmónico del adsorbato con el sustrato

CAPÍTULO 3. FORMALISMO TEÓRICO. 81
no es tan obvia, y esto sigue siendo un problema abierto para el que todavía
no hay una teoría sólida que dé explicación a estos efectos experimentales
observados para el modo T. En la literatura se recogen diferentes modelos
dinámicos [23, 21, 16, 20], los cuales nos permiten una interpretación de la
variación de la anchura energética así como el desplazamiento producido en el
modo T cuando varía la temperatura de superficie. A continuación se va a describir
de forma detallada, la interpretación más directa y más aceptada hasta la fecha que
explican estos dos efectos.
Este modelo ha sido empleado para interpretar el modo T del sistema CO-
Cu(001). Dicho modelo asegura que la anchura se origina debido a la aparición
de un potencial anarmónico CO-Cu. La anchura puede ser calculada desde el
orden más bajo en la expansión anarmónica del potencial como una función del
desplazamiento en una dirección µx:
V (x) =mCO
2ω2
0µ2x(1 + bµ2
x) (3.101)
siendo mCO la masa de la molécula de CO, ω0 la frecuencia fundamental del
modo T y b el parámetro de orden cuártico correspondiente a la anarmonicidad.
Para este potencial, las constantes espectroscópicas en función del parámetro de
anarmonicidad b vienen dadas por [111]:
E(υ) = (υ +1
2)~ω0 − χe(υ +
1
2)2
~ω0 (3.102)
con
χe = − ~b
2mCOω 0
(3.103)
Con este modelo se observa cómo la anarmonicidad conduce a un incremento
lineal en los espaciados de los niveles de energía vibracionales cuando aumenta
la temperatura debido a las contribuciones de los niveles vibracionales νmás altos
del espectro. Las contribuciones sin resolver conducen a una anchura adicional en
el modo T asimétrico, originando un desplazamiento de dicho pico a altas energías

CAPÍTULO 3. FORMALISMO TEÓRICO. 82
[112]:
∆E(T ) =
∑
ν ∆E(υ → υ + 1)e−E(υ)/kBT
∑
υ e−E(υ)/kBT
(3.104)
En el límite de altas temperaturas [kBT >> E(υ)], (3.104) se simplifica:
∆E(T ) = −2χekBT (3.105)
indicar que para la pequeña anarmonicidad que se ha considerado, la asimetría
introducida por la suma de las transiciones anarmónicas de alto orden (υ →υ + 1) resulta ser pequeña para la anchura total del modo T observada
experimentalmente, por tanto el perfil del pico experimental todavía es bien
descrito por una lorenciana. Asumiendo pues lorencianas de anchura constante
γ0 para el perfil del pico correspondiente a los niveles vibracionales individuales
υ, la anchura total energética del pico Γ(T ) viene dada por:
Γ(T ) =
√
∑
υ[∆E(υ → υ + 1) − ∆E(T )]2e−E(υ)/kBT
∑
υ e−E(υ)/kBT
+ γ0 (3.106)
3.7. Difusión y vibración de baja frecuencia.
Los experimentos por STM y FIM no son sensibles al movimiento vibracional
del átomo sobre la superficie. Este no es el caso en QHAS, donde el movimiento
vibracional puede interferir con el pico cuasielástico, haciendo mucho más difícil
interpretar las medidas experimentales. Como ya venimos diciendo a lo largo de
esta memoria, el primer intento de considerar los dos clases de movimientos se
encuentra recogido en el trabajo de Chen y Ying [27]. Una buena alternativa
para entender mejor el acoplamiento de la vibración-difusión ha sido partir de
la función de autocorrelación de velocidades para el oscilador amortiguado[83]:
ψ(t) =< v2∆K > cos(ω2t+ ϕ)e−t/τ2 (3.107)

CAPÍTULO 3. FORMALISMO TEÓRICO. 83
donde ω2 es la frecuencia de oscilación del adsorbato dependiente de la
temperatura, y ϕ es el parámetro de desfase el cual mejora el ajuste de la actual
función de correlación de velocidades, (3.107). A altas temperaturas, la difusión
predomina y τ2 es aproximadamente el coeficiente de difusión dividido por kBT
. A bajas temperaturas, los saltos de las partículas adsorbidas son muy poco
frecuentes y el único movimiento que permanece es el de la vibración.
Tenemos que decir, en contraste con las expresiones obtenidas por estos
autores, que el perfil de línea completo a la que se llega en este modelo no
es una simple suma de dos contribuciones, uno del pico cuasielástico y el otro
de la vibración. Los dos movimientos interfieren, produciendo un efecto de
estrechamiento, y en general no pueden ser separados. Incluso con este modelo
simple, uno realmente encuentra que la deconvolución de las medidas por QHAS
no es trivial. Se puede mejorar ésto haciendo uso de la aproximación gaussiana
considerada en (3.79) para obtener el factor de estructura dinámico de forma
exacta [38]. Pero esto no garantiza que la forma funcional de (3.107) dé una
estimación precisa de la verdadera función de autocorrelación que describe la
dinámica del movimiento del adsorbato. Como ya sabemos, este modelo se
comporta bien, siempre y cuando el proceso sea gaussiano, o dicho de otro modo
el adsorbato se encuentre bajo la acción de un potencial armónico, o no influido
por éste, comportándose como una partícula libre. Por tanto, una completa teoría
que describa de forma correcta la difusión asistida por la vibración se encuentra
en vías de desarrollo.
3.8. Efectos cuánticos.
Experimentalmente, los efectos cuánticos han sido observados en átomos
ligeros tales como hidrógeno y deuterio. Gomer y col. [1], usando el método
por campo de emisión, observaron difusión por efecto túnel de átomos de H
sobre superficie de wolframio [113, 114]. Ellos encontraron que el coeficiente de
difusión llega a ser independiente de la temperatura a muy bajas temperaturas.

CAPÍTULO 3. FORMALISMO TEÓRICO. 84
Resultados más recientes han sido recogidos por Cao y col. [115] utilizando
desorción inducida por láser así como técnicas de difracción óptica para el
estudio de la difusión de átomos de H y D sobre superficies de Ni(111).
Ellos encontraron un débil efecto túnel, sin embargo al dibujar la curva de
Arrhenius del coeficiente de difusión, observaron que la correspondiente al átomo
de deuterio, era sustancialmente más pequeña que la del átomo de hidrógeno.
Estos datos experimentales no han sido analizados teóricamente usando una
dinámica de Langevin. Esto es debido a que estos experimentos sólo han medido
coeficientes de difusión, no pudiéndose obtener información alguna acerca de sus
distribuciones de saltos. La difusión de H y D sobre superficies es un área de
creciente actividad en su estudio, de ahí que un tratamiento de Langevin cuántico
pudiera ser interesante en el futuro. Con respecto a la vibración, tenemos que
decir que estamos desarrollando un método cuántico basado en el formalismo de
la matriz densidad, de forma que pueda aparecer de forma natural los dos efectos,
tanto del desplazamiento como de la anchura del modo T con la temperatura,
sin necesidad de recurrir a efectos anarmónicos. Por tanto, hasta la fecha no se
ha desarrollado una teoría sólida mecanocuántica que explique de forma clara y
conjunta la difusión y vibración de los átomos adsorbidos a la superficie.

Capítulo 4
RESULTADOS.
En este capítulo se van a mostrar los resultados obtenidos en este trabajo de
investigación. Se han obtenido resultados desde el punto de vista numérico y se
han comparado con las ecuaciones analíticas mostradas en el capítulo anterior.
Asimismo, se proponen nuevos modelos teóricos con el fin de reproducir y dar
explicación a las formas de líneas obtenidas experimentalmente por QHAS de los
procesos de difusión y vibración que tienen lugar en el fenómeno de adsorción de
átomos o moléculas sobre las superficies metálicas analizadas.
4.1. Algoritmo numérico para resolver la ecuación
de Langevin.
El movimiento clásico de adsorbatos sobre superficies es considerado como
un proceso estocástico, cuya ecuación de Langevin es la que rige la dinámica del
sistema. Consideramos el adsorbato como una pequeña partícula browniana que
se mueve por la superficie presentando un número pequeño de grados de libertad,
mientras que los átomos que componen la superficie (sustrato) a una determinada
temperatura se comporta como un baño térmico con un número muy elevado
de grados de libertad. Debido a la dificultad de considerar todos los grados de
85

CAPÍTULO 4. RESULTADOS. 86
libertad que constituyen el baño térmico, éstos son reemplazados por suposiciones
adicionales tales como:
1. El tiempo de relajación o amortiguamiento τr que gobierna la velocidad
a la cual el sistema dinámico (adsorbato) alcanza el equilibrio con el baño
térmico (estado estacionario) es mucho mayor que el tiempo de correlación de
las fluctuaciones del baño, τc.
2. El tiempo de interacción entre el sistema y el baño se considera
extremadamente corto comparado con el tiempo de cambio de las propiedades
del sistema, tales como posición y velocidad.
3. El efecto del baño térmico sobre el sistema se traduce en una fuerza aleatoria
fluctuante sobre el adsorbato, que de acuerdo con el teorema de fluctuación-
disipación, es relacionada con la fricción que experimenta el adsorbato al
interaccionar con el baño térmico.
4. Por último, en este problema suponemos que el adsorbato se mueve sobre
una superficie bidimensional, en el que no se considera ninguna interacción con
otros adsorbatos presentes en la superficie.
Con estas suposiciones, el movimiento del adsorbato viene regido por una
ecuación bidimensional de Langevin dentro de una aproximación markoviana:
x = −∂xV (x, y) − γx(t) + ξy(t) (4.1)
y = −∂yV (x, y) − γy(t) + ξy(t) (4.2)
donde x e y son los desplazamientos. Como bien sabemos, los ruidos gaussianos
blancos, ξx y ξy, tienen una media nula y sus correlaciones vienen dadas por:

CAPÍTULO 4. RESULTADOS. 87
< ξx(y)(t)ξx(y)(t′) >= kBTγδ(t− t′) (4.3)
< ξx(t)ξy(t′) >= 0 (4.4)
Las constantes de difusión a lo largo de las direcciones x e y están relacionadas por
el desplazamiento cuadrático medio, como ya se comentó en el capítulo anterior a
través de la relación de Einstein, siendo válida cuando el sistema alcanza el estado
estacionario, por tanto :
Dx =< x2(t) >
2t
∣
∣
∣
∣
t→∞, Dy =
< y2(t) >
2t
∣
∣
∣
∣
t→∞(4.5)
Decir que en un potencial simétrico el coeficiente de difusión D ' Dx ' Dy, y
en el caso de un potencial asimétrico viene dada por (4.5). Estos coeficientes de
difusión también se pueden expresar en términos de la transformada de Laplace
de la función de autocorrelación de velocidades:
Dx =
∫ τ
0
dt < vx(t)vx(0) >τ→∞ (4.6)
Dy =
∫ τ
0
dt < vy(t)vy(0) >τ→∞ (4.7)
Cuando el potencial es simétrico < v(t).v(0 >= 2 < vx(t)vx(0 >. En
estas ecuaciones el promedio térmico estadístico significa un promedio sobre
las condiciones iniciales y sobre las trayectorias estocásticas obtenidas por
integración numérica de las ecuaciones de Langevin. Dicho de otro modo,
significan un promedio en trayectorias y en tiempos, ya que con un doble
promedio se consigue una mejor estadística, y se ha escrito como un corchete
simple < . > por comodidad. Se considera que las fuerzas deterministas ∂xV y
∂yV permanecen aproximadamente constantes en un pequeño intervalo de tiempo
(t, t + dt), las ecuaciones de Langevin pueden ser integradas de la siguiente
forma, obedeciendo el siguiente algoritmo de Verlet, exacto hasta primer orden

CAPÍTULO 4. RESULTADOS. 88
en velocidades y hasta segundo orden en posiciones [116] :
x(t + dt) = x(t) + c1dtvx(t) + c2dt2ax(t) + δxG (4.8)
y(t+ dt) = y(t) + c1dtvy(t) + c2dt2ay(t) + δyG (4.9)
vx(t+ dt) = c0vx(t) + c1dtax(t) + δvGx (4.10)
vy(t+ dt) = c0vy(t) + c1dtay(t) + δvGy (4.11)
donde
c0 = e−γdt, c1 = (1 − c0)/(γdt), c2 = (1 − c1)/(γdt) (4.12)
δxG, δyG y δvGx , δvGy son, respectivamente, las componentes de los desplazamien-
tos y velocidades causados por las fuerzas aleatorias ξx, ξy. Por otro lado, δxGy
δvGx son variables gaussianas y correlacionadas, cuyos elementos diagonales y no
diagonales de la matriz varianza vienen dados por:
< δxGδxG >= dt2kBT
m[2 − (3 − 4c0 + c20]/(γdt)]/(γdt) (4.13)
< δvGx δvGx >=
kBT
m(1 − c20) (4.14)
< δxGδvGx >= dtkBT
m(1 − c0)
2/(γdt) (4.15)
Una forma idéntica se obtiene para los elementos de la matriz varianza de las
variables correlacionadas δyG y δvGy .

CAPÍTULO 4. RESULTADOS. 89
4.2. Superficie de Energía Potencial.
La difusión de átomos de Na a diferentes concentraciones sobre la superficie
de Cu(001) ha sido investigada experimentalmente de forma reciente y de
forma extensiva con la técnica de QHAS, así como teóricamente dentro del
formalismo teórico de Langevin y Fokker-Planck (FP). A baja concentración
de átomos de Na, las interacciones adsorbato-adsorbato no se han tenido en
cuenta y las correspondientes medidas experimentales han sido interpretadas en
términos de una interacción adsorbato-sustrato gobernada por una superficie de
energía potencial no separable, cuyos parámetros fueron ajustados al experimento
mediante una simulación de DM [16, 17].
Esta superficie de energía potencial describe la fuerza adiabática que
experimentan los átomos de Na con el sustrato, y las fuerzas no adiabáticas
vienen descritas por una fuerza de fricción y otra aleatoria debido al intercambio
energético del adsorbato con el sustrato, como consecuencia de las vibraciones
que experimentan los átomos de Cu a una cierta temperatura de superficie T .
Debido a que las vibraciones normales a la superficie de los átomos de Na son
de mayor frecuencia [117] que las vibraciones paralelas a esta (modo frustrado
translacional o modo T), la interacción potencial adiabática se promedia sobre
los modos normales de vibración. Por tanto, la interacción adiabática adsorbato-
sustrato viene representada por una función bidimensional en el plano (x,y):
V (x, y) = V0(x, y) + V1(x, y) + V2(x, y) (4.16)

CAPÍTULO 4. RESULTADOS. 90
El primer término es un potencial coseno separable:
V0(x, y) = V0[2 − cos(2πx/a) − cos(2πy/a)] (4.17)
donde a es la constante de red de la superficie de Cu(001) (a = 2,557Å), y
V0 = 41,4 meV.
El segundo término, se añade para producir las depresiones existentes en los
máximos del potencial, sobre las cuales se encuentran situados los átomos de Cu
de acuerdo con las observaciones:
V1(x, y) = −A∑
m,n
exp(−b[xa− (m+
1
2)]2 + [
y
a− (n+
1
2)]2) (4.18)
con A = 2V0 y b = 11,8.
Finalmente, el tercer término es una parte no separable la cual cambia la
curvatura cerca del mínimo de potencial y cambia la diferencia de potencial
existente entre el mínimo del pozo de potencial y el potencial en los puntos silla:
V2(x, y) = CV0π2∑
m,n
[(x
a−m)2 +(
y
a−n)2] exp[−(
x
a−m)2− (
y
a−n)2] (4.19)
con C = −0,2.
Indicar que para un potencial periódico, las sumas en (4.18) y (4.19) se deben
realizar sobre el conjunto de pares enteros (m,n).
En la práctica, en las simulaciones de trayectorias clásicas, la dinámica se
reduce a una simple celda Wigner-Seitz con unas condiciones de contorno deter-
minadas, y la suma sobre gaussianas es truncada a pocos términos (típicamentem
y n varían entre −10 y 10).
En Fig. 1 se muestra la correspondiente superficie de energía potencial en 3
dimensiones.

CAPÍTULO 4. RESULTADOS. 91
Figura1. Potencial semiempírico bidimensional. El eje cero de energía es tomadoen los siguientes mínimos (x = y = 0 = ±a,...) Las direcciones x, ycorresponden a los ejes azimutales [110] y [110] respectivamente, y la barrerade difusión a lo largo de esta dirección es 74,64 meV. Las direcciones diagonales[100] y [010] tienen una barrera energética de 84,49 meV.
Las direcciones x e y se toman a lo largo de los ejes azimutales con índices de
Miller [110] y [110], respectivamente. La energía cero se toma en el mínimo del
pozo de potencial. La barrera en el punto silla a lo largo de las direcciones x e y
se encuentra a 74,64 meV, y la barrera del punto silla a lo largo de la diagonal
[100] y [010] es de 84,49 meV. El pequeño mínimo en el potencial truncado
corresponde a una serie de depresiones donde se sitúan los átomos de Cu, que se
encuentran a una energía de 82,74 meV. Más tarde se verá cómo estas depresiones
juegan un papel importante en la dinámica clásica pudiendo ser responsables del
comportamiento anómalo debido al alto número de trayectorias que escapan a
través de la diagonal [100] detectado en el experimento [16, 17]. Los máximos de
potencial se encuentran localizados a 85,51 meV. Una característica importante

CAPÍTULO 4. RESULTADOS. 92
en esta SEP es su no separabilidad, diferente a la utilizada para reproducir
experimentos previos [15, 28, 33, 27]. La SEP separable no puede explicar todos
los resultados experimentales obtenidos hasta la fecha por QHAS. En particular,
la marcada anisotropía en la difusión a lo largo de las diferentes direcciones. Esta
no separabilidad del potencial adiabático puede influir de forma considerable en
la aparición de saltos largos y de la dependencia del coeficiente de difusión con la
fricción [118].
4.3. Dinámica caótica.
En esta sección se ha analizado en detalle la dinámica clásica determinista
(no se tiene en cuenta la interacción no adiabática adsorbato-sustrato) del sistema
Na-Cu(001) a diferentes energías. Por debajo de la barrera del punto silla, a
Es = 74,64 meV, se ha localizado únicamente movimientos intra-pozos, debido
claro está a que no tiene energía suficiente para salir del pozo de potencial .
Entre este valor de la energía y la energía correspondiente al máximo de potencial
Emax = 85,51meV, los átomos de Na se difunden a través de las direcciones x
e y con un predominio de largos recorridos interrumpidos por episodios donde la
partícula es atrapada dentro de un pozo de potencial. También se observa que para
una energía E > Emax, la partícula en principio se difunde libremente aunque
se observa también la coexistencia de saltos largos con captura del movimiento
debido a dichos pozos de potencial para algunos rangos de energía. Por último,
si se sigue aumentando la energía, ahora E Emax, nos aproximamos al límite
integrable, donde se produce, ahora sí, a energías superiores a 200 meV, difusión
libre.

CAPÍTULO 4. RESULTADOS. 93
Un mayor entendimiento de lo que realmente está ocurriendo a escala
microscópica, así como por qué la partícula se difunde de diferente forma según
se encuentre en un rango energético u otro, se tiene que buscar en el estudio
minucioso del comportamiento difusivo de la partícula en el espacio de fases.
El conocimiento de la arquitectura del espacio de fases comienza con el análisis
de la sección de Poincaré sobre la superficie de potencial. Está sección se
obtiene explorando el espacio de fases a una energía determinada, en la que se
mantiene constante una de las variables dinámicas, en nuestro caso la posición x.
Seguidamente, se hacen evolucionar un conjunto de trayectorias partiendo de unas
condiciones iniciales preestablecidas; entonces, cada vez que una trayectoria pasa
por la posición x fijada, la otra posición (y en nuestro caso) y el correspondiente
momento conjugado (py) son almacenados. De este modo, finalmente, y después
de un largo tiempo, donde numerosas trayectorias han cruzado la sección de
Poincaré por la posición x fijada, las correspondientes posiciones (y) y momentos
conjugados (py) almacenados constituyen la estructura del espacio de fases para
esa energía determinada . Este Mapa de Poincaré puede expresarse en términos
del flujo Φ que atraviesa la superficie (x =cte) como:
xn+1 = Φ(xn) (4.20)
donde xn son las variables dinámicas intrínsecas a la sección de superficie.
En este espacio de fases (y, py) generado por la sección de Poincaré, el
movimiento periódico se distingue por puntos fijos en el mapa de Poincaré,
xn = Φ(xn) y por tanto, como un punto fijo (y0,py0) en el espacio de fases.
Las trayectorias cuasiperiódicas darán lugar a islas regulares y, por último, las
trayectorias caóticas a puntos distribuidos de forma aleatoria. En la Fig.2 se
pueden ver los diferentes tipos de trayectorias mencionadas.

CAPÍTULO 4. RESULTADOS. 94
Figura 2. Superficie de sección de Poincaré a una energía total de 80 meV, fijandola coordenada x, a) x0 = 0; b) x0 = a/2.
Para obtener una visión más completa de la dinámica a cualquier energía,
se localizan las órbitas periódicas más importantes del adsorbato y se sigue su
evolución, analizando los cambios en su estabilidad y posibles bifurcaciones que
se puedan generar. La estabilidad y posibles bifurcaciones de una órbita periódica
puede ser encontrada calculando la traza de la llamada matriz de estabilidad
o matriz de monodromía M (matriz Jacobiana de la evolución dinámica que
relaciona la posición y momento a un tiempo t, con la posición y momento
inicial)[119].
Por tanto, se dice que una órbita periódica es estable si |Tr M|≤2, e inestable
si |Tr M|>2.
La traza de la matriz de estabilidad después de n iteraciones en el mapa de
Poincaré puede ser relacionada con la traza de una iteración por:
TrMn = 2cos[narc cosTrM1
2] (4.21)

CAPÍTULO 4. RESULTADOS. 95
para órbitas estables; mientras que para órbitas inestables la función coseno es
sustituida por una función coseno hiperbólica. Una nueva órbita periódica de
periodo n puede aparecer (o una que existe puede desaparecer) sólo cuando |Tr
M|=2. Aplicando (4.21) se obtienen las posibles bifurcaciones de periodo n a
partir de un simple movimiento periódico:
TrM1 = 2cos2πm
n(4.22)
donde m es un número entero tal que el argumento del coseno es módulo π.
Las órbitas estables no se bifurcan, pero pueden cambiar su estabilidad. Los
posibles tipos de bifurcaciones son sólo cinco [120], pero para nuestro propósito,
la más importante es la bifurcación de doble periodo: (|Tr M1|= − 2) la cual
cambia la estabilidad del movimiento de periodo uno. Se ha visto que existen tres
movimientos principales periódicos que surgen del mínimo del pozo de potencial
hacia las depresiones de éste.
Estos se pueden ver en la Fig. 3 a E = 80 meV (por encima de la barrera de
difusión) y restringidos a la celda de Wigner-Seitz.
Figura 3. Las principales orbitas periódicas de periodo 1 del sistema a E = 80meV restringidas a la celda unidad de Wigner-Seitz. (También se muestra lasuperficie equipotencial a esta energía).

CAPÍTULO 4. RESULTADOS. 96
Sabemos que la interacción potencial átomo-superficie, las coordenadas x
e y se extienden desde −∞ a ∞, pero debido a su periodicidad, se puede
restringir la dinámica a esta celda unidad imponiendo condiciones de contorno
para las trayectorias clásicas. Así, cada vez que la partícula abandona la celdilla
unidad por uno de sus contornos, esta es reinyectada por el contorno opuesto con
el mismo momento. Haciendo ésto, se consigue una dinámica real únicamente
considerando este tipo de celda unidad. En la Fig. 3 se observa: una órbita
paralela a la dirección x o, equivalentemente, a la dirección y (debido a la simetría
del potencial). Todas las órbitas periódicas con una simetría de reflexión en el
plano x = 0 tienen su homóloga en órbitas con simetría de reflexión y = 0.
Para energías por debajo de la barrera del punto silla, esta órbita representa un
movimiento periódico paralelo translacional frustrado, pero paraE > Es describe
movimientos libres a través de las direcciones x o y. Ya que las frecuencias
de estos dos modos normales son degeneradas, pueden sumarse o restarse, para
originar otros dos modos normales, esta vez a través de las direcciones diagonales
[100] ó [010]. Notar que la SEP presenta un plano de reflexión a x = y o
x = −y. Esta órbita periódica se encuentra localizada a E < 84,49 meV,
presentando un movimiento libre por encima de esta energía. Finalmente, nos
aparece una órbita circular, análogo a un movimiento de rotación frustrado, que
comienzan en las depresiones del potencial. Esta órbita describe un movimiento
localizado dentro del pozo de potencial incluso a energías superiores a Emax,
responsable de la dinámica intra-pozos. Se puede entender el movimiento de
la órbita circular a bajas energías como una combinación de las traslaciones
frustradas a través del eje x e y con diferentes fases, en analogía con el problema
del modo normal de vibración [121]. Las órbitas paralela y circular son estables
dentro del pozo de potencial, en cambio la diagonal es inestable. Cerca del mínimo
de potencial el sistema es casi integrable, y puede obtenerse información acerca
de la frecuencia fundamental de los movimientos localizados por cuantización
clásica de traslaciones paralelas. Esto puede obtenerse mediante la condición de

CAPÍTULO 4. RESULTADOS. 97
cuantización de Einstein-Brillouin-Keller [122] :
1
2Π
∮
(pxdx+ pydy) = ~(n+µ
4) , n = 0, 1, 2, ..., (4.23)
donde las integrales de acción son calculadas a través de caminos cerrados
independientes que topológicamente describen un toro en el espacio de fases, y
µ corresponde al índice de Maslov que únicamente depende de la topología de
la órbita clásica (µ=2 en nuestro caso). Para la traslación paralela, uno de los
momentos siempre es cero, entonces para este movimiento el problema se reduce
a una dimensión, y por tanto la órbita periódica es semiclásicamente cuantizada
calculando su integral de acción a lo largo de un periodo cuando se emplea otra
energía. De este modo, cuando la condición de cuantización alcanza el valor
n = 0, se obtiene un valor estimado de la frecuencia fundamental ω0 = 2E~
. El
valor obtenido es ω0 = 6,4 meV, valor que está en concordancia con el obtenido
experimentalmente para el modo T [16, 17], 6 meV. Notar que, para calcular
las diferentes energías a diferentes valores de n en (4.23), se podría obtener
una estimación de la anarmonicidad ajustando el potencial a una expansión
anarmónica de primer orden a lo largo de las direcciones x o y. Aunque hasta
la fecha no han sido recogidas medidas experimentales de anarmonicidad para
el Na/Cu(001), se han propuesto diversos modelos teóricos que tienen en cuenta
dicha anarmonicidad para explicar la dinámica que tiene lugar en la adsorción de
partículas sobre superficies [20].
Por otro lado, para los tres movimientos periódicos se han calculado las trazas
de sus respectivas matrices de estabilidad, así como se ha seguido su evolución
con la energía para detectar sus bifurcaciones y cambios de estabilidad. Esto puede
verse en la Fig. 4.
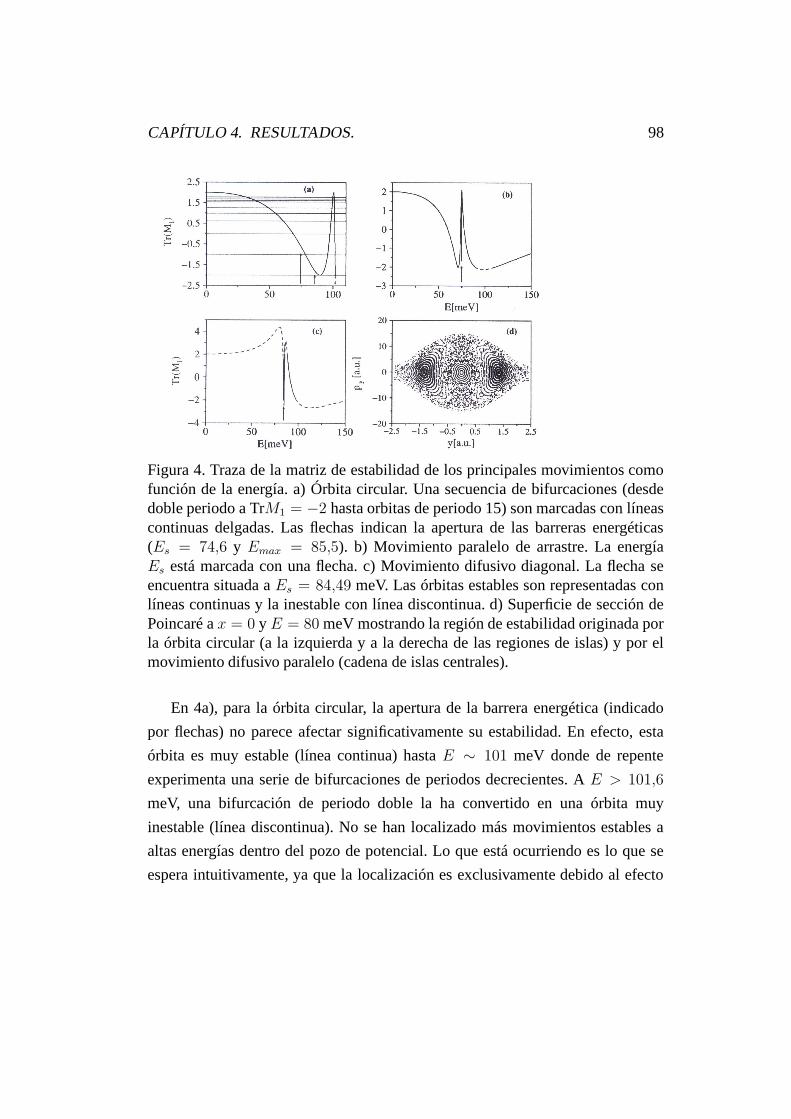
CAPÍTULO 4. RESULTADOS. 98
Figura 4. Traza de la matriz de estabilidad de los principales movimientos comofunción de la energía. a) Órbita circular. Una secuencia de bifurcaciones (desdedoble periodo a TrM1 = −2 hasta orbitas de periodo 15) son marcadas con líneascontinuas delgadas. Las flechas indican la apertura de las barreras energéticas(Es = 74,6 y Emax = 85,5). b) Movimiento paralelo de arrastre. La energíaEs está marcada con una flecha. c) Movimiento difusivo diagonal. La flecha seencuentra situada a Es = 84,49 meV. Las órbitas estables son representadas conlíneas continuas y la inestable con línea discontinua. d) Superficie de sección dePoincaré a x = 0 y E = 80 meV mostrando la región de estabilidad originada porla órbita circular (a la izquierda y a la derecha de las regiones de islas) y por elmovimiento difusivo paralelo (cadena de islas centrales).
En 4a), para la órbita circular, la apertura de la barrera energética (indicado
por flechas) no parece afectar significativamente su estabilidad. En efecto, esta
órbita es muy estable (línea continua) hasta E ∼ 101 meV donde de repente
experimenta una serie de bifurcaciones de periodos decrecientes. A E > 101,6
meV, una bifurcación de periodo doble la ha convertido en una órbita muy
inestable (línea discontinua). No se han localizado más movimientos estables a
altas energías dentro del pozo de potencial. Lo que está ocurriendo es lo que se
espera intuitivamente, ya que la localización es exclusivamente debido al efecto

CAPÍTULO 4. RESULTADOS. 99
del pozo de potencial y cuando se incrementa la energía, estos movimientos
localizados correspondientes a las partículas dentro del pozo, salen fuera del
pozo experimentando movimientos libres. Sin embargo, es interesante ver que
a energías de 15 meV por encima del máximo de potencial Emax los movimientos
localizados permanecen todavía estables. Para los movimientos rectos a través de
los ejes x, y, o diagonal [4b) y 4c) respectivamente], la apertura de la barrera
cambia su comportamiento, de estar localizado a ser difusivo, aunque la topología
de la órbita no cambia. Para el movimiento a lo largo de las direcciones azimutales
[110] y [110], la órbita se encuentra localizada y estable hasta E = Es, donde a
partir de aquí se convierte en un movimiento de arrastre libre. Este cambio, origina
en la SEP una abrupta bifurcación [marcada con flechas en 4b)] originándose de
repente una secuencia infinita de órbitas periódicas de todos los periodos a medida
que la traza de la matriz de estabilidad se va acercando al valor 2, ver (4.22). Por
tanto se generan órbitas de todos los periodos que se difunden a lo largo de las
direcciones x o y. Esto se debe a que la órbita principal permanece estable, y
por tanto se espera que aparezcan en la sección de Poincaré 4d) islas alrededor de
estructura de islas, generando una jerarquía de islas tal y como la “nested cantori”,
que según se ha visto en la literatura es un posible mecanismo para explicar la
difusión anómala en sistemas hamiltonianos [123, 124]. Después de pasar esta
región de transición de bifurcación abrupta, la órbita principal continua estable
hasta E = 89,7 meV, donde aquí sufre un periodo de doble bifurcación. Entre
este valor energético y E = 110 meV ésta permanece inestable, esto se manifiesta
como movimientos de arrastre inestables en la sección de Poincaré. Por encima
de 110 meV esta vuelve a ser estable y permanece así, hasta alcanzar el límite
integrable, tal y como se esperaba. Por último, para el movimiento diagonal a lo
largo de la dirección azimutal [100] 3c) se espera una situación similar.
La diferencia se produce debido al término no separable V2(x, y) alrededor
del mínimo, que origina una órbita altamente no lineal e inestable. La situación
de interés se produce cuando la barrera energética se abre para que esta órbita
se difunda (E = 84,5 meV), ésta coexiste con otra orbita de la misma topología

CAPÍTULO 4. RESULTADOS. 100
originada en los pequeñas depresiones que presentan los máximos del potencial,
pero que es estable.
La abrupta bifurcación origina de nuevo movimientos de arrastre de todos los
periodos, pero ahora a lo largo de las direcciones diagonales. La órbita diagonal
principal se estabiliza por estas depresiones existentes en los máximos, por tanto
para 87,8 meV ≤ E ≤ 97,5 meV es estable, y a partir de este valor energético
comienza a ser inestable, volviendo a la estabilidad a E > 150 meV, que es donde
se alcanza el límite integrable.
La estabilización de este movimiento diagonal originará largos saltos a través
de las direcciones diagonales [100] y [010]. Esto es consistente con la observación
experimental en el que se midió un alto número de partículas que se difundían
por la diagonal comparada con las que se difundían por el punto silla, por las
direcciones [110] y [110], x e y respectivamente.
4.4. Espectro de potencias y difusión anómala.
En esta sección vamos a analizar el espectro de potencias de velocidades a
las diferentes energías en las que se distinguen los diferentes comportamientos
dinámicos discutidos en la sección anterior.
Nuestro análisis refleja que para el pequeño rango de energías entre la barrera
de punto silla y el máximo (74,6 ≤ E ≤ 85,5meV), la difusión anómala domina
debido al mecanismo de captura de trayectorias, como consecuencia de que la
partícula se encuentra rodeada de islas de estabilidad que son las que originan
estos largos movimientos de arrastre.
Después de pasar esta región de transición, alrededor de 90 meV, no se puede
saber qué tipo de difusión domina, si es normal o anómala.
La difusión normal aparece cuando aumenta el caos en el espacio de fases,
manteniéndose hasta una energía E ∼ 125 meV. Aquí se vuelve a obtener
difusión anómala hasta que nos aproximamos al límite integrable, donde aparece
un régimen balístico de difusión (E ∼ 200 meV).
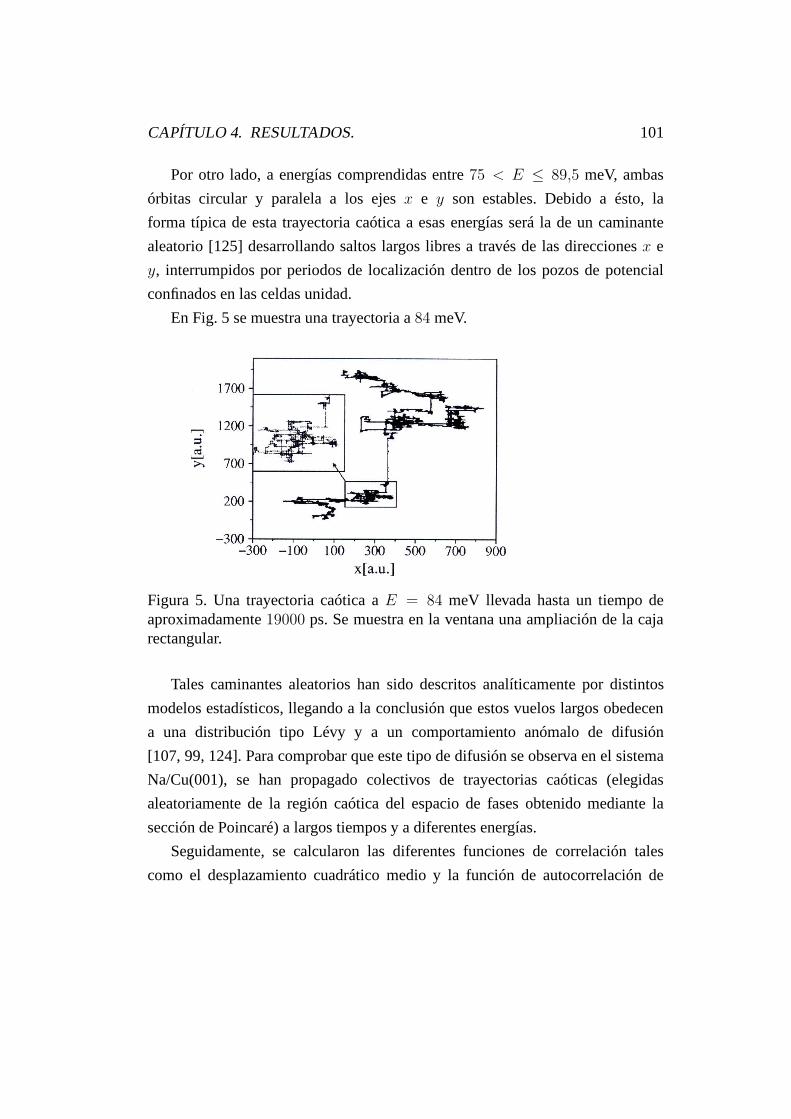
CAPÍTULO 4. RESULTADOS. 101
Por otro lado, a energías comprendidas entre 75 < E ≤ 89,5 meV, ambas
órbitas circular y paralela a los ejes x e y son estables. Debido a ésto, la
forma típica de esta trayectoria caótica a esas energías será la de un caminante
aleatorio [125] desarrollando saltos largos libres a través de las direcciones x e
y, interrumpidos por periodos de localización dentro de los pozos de potencial
confinados en las celdas unidad.
En Fig. 5 se muestra una trayectoria a 84 meV.
Figura 5. Una trayectoria caótica a E = 84 meV llevada hasta un tiempo deaproximadamente 19000 ps. Se muestra en la ventana una ampliación de la cajarectangular.
Tales caminantes aleatorios han sido descritos analíticamente por distintos
modelos estadísticos, llegando a la conclusión que estos vuelos largos obedecen
a una distribución tipo Lévy y a un comportamiento anómalo de difusión
[107, 99, 124]. Para comprobar que este tipo de difusión se observa en el sistema
Na/Cu(001), se han propagado colectivos de trayectorias caóticas (elegidas
aleatoriamente de la región caótica del espacio de fases obtenido mediante la
sección de Poincaré) a largos tiempos y a diferentes energías.
Seguidamente, se calcularon las diferentes funciones de correlación tales
como el desplazamiento cuadrático medio y la función de autocorrelación de

CAPÍTULO 4. RESULTADOS. 102
velocidades, que como ya se vio en el capítulo anterior, ambas están relacionadas
con el coeficiente de difusión, (3.82) y (3.83) respectivamente.
En la Fig. 6 se refleja el espectro de potencias de velocidades (3.87) a dos
energías diferentes, a) E = 84 meV y b) E = 80 meV.
Figura 6. Espectro de potencias de velocidades y el MSD (en las ventanas) a dosenergías diferentes: a) E = 84 meV y b) E = 80 meV. Las frecuencias estándadas en unidades de la frecuencia natural ω0.
A pequeñas frecuencias (las frecuencias han sido reescaladas a la frecuencia
armónica en el pozo de potencial ω0 = 2π√
V0/ma2), se observa el comporta-
miento predicho para la difusión anómala Z(ω)∼ ω−α con α ∼ 1/2 y α ∼ 3/4
para 84 y 80 meV, respectivamente. Por otro lado, se sabe que el MSD se relaciona
con la función de autocorrelación de velocidades a tiempos largos, (3.90). Enton-

CAPÍTULO 4. RESULTADOS. 103
ces, para una divergencia del espectro de potencias a bajas frecuencias como ω−α,
como ya se dijo en el capítulo anterior, cabría esperar una divergencia de el MSD
a tiempos largos del tipo < R2(t) >∼ t1+α para 0 < α < 1, o < R2(t) >∼ t2
(difusión balística) para α ≥ 1. Se puede ver cómo, efectivamente, en la Fig. 6
(ventana superior) la MSD calculada numéricamente se comporta como t3/2 y t7/4
para las energías de 84 meV y 80 meV respectivamente, valores que concuerdan
perfectamente con el valor predicho para la difusión anómala de t1+α. Como ya
se comentó en el capítulo anterior, existen dos modelos basados en la estadística
de Lévy para explicar este comportamiento de difusión anómala, provenientes de
la teoría desarrollada por Geisel, y del formalismo del caminante aleatorio con-
tinuo (CTRW) . En orden a verificar la validez de estos modelos estadísticos en
el sistema, se ha calculado la probabilidad de longitud de saltos φ(l) a energías
donde fue observada la difusión anómala. A estas energías el movimiento difusi-
vo se restringe a las direcciones x e y. Por tanto, se puede definir el comienzo de
un salto en x o y cuando una trayectoria cruza la barrera de activación a través
de estas direcciones, finalizando el salto cuando el momento cambia de signo a lo
largo de dichas direcciones. Entre salto y salto, la partícula se pasa un tiempo de
espera en la celda unidad donde la trayectoria experimenta un movimiento caóti-
co perdiendo así la memoria de sus condiciones iniciales. Debido a ésto, se dice
que dos saltos consecutivos son estadísticamente independientes o, lo que es lo
mismo, la partícula experimenta un proceso markoviano. Este comportamiento es
consistente con los modelos estadísticos ya mencionados.

CAPÍTULO 4. RESULTADOS. 104
Se tiene que indicar que debido a la simetría de la SEP, la distribución de
saltos en x o y es idéntica. En cambio, a altas energías, la partícula puede
moverse libremente a lo largo de cualquier dirección y, por tanto, el criterio para
la terminación de un salto libre por cambio de momento no es tan clara. Por esta
razón, se ha tenido que buscar otra vía más conveniente para contar los saltos. Esta
consiste en el cálculo de la curvatura gaussiana a cada paso de integración de la
trayectoria, y se define como:
κ =|xy − xy|
(x2 + y2)3/2(4.24)
donde el final (comienzo) de un salto se considera aquel punto de la trayectoria
donde el radio de curvatura ρ = κ−1 es menor que el valor crítico ρc. Este valor
es de alguna manera arbitrario, por lo que se elige empíricamente a cada energía
que esté de acuerdo con nuestra idea intuitiva de tiempo de vuelo. Este criterio
de curvatura fue propuesto por Sholl y Skodje cuando investigaron los vuelos de
Lévy en la difusión de Xe sobre Pt [126]. A energías por debajo del máximo de
potencial, se ha encontrado la misma distribución de saltos φ(l) usando el criterio
de cambio de momento y el criterio de curvatura para un ρc = 0,15a. Además,
para obtener una buena estadística, ha sido mejor calcular la distribución de saltos
integrada:
φ(l) =
∫ ∞
l
φ(l′)dl′ (4.25)
donde la correspondiente φ(l) se obtiene por diferenciación de φ(l). En Fig. 7,
se presenta las probabilidades integradas de saltos (se ha usado como longitud
discreta de salto el número de celdas unidad atravesadas en el salto) cuyo valor
de φ(l)∼l−3/2 y φ(l) ∼ l−5/4 corresponden a las energías de 84 y 80 meV,
respectivamente.

CAPÍTULO 4. RESULTADOS. 105
Figura 7. Distribuciones de probabilidades de salto integradas (línea continua) a)E = 84meV y b) E = 80meV. Se muestran únicamente saltos con longitudesmenores que 50 celdas unidad. Las predicciones teóricas dadas por la distribuciónde Lévy son representadas con línea discontinua.
Estas probabilidades integradas numéricamente coinciden bastante bien con
el valor teórico obtenido de los modelos estadísticos que suponen los vuelos de
Lévy para la difusión anómala en el sistema. La razón para la difusión anómala se
sustenta por islas alrededor de estructura de islas en el espacio de fases originadas
por la bifurcación abrupta del movimiento paralelo de arrastre, (ver Fig. 8).

CAPÍTULO 4. RESULTADOS. 106
Figura 8. Sección superficial de Poincaré a x = a/2 y E = 84 meV mostrando laestructura de islas alrededor de islas de la órbita principal paralela que se difunde.La ventana muestra algunas cadenas de islas, ampliación de la caja rectangular.
Ésto causa un efecto de atracción de las trayectorias caóticas por estas islas de
cantor originándose un movimiento difusivo para todo tipo de longitudes de salto.
El último test realizado para corroborar la difusión anómala ha sido calcular
los diferentes factores de estructura dinámicos S(∆K, ω) a las diferentes energías
de 80 y 84 meV respectivamente, recogiendo las correspondientes anchuras
energéticas del pico cuasielástico para un rango pequeño de ∆K, que nos
garantizase un régimen puramente difusivo.
En la Fig. 9, con símbolos se representan las FWHM obtenidas de ajustar una
lorenciana al pico cuasielástico Q obtenido numéricamente, y con línea continua

CAPÍTULO 4. RESULTADOS. 107
se representa el mejor ajuste a la FWHM teórica dada por la expresión D∆Kβ−1,
extraída de (3.100) correspondiente al factor de estructura dinámico válido a
pequeños ∆K obtenido de los modelos estadísticos de Lévy.
Figura 9. FWHM del factor de estructura dinámico numérico a pequeñastransferencias de momento paralelo ∆K, para diferentes energías. Círculos:E = 100 meV, régimen de difusión normal. Triángulos:E = 84 meV. Cuadrados:E = 80 meV. La curva continua nos muestra la ∆K - dependencia dada por(3.100).
Se puede observar que la gráfica con un ajuste de α = 2 corresponde,
efectivamente al régimen de difusión normal a 100 meV, mostrado con círculos.
Por otro lado, para energías superiores a 90 meV (hasta ∼120 meV, todavía
lejos del límite integrable) se ha observado únicamente difusión normal.
Este comportamiento puede verse en la Fig. 10a) y 10b).

CAPÍTULO 4. RESULTADOS. 108
Figura 10. a) Espectro de potencias de velocidad a E = 95 meV, con frecuenciasen unidades de ω0. b) Línea continua: Probabilidad integrada de longitudes desalto a la misma energía. Línea discontinua: probabilidad integrada de duraciónde saltos. Ambas dan un decaimiento exponencial, y sus pendientes coinciden conlas predicciones estadísticas.
En la Fig. 10a, se observa cómo el espectro de potencias converge a un valor
finito cuando ω → 0 o, lo que es lo mismo, el MSD crece linealmente con el
tiempo. Mientras que en Fig. 10b, usando el criterio de curvatura, se ha encontrado
un comportamiento exponencial para las probabilidades de longitudes de salto,

CAPÍTULO 4. RESULTADOS. 109
φ(l) ∼ exp−γl, pudiendo observar cómo coincide con la probabilidad de duración
de salto ϕ(t). El acuerdo entre las probabilidades de longitud de salto y de tiempo
de salto es un argumento adicional a favor de los modelos estadísticos empleados,
ya que ambos suponen que durante el salto la velocidad se mantiene constante. La
difusión normal, encontrada a estas energías, tiene su justificación en el cambio de
estabilidad producida en la órbita paralela principal como puede verse en la Fig.
4 a E ∼ 110 meV. En este rango de energías donde se ha observado la difusión
normal, se ha calculado numéricamente el coeficiente de difusión D usando la
relación de Einstein, (3.82), que la relaciona con la función de autocorrelación de
velocidades, ec. (3.90), para mayor fiabilidad. Puede observarse en Fig. 11 una
trayectoria caótica a E = 100 meV llevada a t ∼ 20000 ps.
Figura 11. Una trayectoria caótica a E = 100 meV integrada hasta un tiempo de20000 ps (tenemos que indicar que en los dos ejes, la longitud de la celda unidades 5 a.u.
Se puede ver que este régimen de difusión normal corresponde a un
movimiento browniano o, lo que es lo mismo, puede simularse por el movimiento
de un caminante aleatorio en el que se caracteriza por saltos cortos entre diferentes
celdas unidad separado de pequeños periodos en los que la partícula se encuentra

CAPÍTULO 4. RESULTADOS. 110
atrapada en en el pozo de potencial de la celda unidad. Por tanto, las longitudes
de salto promedios así como los tiempos de espera dentro de una celda unidad
son finitos. En Fig. 12 se muestra el espectro de potencias de velocidades, y en la
ventana el MSD numéricamente.
Figura 12. Espectro de potencias de velocidades a E = 100 meV. En la ventanase muestra el MSD a la misma energía.
Estas magnitudes se han calculado propagando condiciones iniciales de un
colectivo de ∼1000 trayectorias elegidas aleatoriamente de la región caótica
recogida en la Fig. 13.

CAPÍTULO 4. RESULTADOS. 111
Figura 13. Superficie de sección de Poincaré para x0 = a/2 y E = 100 meV.
Se observa en la Fig. 12 que cuando ω → 0 esta converge a un valor constante
2D, dado por (3.88), mientras que en la ventana se observa que la MSD es lineal
con el tiempo de acuerdo con la relación de Einstein, (3.82).
En Fig. 14 se muestra el factor de estructura dinámico S(∆K, ω) a dos
diferentes valores de transferencia de momento paralelo.
Figura 14. Factores de estructura dinámicos obtenidos de un colectivo microca-nónico de trayectorias a E = 100 meV. Curva continua: ∆K = 0,14 Å
−1. Curva
discontinua: ∆K = 3,34 Å−1
.

CAPÍTULO 4. RESULTADOS. 112
A valores pequeños de ∆K (curva continua) se obtiene un estrecho pico
lorenciano a ω = 0, debido al movimiento de difusión caótico medido a tiempos
largos y por tanto trayectorias que experimentan un largo recorrido libre medio.
Por otro lado, a valores de ∆K grandes (curva discontinua) donde el máximo
recorrido libre medio detectado es inferior a la longitud de una celda unidad a,
la difusión no llega a observarse, pero en su lugar se observan las primeras y
segundas excitaciones inelásticas debido a las vibraciones del adsorbato (modo
T). Por otro lado, en este régimen de difusión normal, el coeficiente de difusiónD
calculado numéricamente, se relaciona con algunas aproximaciones estadísticas
teóricas como la proveniente del formalismo del caminante aleatorio, bajo la
condición markoviana, en la que los saltos no están correlacionados, (3.85) y,
con la proveniente de la teoría cinética de los gases [127] :
D =1
2< v > l (4.26)
donde < v > es la velocidad promedio y l el libre recorrido medio. En la Fig. 15,
se comparan los resultados numéricos y los obtenidos por ambas aproximaciones.
La frecuencia de salto ν ha sido calculada numéricamente como el número total
de saltos dividido por el tiempo total. También decir que para contar un salto, se
requiere que haya atravesado al menos una celda unidad. Puede verse en la Fig.
15 cómo alrededor de E = 110 meV el coeficiente de difusión D experimenta un
incremento abrupto , debido a la estabilización de la órbita paralela principal que
se difunde, que hace que la partícula experimente saltos más largos.

CAPÍTULO 4. RESULTADOS. 113
Figura 15. Coeficientes de difusión microcanónico para un rango de energíacomprendido entre 95 y 120 meV. Curva sólida: Coeficiente de difusiónexacto proveniente de un ajuste a las relaciones de Einstein usando colectivosmicrocanónicos. Curva discontinua: Aproximación utilizando la Ecuación de lateoría cinética de gases. Curva discontinua con puntos: Aproximación utilizandovuelos aleatorios.
4.5. Influencia del ruido gaussiano blanco.
La suposición en la que el sistema (adsorbato) se encuentra en equilibrio
térmico con el sustrato sometido a un ruido gaussiano blanco gaussiano (delta
correlado) [128, 129] y la presencia de dos escalas de tiempos muy diferentes
(τr τc) hacen que podamos resolver la dinámica del sistema mediante
una ecuación de Langevin bajo la aproximación markoviana que consiste en
suponer una fricción constante. Por otro lado, para simular la difusión anómala
puede usarse una ecuación de Langevin, pero ahora, teniendo en cuenta algunas
consideraciones. Por ejemplo, suponer un ruido gaussiano coloreado da lugar a
la superdifusión y subdifusión a tiempos largos [130], incluso en la ausencia
de disipación [131]. En el caso de suponer un ruido coloreado, los tiempos

CAPÍTULO 4. RESULTADOS. 114
característicos ya no son tan diferentes, y por tanto la fricción deja de ser
constante (aproximación no markoviana), dando lugar a una ecuación de Langevin
generalizada [132]. En la mayoría de los casos el ingrediente físico básico
es que el sistema no se encuentre en equilibrio térmico (por ejemplo, si se
tiene una fuente externa de ruido y el teorema de fluctuación-disipación no se
mantiene, o el proceso estocástico aquí considerado se convierte en un proceso
no markoviano debido al tipo de función de memoria que se emplee). Aparte de
este comportamiento asintótico formal, es importante darse cuenta que, incluso
si la solución estacionaria para la densidad de probabilidad de la difusión del
proceso existe, la dinámica de la relajación para alcanzar este proceso estacionario
puede ser muy diferente dependiendo de los parámetros que definan el sistema.
En esta sección se ha tratado de estudiar este dinámica de relajación dentro del
formalismo clásico de Langevin, y clarificar el papel del potencial adiabático así
como entender la dinámica caótica en el proceso de relajación y de la influencia
del ruido y la disipación sobre esta dinámica determinista.
Dicho ésto, se ha comprobado que cuando se introduce disipación, las órbitas
periódicas estables se convierten en sumideros o centros de atracción [133] y
los toros invariantes (islas) son destruidos. Sin embargo, la existencia de una
fuerza externa, incluso si es aleatoria, proporcionará energía suficiente para que
los adsorbatos escapen de este atractor originando un movimiento difusivo. La
adición de un ruido débil a esta dinámica determinista no lineal del sistema
puede ocasionar cambios de consideración en sus propiedades de estabilidad local,
pudiendo conservar la estabilidad global [134]. El ruido también influye en las
propiedades de transporte y puede inducir transiciones entre diferentes estados
estables [135]. Aquí se ha asumido el modelo más simple posible para incluir la
fricción y el ruido (temperatura), que no es más que la inclusión de un ruido blanco
(delta correlado) gaussiano y una fricción constante, o lo que es lo mismo una
función de fricción también delta correlada, que origina un proceso markoviano y
cuya dinámica viene descrita por una ecuación de Langevin ordinaria. En orden
a evitar fuertes perturbaciones en la dinámica determinista, se mantiene baja la
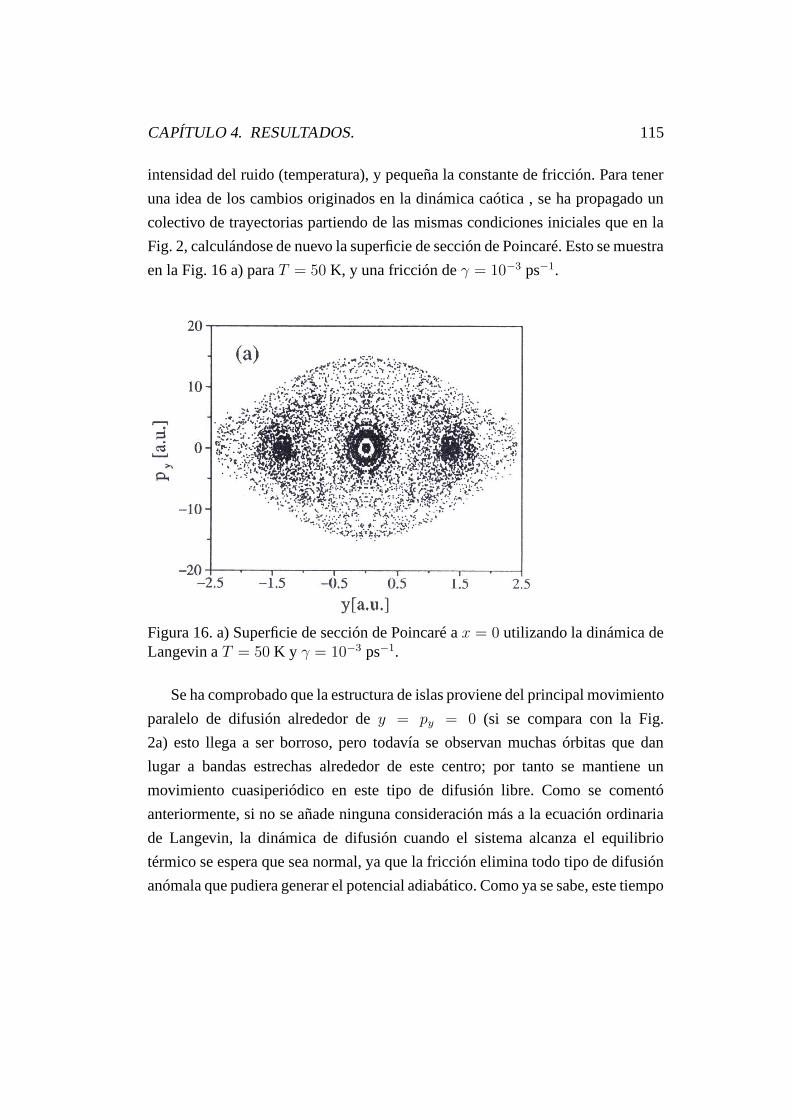
CAPÍTULO 4. RESULTADOS. 115
intensidad del ruido (temperatura), y pequeña la constante de fricción. Para tener
una idea de los cambios originados en la dinámica caótica , se ha propagado un
colectivo de trayectorias partiendo de las mismas condiciones iniciales que en la
Fig. 2, calculándose de nuevo la superficie de sección de Poincaré. Esto se muestra
en la Fig. 16 a) para T = 50 K, y una fricción de γ = 10−3 ps−1.
Figura 16. a) Superficie de sección de Poincaré a x = 0 utilizando la dinámica deLangevin a T = 50 K y γ = 10−3 ps−1.
Se ha comprobado que la estructura de islas proviene del principal movimiento
paralelo de difusión alrededor de y = py = 0 (si se compara con la Fig.
2a) esto llega a ser borroso, pero todavía se observan muchas órbitas que dan
lugar a bandas estrechas alrededor de este centro; por tanto se mantiene un
movimiento cuasiperiódico en este tipo de difusión libre. Como se comentó
anteriormente, si no se añade ninguna consideración más a la ecuación ordinaria
de Langevin, la dinámica de difusión cuando el sistema alcanza el equilibrio
térmico se espera que sea normal, ya que la fricción elimina todo tipo de difusión
anómala que pudiera generar el potencial adiabático. Como ya se sabe, este tiempo

CAPÍTULO 4. RESULTADOS. 116
de relajación al equilibrio viene dado por τr = γ−1. Si la constante de fricción
es bastante pequeña, es posible que el tiempo de equilibrio térmico sea muy
superior al tiempo en el que se equilibre la dinámica caótica, por lo que, bajo
estas circunstancias, se podría observar una región transitoria en el MSD o en el
espectro de potencias donde el potencial adiabático jugaría un papel importante.
Por tanto, para comprobar esto, se ha calculado el MSD y espectro de potencias
de un colectivo canónico de trayectorias con los valores de temperatura y fricción
usados en Fig. 16 a) y partiendo de una distribución inicial de Maxwell de
velocidades, (ver Fig. 16 b).
Figura 16. b) En la ventana se muestra el espectro de potencias de velocidades yla MSD para un colectivo de 1000 trayectorias con una distribución de Maxwellde velocidades a la misma temperatura.
Por tanto, se sabe que el tiempo de equilibrio térmico es del orden de 103 ps,
mientras que el equilibrio dinámico se alcanza en 100 ps. Esto puede observarse
claramente en la ventana, donde se distinguen dos pendientes claramente
diferenciadas para el MSD, la primera da una difusión anómala pasajera generada
por la influencia del potencial adiabático, y la otra la difusión normal, como
consecuencia de que el adsorbato ha alcanzado el equilibrio térmico, y por lo tanto

CAPÍTULO 4. RESULTADOS. 117
la fricción a partir de este tiempo llega a ser importante, neutralizando todo efecto
dinámico ocasionado inicialmente por el potencial. Este mismo comportamiento
se observa en el espectro de potencias. Ya que la escala de tiempos en el
experimento es finita, esta difusión anómala pasajera podría ser observada en
algunos sistemas (en nuestro caso, γ fue medido a 1 ps−1[16, 17], por tanto la
difusión anómala no puede ser vista).
4.6. Teoría de Kramers aplicada a la difusión de
adsorbatos por superficies.
En esta sección vamos a mostrar como la teoría de Kramers se puede
generalizar a la difusión de adsorbatos en superficies, proporcionando un buen
método teórico para analizar los resultados experimentales por QHAS.
4.6.1. Teoría unidimensional.
En [136, 137, 9] ha sido desarrollada una teoría clásica y semiclásica para
explicar la difusión activada en superficies en una dimensión. Esta es una teoría
que generaliza la solución de Kramers para el problema de escape de una partícula
dentro de un potencial metaestable [7], extendiéndola a una superficie [138, 9, 8].
La teoría es válida para todo el rango de fricción, desde el régimen de difusión
energética al régimen de difusión espacial (alta fricción) bajo las siguientes
condiciones:
1) La dinámica es descrita por una ecuación de Langevin.
2) Las barreras energéticas del potencial adiabático son altas (V ‡/(KBT ) 1).
3) La forma del potencial en el máximo de la barrera energética es parabólica,
con un frecuencia ω‡.
4) La pérdida de energía debido a la interacción de la partícula con el baño
térmico cuando ésta cruza la SEP desde una barrera hasta la siguiente viene dada

CAPÍTULO 4. RESULTADOS. 118
por la mecánica clásica.
Teniendo en cuenta estas consideraciones, el punto de partida para la
evaluación de la frecuencia de salto, distribución de salto y el coeficiente de
difusión proviene de la siguiente ecuación del flujo estacionario de partículas que
escapan de un pozo de potencial para caer en otro:
f+j (ε) =
∫ ∞
−∞dε′P (ε|ε′)[θ(−ε′)f−
j (ε′) + f+j−1(ε
′)θ(ε′)] (4.27)
donde f+j (f−
j ) representa el número de partículas por unidad de energía y por
unidad de tiempo golpeando la barrera derecha (izquierda) del jth pozo con
velocidad positiva (negativa), θ(x) la función escalón, y el núcleo P(ε|ε′) da la
probabilidad de que la partícula cambie su energía de ε a ε′ cuando ésta pasa de
un pozo de potencial al siguiente. Se ha demostrado que el núcleo presenta una
forma gaussiana [138, 9]:
P (ε|ε′) =1√4πδ
exp[−(ε− ε′ + δ)2
4δ] (4.28)
donde δ es la pérdida de energía promedio de la partícula al baño. Para un
amortiguamiento y altura de la barrera de primer orden, la perdida de energía
es simplemente δ = γS/(kBT ), donde S es la acción clásica de la trayectoria la
cual cruza una celda unidad con una energía igual a la barrera energética. Para la
SEP adiabática dada por (4.17) con altura de la barrera V ‡ = 2V0, la pérdida en
energía viene dada por:
δ =8V0γ
kBTω0(4.29)
donde ω0 es la frecuencia armónica de oscilación en el pozo de potencial
(frecuencia natural del sistema). Por otro lado, para obtener velocidades de escape
y distribuciones de salto, las frecuencias parciales Γj se definen como el número
de partículas por unidad de tiempo que salen de un pozo j = 0 y son atrapadas en
el pozo jth. Estas velocidades se obtienen como la diferencia entre el flujo entrante
y saliente de un pozo:

CAPÍTULO 4. RESULTADOS. 119
Γj =
∫ ∞
0
dε[f+j−1(ε) + f−
j+1(ε) − f−j (ε) − f+
j (ε)] (4.30)
Finalmente, uno necesita resolver (4.27) y (4.30) sujetas a la condición de
contorno:
f+j (ε) ' δj0
ω0λ‡
2πω‡ e−(ε+V ‡) ε→ −∞ (4.31)
la cual implica que, inicialmente, todas las partículas se encuentran en el pozo
j = 0 con una distribución de energía térmica situada en el mínimo de potencial.
Aquí ω‡ representa la frecuencia en el punto máximo de la barrera armónica V ‡.
Notar también que el prefactor de Kramers-Grote-Hynes viene dado por:
λ‡
ω‡ =
√
1 +γ2
4ω‡ −γ
2ω‡ (4.32)
donde aparece como un factor de normalización sin tener en cuenta el recruza-
miento, es decir, que la partícula una vez que escapa la celda unidad ya no vuelve.
Esta suposición es siempre válida si se trabaja en sistemas de coordenadas nor-
males para la partícula que se difunde y para el baño térmico [138]. Por otro lado,
(4.27) y (4.30) con la condición de contorno (4.31) se resuelven por transforma-
da de Fourier discreta seguida por una transformada de Laplace en energía [136].
Obteniéndose finalmente la siguiente expresión para la frecuencia parcial:
Γj = −ΓsdΠ
∫ 2π
0
dk sin2(k
2) cos(jk)exp 2
π
∫ π/2
0
dx ln[1 − P 2(x)
1 + P 2(x) cos(k)]
(4.33)
donde Γsd es la velocidad de escape de difusión espacial [7]:
Γsd =λ‡
ω‡ω0
πexp[−V ‡/(kBT )] (4.34)
donde el factor ΓTST = ω0
πexp[−V ‡/(kBT )] es la denominada velocidad de
escape dada por la teoría del estado de transición (TST) [65] , y la función P (x)

CAPÍTULO 4. RESULTADOS. 120
viene dada por:
P (x) = exp[− δ
4 cos2(x)] (4.35)
La velocidad de escape desde el pozo de potencial (j = 0) es:
κ = −Γ0 (4.36)
Entonces, la probabilidad relativa para un salto de longitud j está dada por la
probabilidad de que la partícula sea atrapada en el pozo de potencial jth:
Pj =Γjκ
(4.37)
Para un potencial unidimensional, el coeficiente de difusión está relacionado con
la velocidad de escape [139] por:
D =1
2κ < l2 >=
1
2a2
∞∑
j=−∞j2Γj (4.38)
donde < l2 > es la longitud del recorrido libre medio. Introduciendo (4.33) en
(4.38) , el coeficiente de difusión adopta la forma:
D = DsdΥ−1 exp 2
Π
∫ Π/2
0
dx ln[1 + P (x)] (4.39)
donde Dsd ≡ 1/2a2Γsd es el coeficiente de difusión en el régimen de difusión
espacial, y Υ es el factor de población para el pozo metaestable dado por
Mel’nikov [9]:
Υ = exp 2
π
∫ π/2
0
dx ln[1 − P (x)] (4.40)
En analogía con el modelo de Chudley-Elliot, se puede obtener una expresión
analítica para el factor de estructura dinámico imponiendo una ecuación maestra
para la función dependiente del tiempo Gl(t), la cual nos da la probabilidad que

CAPÍTULO 4. RESULTADOS. 121
la partícula esté a un tiempo t en el sitio l, encontrándose inicialmente a t = 0
en el pozo j = 0 de potencial. Usando las velocidades parciales Γj , esta ecuación
maestra adopta la forma [137]:
dGl(t)
dt=
∞∑
j=−∞ΓjGl−j(t) (4.41)
la cual es resuelta por transformada de Fourier. Por tanto, definiendo:
Γ(κ) =
∞∑
j=−∞Γje
ikj (4.42)
y usando (4.33), obtenemos finalmente una anchura angular dada por:
Γ(κ) = 4Γsd sin2(κ
2) exp 2
π
∫ π/2
0
dx ln[1 − P 2(x)
1 + P 2(x) − 2P (x)cos(κ)] (4.43)
La ec.(4.43) es importante en el sentido que, asumiendo la validez del modelo de
Kramers y la aproximación de la ecuación maestra, nos permite una comparación
directa con los resultados experimentales y por tanto una estimación de la
velocidad de difusión espacial Γsd, y de la pérdida energética δ. Por tanto a partir
de estos parámetros y de su dependencia con la temperatura se puede obtener la
altura de la barrera V ‡, el coeficiente de fricción γ , y la frecuencia en la barrera
de potencial ω‡.
4.6.2. Teoría multidimensional.
La teoría de Kramers ha sido generalizada bajo ciertas limitaciones a muchas
dimensiones, [140, 141]. Las ecuaciones finales son formalmente equivalentes
a las del caso unidimensional, pero difieren en dos aspectos. El primero, la
velocidad de difusión espacial depende de las matrices de constantes de fuerza
en la barrera y en el pozo, denotadas por Ω‡ y Ω0, respectivamente:

CAPÍTULO 4. RESULTADOS. 122
Γ2Dsd =
1
π[det(Ω0)
det(Ω‡)]1/2λ exp[−V ‡/(kBT )] (4.44)
La frecuencia de la barrera de potencial en la representación de coordenadas de
modos normales λ‡ es la solución positiva de la ecuación secular:
det(λ‡2
I + λ‡γ + Ω‡) = 0 (4.45)
donde I es la matriz identidad 2 × 2 y γ es la matriz diagonal de fricción,
cuyos elementos son los coeficientes de fricción a lo largo de las direcciones
estables e inestables. La otra diferencia se encuentra en la pérdida energética:
el parámetro δ que aparece en el núcleo gaussiano en (4.28) depende ahora de las
condiciones iniciales. Estrictamente hablando, la pérdida energética de los estados
iniciales y finales en las barreras energéticas de la celda unidad, ε y ε′ deberían ser
promediadas sobre todas las trayectorias iniciadas en la barrera de salida con una
distribución térmica de energía preestablecida. Por tanto, para aplicar la teoría
multidimensional de Kramers, tenemos que calcular numéricamente el parámetro
de pérdida energética, el cual puede ser igual de costoso computacionalmente que
resolver la velocidad con la ecuación exacta de Langevin. Por otro lado, se puede
establecer una estimación de este parámetro de pérdida energética en dos límites.
En el primer límite, los dos grados de libertad son fuertemente acoplados a través
de la SEP. El movimiento en la región del pozo es ergódica y entonces la pérdida
de energía resulta ser proporcional a [V ‡/(kBT )]2. El segundo límite es suponer
un acoplamiento débil, en el que la pérdida energética para el potencial separable
dada por (4.29) proporcional a V ‡/(kBT ) es una buena aproximación.
4.6.3. Corrección de barrera finita.
Cuando la altura de la barrera del potencial es V ‡/(kBT ) ≤ 5, se debe incluir
en la teoría correcciones de barrera finita (fbc) para las expresiones de la velocidad
[142, 143, 144]. Las correcciones para la velocidad de difusión espacial son
obtenidas usando el método de flujo reactivo en el cual la elección de división de

CAPÍTULO 4. RESULTADOS. 123
la superficie se elige minimizando el flujo en el estado de transición. Los detalles
de esta derivación se pueden ver nuevamente en los trabajos[142, 143, 144]. Aquí
se proporcionan las fórmulas 2D para los potenciales separables y no separables.
Por tanto para el potencial unidimensional, o para el potencial separable en dos
dimensiones, se tiene que:
Γfbc 'λ‡
ω‡ω0
πe−[V ‡/(kBT )][1− 1
8β(
1
χ2
V(4)x (x = a/2)
[V(2)x (x = a/2)]2
− V(4)x (x = 0)
[V(2)x (x = 0)]2
)] (4.46)
donde V (n)x denota la nth derivada parcial del potencial a través de la coordenada
de la reacción x, y χse define como un parámetro de no linealidad [30]. En un
camino similar, para un potencial 2D no separable, uno encuentra que el término
de corrección de barrera finita más importante viene dado por:
Γ2Dfbc ' Γ2D
sd [1− 1
4β(
V(4)x (a/2, 0)
2χ2[V(2)x (a/2, 0)]2
+V
(4)y (a/2, 0)
2χ2[V(2)y (a/2, 0)]2
+V
(2,2)x,y (a/2, 0)
3χV(2)x (a/2, 0)V
(2)y (a/2, 0)
−
(4.47)
V(4)x (0, 0)
2[V(2)x (0, 0)]2
− V(4)y (0, 0)
2[V(2)y (0, 0)]2
− V(2,2)x,y (0, 0)
3χV(2)x (0, 0)V
(2)y (0, 0)
]
donde ahora V (2,2)x,y denota la derivada parcial cruzada de segundo orden en ambas
direcciones. Excepto donde se manifiesta de otra forma, todos los resultados de
Kramers en este trabajo llevan incluidos las correcciones de barrera finita para la
velocidad de difusión espacial. Por último decir que en el régimen de difusión
energética, es decir para V ‡/(kBT )≥ 3, tales correcciones son pequeñas y por
tanto no se tienen en cuenta.

CAPÍTULO 4. RESULTADOS. 124
4.6.4. Resultados Numéricos.
4.6.4.1. Potencial separable.
Como una primera demostración de la relevancia de la teoría de Kramers, se
ha empleado como potencial el coseno separable dado en (4.17) para las primeras
simulaciones numéricas con una barrera de potencial de 67 meV [27, 15]. Se ha
calculado las velocidades de escape, los coeficientes de difusión, distribuciones
de salto y el factor de estructura dinámico resolviendo la ecuación de Langevin
partiendo de un colectivo inicial de partículas termalizadas en el mínimo del pozo
de potencial. Se han usado dos aproximaciones numéricas para determinar tales
velocidades:
1. La primera está basada en el primer tiempo de pasaje promedio τMFPT
de trayectorias que están sujetas a unas condiciones de contorno previamente
establecidas. Dicho de otro modo, se sabe que en una dimensión, el potencial
periódico presenta dos salidas equivalentes por donde pueden escapar las
partículas encerradas en un pozo de potencial. Esta velocidad de escape está
relacionado con el τMFPT por la siguiente relación:
κ =1
2τMFPT
(4.48)
Esta relación entre el τMFPT y la velocidad de escape, ha sido demostrada para
el caso especial en el que se considera un ruido blanco [8], como el caso que nos
ocupa.
2. La otra estrategia numérica consiste en determinar los saltos de forma
explícita. Las velocidades son entonces obtenidas como el número total de saltos
del colectivo, dividido por el tiempo total de propagación. Numéricamente, se ha
empleado el criterio de que un salto comienza cuando la partícula deja la celda
unidad, y para la conclusión de dicho salto se han empleado dos criterios:
a) El salto finaliza cuando la energía de la partícula es mucho menor que una
energía preestablecida por debajo del pozo de potencial [118, 36].
b) El salto termina cuando el tiempo de residencia dentro del pozo de potencial

CAPÍTULO 4. RESULTADOS. 125
es mayor que el tiempo de relajación dentro de dicho pozo.
Estos criterios se eligen para asegurar una termalización de las partículas
dentro del pozo de potencial antes de comenzar un nuevo salto. En Fig. 17 se
muestran las velocidades de escape calculadas a dos temperaturas diferentes: a
T = 110 K, V ‡/(kBT )∼ 7 y a T = 200 K, V ‡/(kBT ) ∼ 3,9, ambos casos se
encuentran dentro del régimen activado.
Figura 17. Velocidades de escape (en ps−1) como una función de la fricciónescalada para el potencial separable coseno, a dos temperaturas de superficie:panel izquierdo , T = 110 K, y panel derecho, T = 200 K. Los resultadosde Kramers (línea continua y discontinua) son obtenidos por ecs.(4.33)-(4.36)sin y con correcciones de barrera finita, respectivamente. Los resultados porτMFPT son representados con círculos abiertos y los cuadrados negros son lascorrespondientes velocidades numéricas obtenidas por el criterio de conteo desaltos.
Los resultados de τMFPT se muestran con círculos abiertos, los cuadrados
negros con barras de error muestran las velocidades obtenidas por conteo de
saltos, la línea sólida son los resultados teóricos predichos por (4.33) y (4.36),
y la línea discontinua la corrección de barrera finita. Se puede apreciar una
ligera diferencia pero importante entre la teoría de Kramers (línea continua) y
la simulación numérica (círculos y cuadrados). Esta diferencia es debida a que la
altura de la barrera adiabática no es suficientemente grande y uno debe tomar
correcciones de barrera finita (línea discontinua) para el régimen de difusión
espacial. Como se puede ver, incluyendo esta corrección de barrera finita la

CAPÍTULO 4. RESULTADOS. 126
teoría coincide bastante bien con los resultados numéricos para todo el rango de
fricción.También se ha investigado los coeficientes de difusión para este amplio
rango de fricción. Las predicciones analíticas para el coeficiente de difusión en
un potencial periódico, (4.39), se encuentran sólo disponibles para el régimen de
difusión espacial [145, 146, 147, 148, 115], en el que se supone únicamente saltos
a los vecinos más cercanos. En la Fig. 18 la predicción teórica dada por (4.39) es
representada con línea continua.
Figura 18. Coeficientes de difusión (en cm2/s) como una función de la funciónescalada para el potencial coseno separable a dos temperaturas de superficie, 110 y200 K . Línea continua: Resultados de Kramers dados por (4.39) con correccionesde barrera finita. Línea discontinua: Estimación analítica a alta fricción, (4.49).Cuadrados negros: Resultados numéricos usando (4.38) y los datos de Langevinpara la obtención de la velocidad de escape y longitud de recorrido cuadráticomedio.
Por otro lado, los coeficientes de difusión numéricos son obtenidos de dos
formas posibles: a través de las velocidades y las longitudes del recorrido libre
medio, (4.38) (cuadrados negros), y usando la relación de Einstein a tiempos
largos, (3.82) (círculos abiertos). Además, también se ha mostrado con la línea
discontinua la predicción analítica válida en el régimen de Smoluchowski [145,
146, 115]:
Dx =D0a
2
∫ a
0dxeβV (x)
∫ a
0dxe−βV (x)
, (4.49)
donde D0 es el coeficiente de difusión en la ausencia de potencial, D0 =
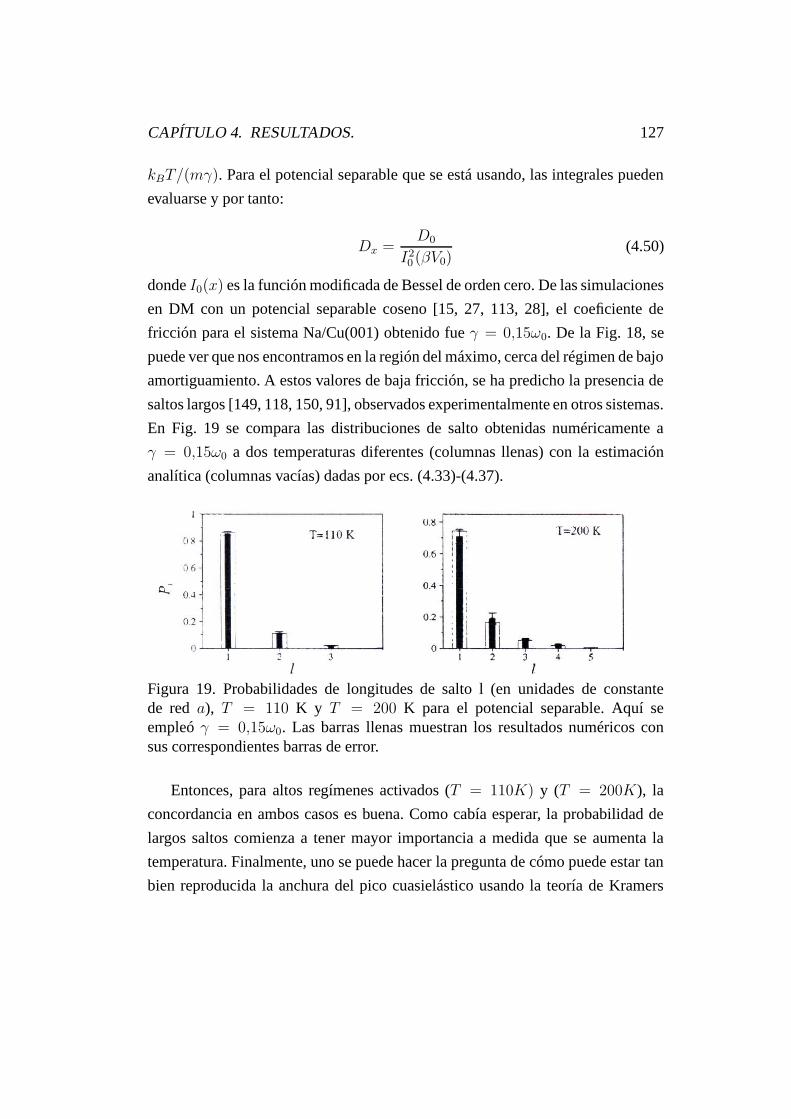
CAPÍTULO 4. RESULTADOS. 127
kBT/(mγ). Para el potencial separable que se está usando, las integrales pueden
evaluarse y por tanto:
Dx =D0
I20 (βV0)
(4.50)
donde I0(x) es la función modificada de Bessel de orden cero. De las simulaciones
en DM con un potencial separable coseno [15, 27, 113, 28], el coeficiente de
fricción para el sistema Na/Cu(001) obtenido fue γ = 0,15ω0. De la Fig. 18, se
puede ver que nos encontramos en la región del máximo, cerca del régimen de bajo
amortiguamiento. A estos valores de baja fricción, se ha predicho la presencia de
saltos largos [149, 118, 150, 91], observados experimentalmente en otros sistemas.
En Fig. 19 se compara las distribuciones de salto obtenidas numéricamente a
γ = 0,15ω0 a dos temperaturas diferentes (columnas llenas) con la estimación
analítica (columnas vacías) dadas por ecs. (4.33)-(4.37).
Figura 19. Probabilidades de longitudes de salto l (en unidades de constantede red a), T = 110 K y T = 200 K para el potencial separable. Aquí seempleó γ = 0,15ω0. Las barras llenas muestran los resultados numéricos consus correspondientes barras de error.
Entonces, para altos regímenes activados (T = 110K) y (T = 200K), la
concordancia en ambos casos es buena. Como cabía esperar, la probabilidad de
largos saltos comienza a tener mayor importancia a medida que se aumenta la
temperatura. Finalmente, uno se puede hacer la pregunta de cómo puede estar tan
bien reproducida la anchura del pico cuasielástico usando la teoría de Kramers

CAPÍTULO 4. RESULTADOS. 128
Figura 20. FWHM (en µeV ) del factor de estructura dinámico como unafunción de la transferencia del vector de onda a lo largo de la dirección x,Kx para el potencial separable coseno a T = 110 y 200 K y γ = 0,15ω0.Línea continua: resultados de Kramers, ec. (4.43). Línea discontinua: modelode Chudley-Elliot, ec. (3.75) utilizando las velocidades y distribuciones de saltocalculadas numéricamente. Círculos cerrados: resultados numéricos obtenidosajustando el factor de estructura dinámico , ec. (3.65) a un perfil lorenciano.
y el modelo de difusión de saltos. Los resultados son presentados en Fig. 20 a
γ = 0,15ω0 a las diferentes temperaturas ya mencionadas anteriormente.
Las líneas continuas son las predicciones teóricas y las líneas discontinuas
corresponden al modelo de Chudley-Elliot, utilizando las velocidades de salto
y distribuciones de salto obtenidas numéricamente. Con círculos cerrados se
representa la FWHM obtenida del mejor ajuste lorenciano al factor de estructura
dinámico obtenido numéricamente. El buen acuerdo entre la anchura “exacta” y
el modelo de Chudley-Elliot a T = 110 K nos demuestra que para altas barreras
energéticas la aproximación de difusión discreta por saltos es bastante buena. En
cambio, cuando V ‡/kBT < 4 esta aproximación se deteriora, como se observa
en la figura a 200 K. Además, a valores de transferencia de momento paralelo
grandes, se aprecia que no se puede obtener difusión pura, pues la contribución
vibracional comienza a influir bastante. Debido a ésto, a ambas temperaturas se
observa una pequeña discrepancia entorno a kx = π, además que a partir de este
valor, y debido al solapamiento del modo T con el pico Q, un ajuste lorenciano
a este pico cuasielástico es más que cuestionable. Por último atribuir, que la no

CAPÍTULO 4. RESULTADOS. 129
tan buena concordancia a 200 K entre el modelo de Chudley-Elliot y el ajuste
lorenciano puede deberse a la aparición considerable de saltos dobles, no tenidos
en cuenta en el modelo.
4.6.4.2. Potencial no separable.
De la sección previa se concluye que, para V ‡ ≥ 4kBT , la teoría de Kramers
nos proporciona una buena aproximación a la dinámica exacta obtenida de la
simulación numérica por Langevin. Todas las cantidades las cuales pueden ser
de importancia experimental son precisamente estimadas siempre y cuando la
aproximación mediante la ecuación de Langevin con fricción óhmica presente una
buena descripción de la dinámica adsorbato-sustrato, como ocurre en el sistema
bajo estudio. En esta sección se va a analizar la teoría para el problema en dos
dimensiones, esto es, cuando los grados de libertad x e y están acoplados. El
potencial que se emplea en el sistema es el que se utiliza para las comparaciones
numéricas y analíticas. Como ya se mencionó, la aplicación de la teoría de
Kramers al caso multidimensional no es tan satisfactoria como para el caso de
una dimensión. Para aplicar la teoría de Kramers, se necesita una estimación de
la pérdida de energía promedio δ que aparece en el núcleo de la probabilidad
gaussiana. La teoría es realmente aplicable sólo si el acoplamiento entre los
adsorbatos y el sustrato es débil o fuerte. Para este fin es necesario entender la
dinámica clásica en la ausencia de fricción. Como ya se vio en una sección previa,
un estudio detallado de la dinámica clásica de las partículas de Na moviéndose
en esta particular SEP ha mostrado que para energías por encima del punto silla
la dinámica caótica juega un papel importante [14, 35]. Por tanto, encontrando
las órbitas periódicas más simples del sistema y estudiando su evolución con
la energía se puede conseguir una buena estimación de la irregularidad de la
dinámica clásica a cualquier energía.

CAPÍTULO 4. RESULTADOS. 130
Como ya se sabe, los principales movimientos en este sistema son tres : dos
translacionales, correspondientes a órbitas paralelas a las direcciones x e y, dos
movimientos diagonales, y un movimiento circular, (ver Fig. 21a).
Seguidamente se estudia la evolución de estas trayectorias a la energía de
interés, que en este caso es la correspondiente a la energía en el punto silla, 74,64
meV.
Podemos ver en Fig. 21b) como la superficie de sección de Poincaré a x = 0
nos muestra la estabilidad en las órbitas paralelas a las direcciones x e y, debido
a la cadena central de islas de estabilidad correspondiente a este movimiento de
arrastre a lo largo del eje x.
Fig. 21. a) Órbitas periódicas principales del Na/Cu(001) con el potencial noseparable a la energía del punto silla de 74,64 meV. La línea continua indicaorbitas estables y las líneas discontinuas las inestables. La superficie equipotenciala esta energía también es mostrada como un línea continua delgada a la alturade nuestros ojos. b) Superficie de sección de Poincaré a la mitad del pozo depotencial, fijando x = 0 , para un colectivo de trayectorias clásicas con una energíacorrespondiente a la barrera de potencial.
Por tanto, es razonable esperar que la aproximación separable para calcular la
pérdida energética a lo largo de esta dirección será buena.

CAPÍTULO 4. RESULTADOS. 131
En efecto, se comprueba que la acción que cruza una celda unidad calculada
numéricamente para la órbita paralela en x o y es S ∼ 46,4 a.u, mientras que la
acción para el potencial separable coseno, S = 4a√
mV ‡/2/π, con una altura de
barrera adiabática V ‡ = 74,64 meV produce el valor de 46,4 a.u.
En un camino similar, para el caso de fricción débil, el movimiento
perpendicular a la coordenada de reacción x es integrable.
Por lo tanto se puede ignorar los términos de corrección de barrera finita
provenientes de la dirección perpendicular, incluyendo en (4.47) sólo las
correcciones de barrera finita a lo largo de la coordenada de reacción. Incluyendo
las correcciones de barrera finita para la dirección perpendicular (dirección y)
se obtienen resultados cualitativos erróneos, debido a la poca profundidad que
presenta el potencial en su máximo a lo largo de la dirección y.
Se tiene que recalcar que esta dificultad de obtener de la teoría la velocidad de
difusión espacial no significa que tal velocidad no exista.
Esto sólo significa que debido a la relativa alta temperatura, es difícil
estimar dicha velocidad de la teoría sin ninguna suposición previa, tal como la
integrabilidad del movimiento a lo largo de esta coordenada.
En la Fig. 21 se muestran los resultados a) para velocidad a lo largo del eje x, b)
el coeficiente de difusión, c) las probabilidades de salto, y d) la anchura energética
del pico Q del factor de estructura dinámico a T = 200K o V ‡/(kBT )∼ 4,3.

CAPÍTULO 4. RESULTADOS. 132
Figura 22. Resultados para el potencial no separable, ecs. (4.16)-(4.19), a T =200 K. a) Velocidades de escape: líneas continuas y discontinuas , resultadosde Kramers, (4.33), sin y con correcciones de barrera finita, respectivamente.Círculos abiertos: cálculos numéricos de Langevin con barras de error. b)Coeficientes de difusión : línea continua, (4.39); Línea discontinua, aproximacióncuasi-2D, (4.52); cuadrados llenos, resultados numéricos de ec. (4.38). Círculosabiertos: relación de Einstein (3.82). c) Distribuciones de salto: Barras llenas,resultados de Langevin con barrras de error; barras vacías: ecs. (4.33) y (4.37)con γ = 0,1ω0. d) FWHM del factor de estructura dinámico con γ = 0,1ω0;línea continua, resultados de Kramers obtenidos por ec. (4.43); línea discontinua,modelo de Chudley-Elliot, ec. (3.75) con velocidades y distribuciones de saltoobtenidos numéricamente: círculos cerrados, resultados numéricos del factorde estructura dinámico, ec.(3.65) con barras de error. Cuadrados llenos: datosexperimentales [16, 17].
Para las velocidades de escape, se incluye la corrección multidimensional de
barrera finita [142] a lo largo de la coordenada de reacción. La distribución de
saltos y la anchura energética han sido calculadas para el coeficiente de fricción

CAPÍTULO 4. RESULTADOS. 133
γ = 0,1ω0, con ω0 = 9THz del modo T. Este es el valor dado por [16, 17] para
este particular SEP después de unas extensivas simulaciones y comparación con
experimentos. Los resultados analíticos se han obtenido considerando una pérdida
energética promedio:
δ ∼ 4γa
πkBT
√
mV ‡
2(4.51)
Por otro lado, el coeficiente de difusión se ha podido comparar con una expresión
similar a (4.47) generalizada ahora a dos dimensiones, denominada aproximación
cuasi-bidimensional :
D = D0a2
∫ a
0dy[
∫ a
0dx exp(βV (x, y))]−1
∫ a
0dy
∫ a
0dx exp(−βV (x, y))
(4.52)
Esta expresión es válida para el potencial separable coseno, pero para el potencial
no separable tiene sus inconvenientes debido a que el acoplamiento, aunque débil,
influye en la dinámica del proceso de difusión. Por ejemplo, se puede ver en
la Fig. 22d) una comparación entre los resultados numéricos y analíticos de la
anchura energética con la obtenida experimentalmente a 200 K, [16, 17]. Para
comparar estos resultados experimentales con el modelo de Chudley-Elliot y los
resultados de la teoría de Kramers, hemos seguido un razonamiento similar a los
experimentalistas: si sólo son posibles saltos en la dirección x e y , la anchura
energética en la dirección diagonal [100] en el máximo deberá ser dos veces el
valor de los saltos a lo largo de una dirección paralela, bien en x o y. Sin embargo,
la relación que se mide en el máximo es 1,4. Esto indica que se tiene un porcentaje
mayor de saltos en la dirección diagonal que en las direcciones paralelas. Por
tanto, los resultados analíticos en la Fig. 22d), velocidad y distribución de saltos
fueron calculados en la dirección x, y entonces multiplicados por el factor 1.4.
Pero se observa, que aún con ésto, los resultados numéricos y los analíticos
no concuerdan tan bien. Además, variando la barrera adiabática se obtiene un
mayor acuerdo entre energía y experimento, encontrando la concordancia óptima
a V ‡ = 72 meV, sólo ∼ 2,5 meV más bajo que el valor estimado por simulación

CAPÍTULO 4. RESULTADOS. 134
numérica de Langevin.
4.6.4.3. Potencial separable y no separable.
Como se ha podido comprobar por nuestros resultados numéricos desde el
punto de vista de una dinámica clásica, considerando el sistema a baja fricción
y temperatura, ambos modelos de potencial separable y no separable presentan
diferencias notables y bastante importantes. Si en un modelo separable se obtiene
un hamiltoniano integrable en el que la dinámica clásica consiste en órbitas
estables, por debajo y por encima del punto silla (barrera de potencial en el caso
unidimensional). Y en el que, por encima de esta barrera , las trayectorias a lo
largo de las direcciones x e y se propagan de forma balística, difundiéndose
libremente, y sin cambiar de dirección, no quedando capturada en ningún pozo
de potencial. El modelo de potencial no separable origina inestabilidad y caos.
Trayectorias por encima de la barrera de difusión pueden quedar atrapadas
tiempos largos en un pozo, debido a la existencia de órbitas rotacionales estables
no presentes en el caso integrable [14, 35]. Hemos visto, del análisis de los
colectivos microcanónicos como dependiendo de la energía que empleemos,
podemos obtener transporte, normal o anómalo. El acoplamiento también influye
en la velocidad de escape y coeficientes de difusión. En la Fig. 23, las velocidades
son calculadas por el método de τMFPT a T = 200 K para el potencial sin
acoplamiento (línea discontinua) y con acoplamiento (línea continua). Para hacer
una buena comparación, las alturas de las barreras de potencial se han elegido las
mismas 74,64 meV, por lo que las diferencias entre estas dos curvas son debidas
al acoplamiento.
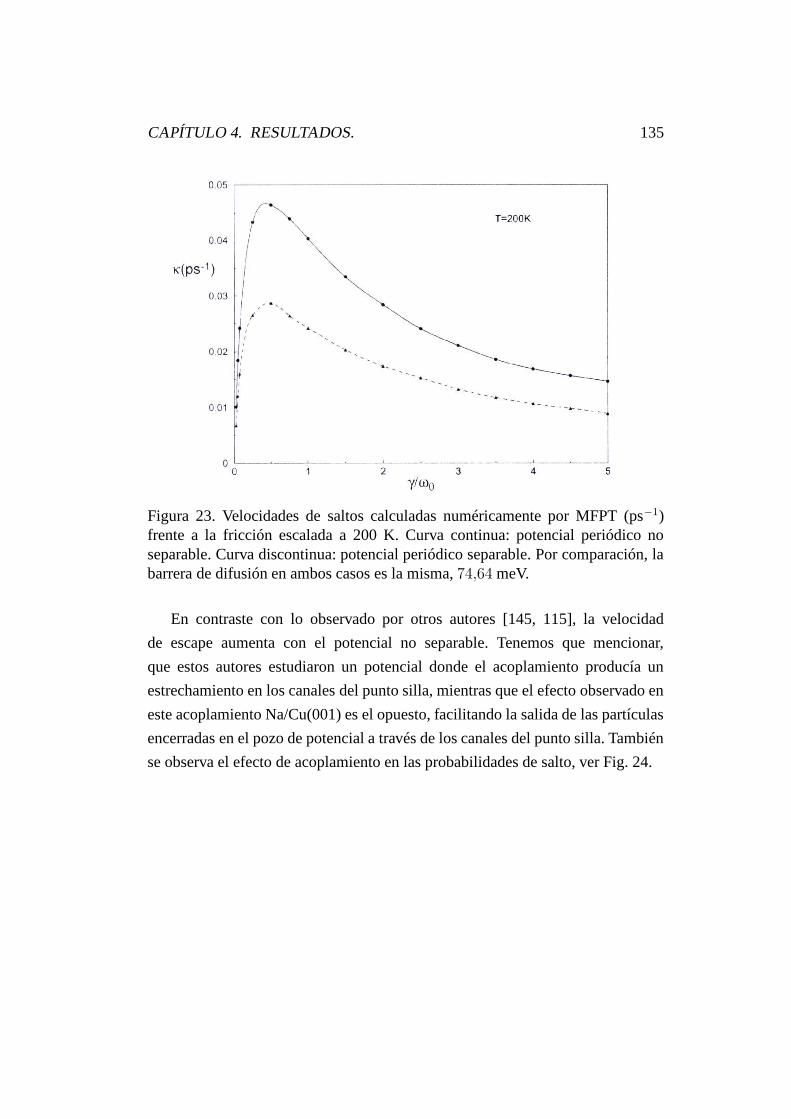
CAPÍTULO 4. RESULTADOS. 135
Figura 23. Velocidades de saltos calculadas numéricamente por MFPT (ps−1)frente a la fricción escalada a 200 K. Curva continua: potencial periódico noseparable. Curva discontinua: potencial periódico separable. Por comparación, labarrera de difusión en ambos casos es la misma, 74,64 meV.
En contraste con lo observado por otros autores [145, 115], la velocidad
de escape aumenta con el potencial no separable. Tenemos que mencionar,
que estos autores estudiaron un potencial donde el acoplamiento producía un
estrechamiento en los canales del punto silla, mientras que el efecto observado en
este acoplamiento Na/Cu(001) es el opuesto, facilitando la salida de las partículas
encerradas en el pozo de potencial a través de los canales del punto silla. También
se observa el efecto de acoplamiento en las probabilidades de salto, ver Fig. 24.

CAPÍTULO 4. RESULTADOS. 136
Figura 24. Probabilidades de salto frente a distancia de salto (en unidades deconstante de red) para el potencial separable (línea continua) y no separable (líneadiscontinua) a dos temperaturas diferentes. Círculos, 100 K; Triángulos, 200K.
Aquí se combinan el efecto de la temperatura y el acoplamiento de potencial.
Se muestran distribuciones de saltos a dos temperaturas diferentes, T = 110 K
(círculos) y T = 200 K (triángulos) para el potencial separable (línea continua) y
para el no separable (línea discontinua) a una fricción experimental de 0,1ω0. La
fracción de doble y triple salto obtenida en el potencial no separable son del 13 %
y 4 % a T = 110 K, y del 14 % y 8 % a 200 K, respectivamente. Como puede verse,
no sólo el incremento de temperatura influye en la aparición de saltos múltiples,
sino que el acoplamiento juega también un papel importante. En Fig. 25 se puede
observar la influencia de este acoplamiento sobre otras propiedades de transporte
como es el caso del coeficiente de difusión.

CAPÍTULO 4. RESULTADOS. 137
Figura 25. Coeficiente de difusión en unidades atómicas como una función de lafricción escalada a 200 K para el potencial separable (cuadrados blancos y negros)y no separable (círculos blancos y negros). curva sólida: Relación de Einstein.Curva de puntos: Se usa el criterio de la energía para la velocidad de salto, 4.38.
Se ha representado el coeficiente de difusión para el potencial separable
(cuadrados) y para el no separable (círculos) en función del coeficiente de fricción
sin dimensiones. Este ha sido calculado empleando la relación de Einstein a
tiempos largos (curva continua) y usando la ec. (4.38) donde la velocidad de
escape κ ha sido obtenida numéricamente mediante el conteo de saltos empleando
el criterio de la energía (curva de puntos), observando un buen acuerdo en ambos
casos.

CAPÍTULO 4. RESULTADOS. 138
Por último decir que Chen y Ying [113] usando un formalismo de proyección
de Mori y un modelo de saltos instantáneos , calcularon las velocidades de salto
para el potencial separable, para un rango de temperaturas comprendidos entre
200 y 300K encontrando un comportamiento de Arrhenius activado cuyo prefactor
fue ∼0,1. En Fig. 26a) se ha encontrado el mismo comportamiento activado
para el mismo rango de temperaturas pero ahora obteniendo un prefactor de ∼0,22. Los correspondientes cálculos numéricos han sido llevados a cabo para una
fricción de 0,15ω0 con ω0 = 9,31 THz. La barrera de difusión efectiva para este
potencial separable fue de 52meV el cual está bastante de acuerdo con el valor de
V ‡ = 51 ± 6 meV obtenido experimentalmente [151, 15].
Para el potencial no separable , el prefactor de Arrhenius fue extraído
directamente de un ajuste al pico cuasielástico experimental con una lorenciana
a pequeños valores de ∆K, representándose las diferentes anchuras energéticas
obtenidas a diferentes temperaturas (entre 150 − 250K) dándonos un resultado
de 3,3 ± 0,3 frente al resultado de dado por la TST. Este resultado obtenido es
bastante sorprendente ya que se tiene que recordar que la TST usualmente da un
prefactor que concuerda bastante bien con su valor real. Además, en la Fig. 26b) se
ha dibujado nuestros resultados numéricos calculados a una fricción experimental
de 0,1ω0 con ω0 = 9Thz y el correspondiente prefactor hallado es de ∼ 0,23 y
una barrera efectiva de V ‡ ∼ 55 meV.

CAPÍTULO 4. RESULTADOS. 139
Figura 26. Dibujo de Arrhenius para a) potencial separable y b) potencial noseparable. Líneas sólidas son los MFPT resultados y las líneas discontinuasrecogen los resultados numéricos con el criterio de energía.
En nuestra opinión, la razón de esta fuerte discrepancia es que estos autores
obtienen el prefactor f0 del coeficiente de difusión dado por ec. (4.38): D =
0,5.f0.ω0a2/π, mientras que a estas bajas fricciones la contribución de saltos
múltiples son importantes y las longitudes de saltos cuadráticas medias < l2 >a2 y, como puede verse en [152, 3] , estos autores recogen una fracción del 20 %
de saltos dobles y alrededor de un 5 % en saltos triples.

CAPÍTULO 4. RESULTADOS. 140
4.7. Teoría hamiltoniana de línea vibracional de
átomos adsorbidos sobre superficies.
4.7.1. Formalismo hamiltoniano.
La posición de un adsorbato de masa m sobre una superficie puede ser
considerado en su forma más general como un proceso estocástico que obedece a
una ecuación de Langevin generalizada de la forma (q =√mx) :
q +∂V (q)
∂q+
∫ t
0
dτγ(t− τ)q(τ) = Fr(t) (4.53)
donde V (q) es el potencial de interacción adiabática y Fr(t) la fuerza aleatoria
proveniente del baño fonónico. Es bien conocido que esta ecuación de Langevin
se deriva del siguiente hamiltoniano de Caldeira-Legget [153]:
H =p2q
2+ V (q) +
N∑
j=1
[p2xj
2+
1
2(ωjxj −
cjωjq)2] (4.54)
donde el modo jth del baño armónico está caracterizado por la coordenada de
masa ponderada xj , momento pxj, y frecuencia ωj. La solución de las ecuaciones
de Hamilton del movimiento para los modos del baño son expresadas en términos
de la coordenada del sistema q y sus condiciones iniciales. Esto conduce a la
ecuación de Langevin (4.53) y donde la función de fricción se identifica como:
γ(t) =
N∑
j=1
c2jω2j
cos(ωjt) (4.55)
Fr(t) se encuentra distribuida de forma gaussiana con media cero y obedece
al teorema de fluctuación-disipación. El límite al continuo en el formalismo
hamiltoniano puede llevarse a cabo usando una densidad espectral dada por [153]:
J(ω) =π
2
N∑
j=1
c2jωj
[δ(ω − ωj) − δ(ω + ωj)] (4.56)

CAPÍTULO 4. RESULTADOS. 141
La formulación hamiltoniana presenta algunas ventajas sobre la formulación
fenomenológica de Langevin: primero, la fricción de memoria y óhmica son
tratadas de la misma forma, y los resultados obtenidos pueden ser extendidos
a cualquier función de fricción siempre y cuando obedezca al teorema de
fluctuación disipación. Segundo, el tratamiento cuántico es sencillo, todo lo que
se necesita es tratar el hamiltoniano de (4.54) como un operador en el espacio de
Hilbert pertinente [154].
Si la interacción potencial es puramente armónica, V (q) = ω20q
2/2,el
hamiltoniano (4.54) puede ser separado via transformación a modos normales
[155, 156, 157]. La forma a modos normales es:
HNM =P 2ρ
2+
1
2λ2
0ρ2 +
N∑
j=1
(p2yj
2+
1
2λ2jy
2j ) (4.57)
donde las coordenadas de los modos normales del sistema ρ, y modos del baño
yj están relacionadas con las coordenadas de masa ponderada q y xj por la matriz
ortogonal de transformación U. En particular:
q = u00ρ+
N∑
j=1
uj0yj (4.58)
Las frecuencias del nuevo sistema, λ0 y del baño λj, pueden ser expresadas en
términos de las antiguas frecuencias ω0 y ωj. También es conveniente definir la
función de fricción en modo normal:
K(t) =
N∑
j=0
u2j0 cos(λjt) (4.59)
y la densidad espectral como:
Υ(λ) =π
2
N∑
j=1
u2j0
λj[δ(λ− λj) − δ(λ+ λj)] (4.60)
donde el indice 0 se refiere al modo normal del sistema ρ. La transformación de

CAPÍTULO 4. RESULTADOS. 142
modo normal tiene la propiedad que para cualquier s [158]:
N∑
j=0
u2j0
s2 + λ2j
=1
s2 + sγ(s) + ω20
(4.61)
donde γ(s) es la transformada de Laplace de la función de fricción. De esta
relación, usando la descomposición de Fourier de la función delta de Dirac, se
puede deducir el límite continuo de la densidad espectral de modos normales:
λΥ(λ) = Re [K(iλ)] = Re[iλ
ω20 − λ2 + iλγ(iλ)
] (4.62)
Estas identidades serán empleadas más tarde. Para futuras referencias, se tiene que
mencionar que la frecuencia del modo normal del sistema λ0 y el coeficiente µ00
puede ser relacionado con la función de fricción por las siguientes expresiones:
λ20 = ω2
0[1 +γ(iλ0)
iλ0
]−1 (4.63)
µ200 = [1 +
1
2(γ(iλ0)
iλ0+dγ(s)
ds|s=iλ0
)]−1 (4.64)
Viendo (4.63), λ0 es en general una frecuencia compleja. La parte real da el
desplazamiento en frecuencia y la parte imaginaria la velocidad de transferencia
de energía al baño. Por ejemplo, cuando la fricción es óhmica [J(ω) = γω] se
obtiene que:
λ0 = ω1 + iγ/2 (4.65)
con
ω1 ≡√
ω0 −γ2
4(4.66)
Entonces, como es bien conocido, para el oscilador armónico, con disipación
óhmica, la frecuencia del oscilador decrece con el incremento de la fricción γ

CAPÍTULO 4. RESULTADOS. 143
pero es independiente de la temperatura.
4.7.2. Forma de línea para el modo vibracional.
4.7.2.1. Oscilador armónico.
Un representación simple de la interacción potencial adsorbato-sustrato para
una superficie simétrica es de la forma coseno [15]:
V (q) = V0[1 − cos(2π
aq)], (4.67)
El primer término en la expansión de Taylor alrededor de q = 0 da un potencial
parabólico con frecuencia de oscilación ω0. En coordenadas normales, la función
de autocorrelación de velocidades viene dada por:
Cv(t) =< [µ00pρ(t) +N
∑
j=1
µj0pyj(t)].[µ00pρ(0) +
N∑
j=1
µj0pyj(0)] > (4.68)
Las ecuaciones de movimiento para los modos normales yj son las correspodien-
tes a las de un oscilador armónico con frecuencia λj , así que:
yj(t) = yj(0)cos(λjt) +pyj
(0)
λjsin(λjt) (4.69)
Las posiciones iniciales yj(0) y velocidades pyj(0) de los modos del baño se
encuentran distribuidas térmicamente, es decir:
< λ2jy
2j (0) >=< p2
j(0) >= β−1 (4.70)
donde β ≡ 1/(kBT ). Diferenciando (4.63) con respecto al tiempo y usando el
promedio térmico (4.64) se obtiene:
Cv(t) =1
βK(t) (4.71)

CAPÍTULO 4. RESULTADOS. 144
También se sabe que la inversa de la transformada de Laplace (4.62) para
la fricción óhmica [ ˆγ(s) = γ], da el resultado estándar para la función de
autocorrelación de velocidades:
Cv(t) =e−(γ/2)t
β[cos(ω1t) −
γ
2ω 1sin(ω1t)] (4.72)
Sustituyendo la función de autocorrelación de velocidades exacta, (4.72) en
(3.77), la función de dispersión intermedia se puede escribir como:
I(∆K, t) = e−2$e2$f0(t)
f0(t) = Cv(t) = e−γt/2[cos(ω1t) − γ2ω 1
sin(ω1t)](4.73)
y 2$ = ∆K2/mβω20 . Tenemos que notar que 2$ es el factor de atenuación de
Debye-Waller, ya que:
2$ = ∆K2 < q(t)2 + q(0)2 > /2 (4.74)
Por otro lado, la segunda exponencial en (4.73) puede ser expandida en series
de potencias en la variable 2$. La forma de línea es entonces obtenida por la
transformada de Fourier, (3.65) . Despreciando los términos no resonantes uno
encuentra que:
S(∆K, ω) ' e−2$∑
j
2j$j
(j − 1)!(
γ
(ω + ω1j)2 + γ2j2/4+
γ
(ω − ω1j)2 + γ2j2/4)
(4.75)
Este expresión está formada por una serie de lorencianas centradas a frecuencias
±ω1 y sus armónicos, con intensidades decrecientes (típicamente, sólo para el
caso j = 1 esta forma de línea es la que se observa experimentalmente). La
anchura del primer pico corresponde al coeficiente de fricción γ. Notar también
que el pico no se encuentra centrado a la frecuencia del oscilador ω0, en cambio
sí a ω1, esto es debido al acoplamiento con el baño térmico. Experimentalmente,
el valor del coeficiente de fricción es usualmente estimado por extrapolación de la

CAPÍTULO 4. RESULTADOS. 145
FWHM del modo T a temperatura cero, un procedimiento el cual se justifica con
(4.75).
4.7.2.2. Correcciones anarmónicas.
Si ahora se supone que el potencial coseno se expande hasta la primera
corrección anarmónica, tal que el hamiltoniano sistema-baño (4.54) puede ser
expresado en términos de modos normales:
H = HNM +K4(u00ρ+
N∑
j=1
uj0yj)4 (4.76)
dondeHNM es el hamiltoniano en modo normal (4.57), y para el potencial coseno
K4 ≡ V (4)(0)/24m2 = −ω40/24V0 es negativo. La dependencia temporal de
las coordenadas de los modos normales es resuelta por teoría de perturbaciones,
entonces con la aproximación a primer orden en K4 se tiene:
yj(t) = y(0)j (t) + 4K4y
(1)j (t) (4.77)
donde y(0)j (t) es la solución para el oscilador armónico dada en (4.63), y la
corrección de primer orden y(1)j (t) viene dada por:
y(1)j (t) = −uj0
λj
∫ t
0
sin[λj(t− t′)][
N∑
j=0
uj0y(0)j (t′)]3dt′ (4.78)
Para calcular la función de autocorrelación de velocidades, se emplea el promedio
térmico:
< y2j (0)
p2k(0)
λk>=<
p2j(0)
λ2j
p2k(0)
λk>=
1
β2λ2jλk
(4.79)
Aquí señalamos que se debería usar estrictamente el hamiltoniano total anarmó-
nico para el promedio, mientras que (4.79) es el resultado de un promedio sobre
el hamiltoniano armónico. Sin embargo, para el rango de parámetros usados en

CAPÍTULO 4. RESULTADOS. 146
nuestro sistema, la corrección estática es pequeña y por tanto puede ser despre-
ciada. El resultado ahora para la función de autocorrelación de velocidades viene
dada por:
Cv(t) =1
βK(t) − 12K4
β2ω20
∫ t
0
K(t− t′)
∫ t′
0
K(t′′)dt′′ (4.80)
El primer término corresponde a la contribución del oscilador armónico, mientras
que el segundo término es una convolución integral con la función de fricción
K(t) en la representación de modos normales. Mediante la transformación por
Laplace, y usando la identidad (4.61) produce el siguiente resultado:
Cv(s) =1
β
s
s2 + sγ(s) + ω20
− 12K4
β2ω20
s
[s2 + sγ(s) + ω20]
(4.81)
En el caso que nos ocupa de una fricción óhmica, la inversa de Laplace da la
primera corrección anarmónica para la función de autocorrelación de velocidades:
Cv(t) = C(0)v (t)− 12K4
β2ω20
× e−(γ/2)t[γω1t cos(ω1t) + (2ω21t− γ) sin(ω1t)]
4ω31
(4.82)
donde C(0)v (t) está dado por (4.72). De (3.77), la función dispersión intermedia
puede ser escrita como:
I(∆K, t) = I (0)(∆K, t)I (1)(∆K, t) (4.83)
Aquí, I (0)(∆K, t) es la función de dispersión intermedia , y I (1)(∆K, t) es la
corrección anarmónica de primer orden para el potencial coseno, cuya expresión
viene dada por:
I(1)(∆K, t) = e−2$/(βV ‡)e[2$/(βV‡)]f1(t) (4.84)
y

CAPÍTULO 4. RESULTADOS. 147
f1(t) = e−γt/2[(1 − γω20
4ω21
t) cos(ω1t) + (6γω2
0 − γ3
8ω31
+ω2
0
2ω1
) sin(ω1t)] (4.85)
Antes de comparar con nuestros cálculos numéricos se debe notar que aparte del
término puramente sinusoidal en el exponente similar al que aparece en (4.73),
hay términos que son lineales en t. Si se procede en el mismo camino con el que
se derivó (4.75) , puede verse que ellos conducen a perfiles tipo Fano centrados
a las frecuencias armónicas. Esto causa una distorsión del modo T cambiando
su anchura y posición. Segundo, la magnitud de la corrección de primer orden
depende de la altura de la barrera βV ‡ . Para βV ‡ 1 la aproximación armónica
permanece válida. Esto era de esperar ya que a bajas temperaturas y altas barreras
(altas frecuencias vibracionales) las partículas permanecen cerca del mínimo del
pozo de potencial. En orden a establecer el rango de validez de la teoría basada
sobre la primera corrección de primer orden, se ha resuelto numéricamente la
ecuación de Langevin con fricción óhmica. Los parámetros han sido tomados
del modelo de átomos de Na adsorbidos moviéndose a través de un potencial
coseno que es el que simula la superficie en una dimensión. En la Fig. 27 se
muestra la función de autocorrelación de velocidades para este sistema a dos
valores de fricción: baja, γ = 0,1ω0, y moderada: γ = 0,5ω0, y dos diferentes
alturas de barreras V ‡, comparándolas con las aproximaciones armónicas (4.72),
y anarmónicas (4.82).

CAPÍTULO 4. RESULTADOS. 148
Figura 27. Funciones de velocidad de autocorrelación numérica y analítica enun potencial coseno a T = 100 K con diferentes coeficientes de fricción γ yaltura de barreras V ‡ . (a)-(c) γ = 0,1ω0. V ‡ = 67 y 134 meV, respectivamente.(b)-(d) γ = 0,5ω0. V ‡ = 67 y 134 meV. Línea continua gruesa: función deautocorrelación numérica. Línea continua fina: aproximación armónica , (4.72).Línea discontinua: corrección anarmónica, (4.82).
Como esperábamos, doblando la altura de la barrera (el mismo efecto,
podría haberse obtenido disminuyendo la temperatura) se mejora el acuerdo
entre las funciones de autocorrelación analítica y numérica, Fig. 27 (c)-(d).
Notar que los desplazamientos, resultado de las correcciones anarmónicas , se
dirigen a las frecuencias correctas, pero esto falla a baja barrera y fricción, Fig.
27(a), reproduciéndose únicamente de forma correcta las primeras dos o tres
oscilaciones. Claramente, la primera corrección anarmónica no es suficiente para
reproducir este caso. En cambio, a alta fricción, Fig. 27 (b)-(d), la partícula
permanece cerca del mínimo del pozo de potencial, y la corrección anarmónica
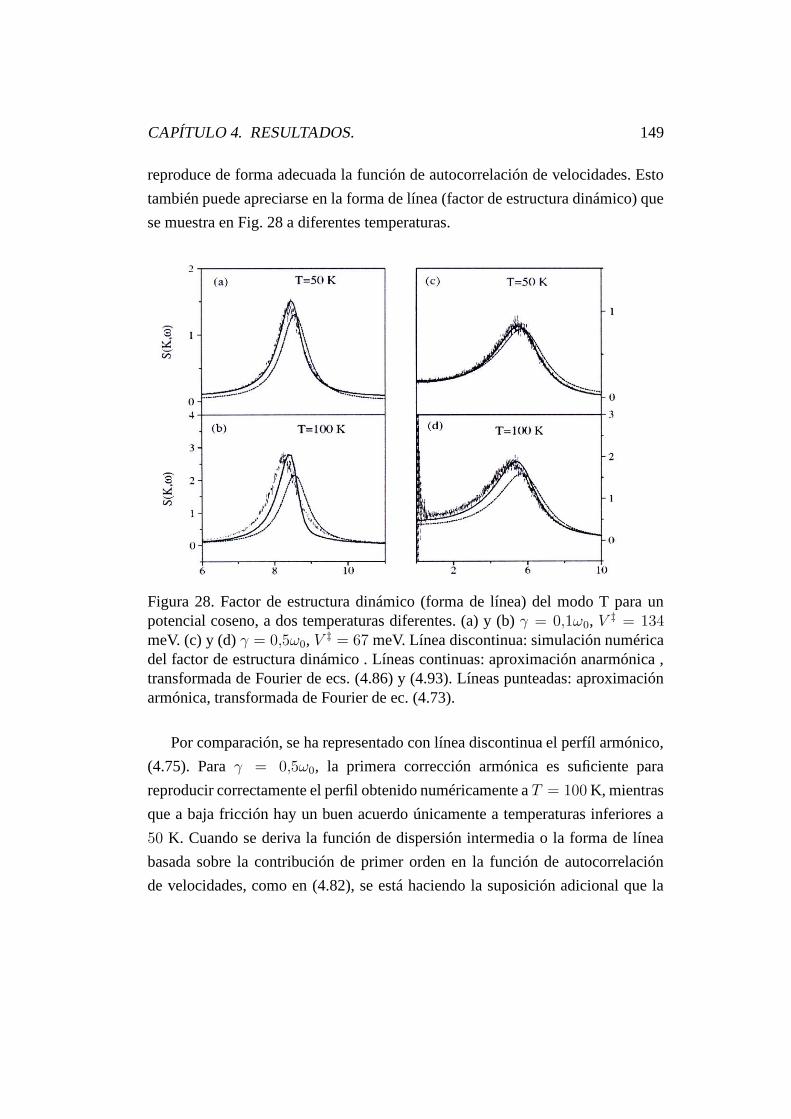
CAPÍTULO 4. RESULTADOS. 149
reproduce de forma adecuada la función de autocorrelación de velocidades. Esto
también puede apreciarse en la forma de línea (factor de estructura dinámico) que
se muestra en Fig. 28 a diferentes temperaturas.
Figura 28. Factor de estructura dinámico (forma de línea) del modo T para unpotencial coseno, a dos temperaturas diferentes. (a) y (b) γ = 0,1ω0, V ‡ = 134meV. (c) y (d) γ = 0,5ω0, V ‡ = 67 meV. Línea discontinua: simulación numéricadel factor de estructura dinámico . Líneas continuas: aproximación anarmónica ,transformada de Fourier de ecs. (4.86) y (4.93). Líneas punteadas: aproximaciónarmónica, transformada de Fourier de ec. (4.73).
Por comparación, se ha representado con línea discontinua el perfíl armónico,
(4.75). Para γ = 0,5ω0, la primera corrección armónica es suficiente para
reproducir correctamente el perfil obtenido numéricamente a T = 100 K, mientras
que a baja fricción hay un buen acuerdo únicamente a temperaturas inferiores a
50 K. Cuando se deriva la función de dispersión intermedia o la forma de línea
basada sobre la contribución de primer orden en la función de autocorrelación
de velocidades, como en (4.82), se está haciendo la suposición adicional que la

CAPÍTULO 4. RESULTADOS. 150
posición y velocidad son procesos gaussianos (truncamiento en la expansión de
cumulantes). En orden a revisar esta aproximación , se calcula numéricamente la
función de dispersión intermedia exacta (3.77) bajo la aproximación gaussiana.
Como se ve en Fig. 29, la aproximación gaussiana es excelente incluso en el caso
de valores a baja fricción y bajas barreras de potencial.
Figura 29. Función de dispersión intermedia a T = 100 K, γ = 0,1ω0 yV ‡ = 67 meV. Se ha utilizado una transferencia de momento paraleloK = 1 Å
−1.
Línea discontinua: simulación numérica de la función de dispersión intermedia.Línea continua: aproximación gaussiana, ec. (3.77) empleando las funcionesde correlación numéricas. Línea de puntos: simulación numérica empleando unoscilador cuártico (expansión de cuarto orden del potencial coseno). No aparecentrayectorias que se difunden en las funciones de correlación. En cambio, enla ventana, tanto en la simulación numérica (línea discontinua) como en laaproximación gaussiana teórica (línea continua) aparecen trayectorias difusivas.
También, con líneas de puntos, se muestra la función de dispersión intermedia
calculada para un potencial cuártico:
V (q) =1
2ω2
0q2 +K4q
4 (4.86)

CAPÍTULO 4. RESULTADOS. 151
El buen acuerdo sugiere que en efecto, el primer término anarmónico en la
expansión de Taylor del potencial adiabático es capaz de tener en cuenta el
desplazamiento y anchura en el modo T, y que el fallo del modelo analítico a
bajas barreras de potencial (alta temperatura) es principalmente debido al hecho
de despreciar los términos de mayor orden en nuestra solución perturbativa.
Las curvas en Figs. 27 y 29 han sido obtenidas sólo con trayectorias donde
únicamente se da el proceso de vibración. Para los parámetros usados en Fig.
29, especialmente a tiempos largos, muchas de las trayectorias vencen la barrera
de potencial comenzando a difundirse libremente por la superficie originándose
el pico cuasielástico. Por otro lado, en la ventana de la Fig. 29 se muestra como
la aproximación gaussiana falla para las trayectorias que se difunden. Además,
esta aproximación depende ahora del valor de ∆K, (que como sabemos, es mejor
a valores de ∆K pequeños). Además, la función de dispersión intermedia para
el movimiento vibracional no depende de ∆K. Sólo su amplitud, y entonces la
intensidad del modo T es la que se ve afectada, no la posición ni la forma de línea.
De la Fig. 28 notar que el factor de estructura dinámico anarmónico presenta
un desplazamiento con la temperatura hacia frecuencias más bajas. Un cálculo
de la anchura de la forma de línea analítica, sin embargo, muestra un efecto no
significativo con la temperatura incluso en el rango donde ésto compara bien con
las simulaciones numéricas.
4.7.2.3. Correcciones cuánticas.
Como un ejemplo, se va a calcular una forma de línea cuántica para
el oscilador armónico disipativo. Ya que las correcciones cuánticas son sólo
significativas a bajas temperaturas, especialmente para adsorbatos de gran masa,
las aproximaciones armónicas ilustrarán de forma suficiente la importancia de los
efectos cuánticos. Es conveniente expresar la función de dispersión intermedia
gaussiana exactamente [159] en términos de la función de autocorrelación de
posiciones [160] como:

CAPÍTULO 4. RESULTADOS. 152
I(K, t) = e−2$e∆K2<q(t)q(0)> (4.87)
El factor de estructura resultante será real, mostrando dos picos a frecuencias
positivas y negativas (teniendo en cuenta la creación cuántica de creación y de
aniquilación). Estrictamente hablando, S(∆K, ω) y S(∆K,−ω) se encuentran
relacionados por un balance detallado de la condición impedida de aniquilación
a T = 0 K [159], por tanto a bajas temperaturas el factor de estructura dinámico
es asimétrico. Los dos picos, sin embargo, tendrán casi la misma forma de línea
(aunque sus magnitudes pueden ser diferentes). Entonces se puede calcular un
promedio de formas de líneas reemplazando < q(t)q(0) > con su parte real
Cq(t) = 1/2[< q(0)q(t) > + < q(t)q(0) >]. Para obtener la función de
autocorrelación cuántica de posiciones hay que indicar que el promedio cuántico
térmico de la energía cinética y potencial viene dado por:
1
2< λ2
jy2j (0) >=
1
2< p2
j(0) >=~
4λj coth(~βλj/2) (4.88)
Usando la representación de modos normales del hamiltoniano, uno encuentra
que:
Cq(t) =
N∑
j=0
u2j0 < y2
j (0) > cos(λjt) =~
π
∫ ∞
0
dλΥ(λ) coth(~βλ/2) cos(λt)
(4.89)
donde se ha utilizado la densidad espectral de los modos normales dada en (4.60).

CAPÍTULO 4. RESULTADOS. 153
Para la fricción óhmica la densidad espectral utilizada viene dada por (4.62),
obteniéndose:
Cq(t) =~γ
π
∫ ∞
0
dλ(λ coth(~βλ/2) cos(λt)
(ω20 − λ2)2 + γ2λ2
) (4.90)
Decir que el uso de Cq(t) es consistente con el teorema de fluctuación-disipación
cuántica [153, 154]:
γ(t) =N
∑
j=1
1
2< Frj (t)Frj (0) + Frj (0)Frj(t) >
2 tanh(~ωjβ/2)
~ωj(4.91)
El factor de Debye-Waller para el oscilador armónico se obtiene realmente
teniendo en cuenta esta relación:
< q2(t) >=< q2(0) >= Cq(0) (4.92)
En Fig. 30 se compara la aproximación harmónica para el factor de estructura
dinámico clásico y cuántico del átomo de Na moviéndose en un potencial coseno.
Los parámetros elegidos son V ‡ = 67 meV y γ = 0,5ω0.

CAPÍTULO 4. RESULTADOS. 154
Figura 30. Factor de estructura dinámico cuántico bajo un aproximaciónanarmónica, de ecs. (4.93) y (4.96), para los mismos parámetros como en Figs.28(c) y (d), γ = 0,5ω0, V ‡ = 67 meV, y distintas temperaturas: línea continua,resultado clásico a T = 100 K. Resultados cuánticos: línea discontinua, T = 100K: línea de puntos, T = 50 K: larga línea discontinua, T = 25 K; línea discontinuacon puntos, T = 10 K. La transferencia de momento paralelo K = 1Å
−1.
Los resultados se muestran a diferentes temperaturas. Uno nota que a medida
que baja la temperatura se induce un corrimiento hacia el azul en el modo T. El
corrimiento al rojo experimentalmente observado es debido a la anarmonicidad
como se mostrará en la siguiente sección. Los efectos cuánticos decrecen cuando
el coeficiente de fricción decrece, para γ = 0,1ω0, por tanto cualquier efecto
cuántico no es observable para temperaturas por encima de 10 K.

CAPÍTULO 4. RESULTADOS. 155
4.7.2.4. Desplazamiento y anchura del modo T dependiente de la tempera-
tura.
El desplazamiento en frecuencias puede ser obtenido directamente con el
formalismo precedente sin calculo del factor de estructura dinámico. De la ec.
(4.76) uno nota que la anarmonicidad causará un desplazamiento del mínimo
de potencial y frecuencia del modo normal del sistema. Específicamente, la
frecuencia instantánea para el primer orden en el parámetro de anarmonicidad
K4 es:
λ0(t) ≡ (∂2H
∂ρ2)1/2 = [λ2
0 + 12K4µ200(
N∑
j=0
uj0yj)2]1/2 ∼ (4.93)
λ0 +6K4u
200
λ0
N∑
j=0
uj0yj)2 + o(K2
4 )
El desplazamiento en frecuencia se obtiene del promedio térmico < λ0(t) > .
Introduciendo la solución del oscilador armónico, ec. (4.69), para la coordenada
del modo normal yj en el último término en ec. (4.93), y desarrollando los
promedios (ignorando la anarmonicidad en la función de partición) se encuentra
una contribución al desplazamiento en frecuencias dependiente de la temperatura:
∆λ0 ≡< λ0(t) > −λ0 =6K4
βω20ω1
(4.94)
Como ya se mencionó anteriormente, para el potencial coseno,K4 es negativo, por
tanto la anarmonicidad produce un desplazamiento al rojo con una dependencia
lineal con la temperatura. En la Fig. 31 se compara el desplazamiento obtenido
del promedio en frecuencias del modo normal, ec. (4.94), con los resultados
numéricos (línea continua) y experimentales (círculos abiertos) obtenidos para
el sistema Na/Cu(001). El valor de la barrera y coeficiente de fricción estimados
de las medidas experimentales son V ‡ ∼ 75 meV y γ/ω0 = 0,1.

CAPÍTULO 4. RESULTADOS. 156
Figura 31. Posición del modo T como una función de la temperatura parael sistema Na/Cu(001). Círculos: resultados experimentales, [16, 17]. Líneacontinua: resultados numéricos obtenido con la ecuación de Langevin con unpotencial coseno en 1D, (4.67) . Los parámetros utilizados han sido, γ = 0,1ω0,V ‡ = 75meV , estimados de los experimentos. Línea discontinua: predicciónteórica ec. (4.94), usando la correcciones anarmónicas de primer orden.
El acuerdo es bastante bueno para temperaturas por debajo de T = 150
K. Se demostrará más adelante, que debido a la baja fricción del sistema y
la relativa barrera pequeña, las correcciones a segundo orden son necesarias
para altas temperaturas. En la estimación analítica de la forma de línea dada
con anterioridad, se observó un desplazamiento en frecuencias, pero no así
una anchura de línea con el aumento de la temperatura. Esto puede entenderse
calculando la varianza en la frecuencia del sistema < λ20(t) > − < λ0(t) >
2,
la cual es proporcional a la anchura del modo T. Uno inmediatamente ve que,
a primer orden en K4 la varianza es despreciable. Por tanto, las correcciones
de segundo orden en la anarmonicidad son necesarias para obtener la anchura

CAPÍTULO 4. RESULTADOS. 157
dependiente de la temperatura. Un calculo directo del factor de estructura
dinámico a través de las líneas discutidas en la sección 4.7.2.2 , usando una teoría
de perturbación de segundo orden, es bastante complicado. En su lugar se ha
recurrido a un modelo más simple, el cual, sin embargo, conserva los ingredientes
de la dinámica básica: el oscilador de Kubo [161, 74].
Primero se expande la función de dispersión intermedia en momentos, en lugar
de cumulantes. Si nos quedamos a orden dos, esto es equivalente a la expansión
en Taylor hasta segundo orden de la ec. (4.93) alrededor de ∆K = 0. Entonces,
la función de dispersión intermedia será esencialmente proporcional a la función
de autocorrelación de posiciones, como en la teoría de dispersión Raman [162],
y se puede utilizar el modelo del oscilador de Kubo. También se supone que
el oscilador de modos normales con frecuencia λ0 presenta una componente de
frecuencia aleatoria debido a la anarmonicidad, y se define:
λ0(t) = λ0 + λ1(t) (4.95)
Entonces, usando las ecuaciones de movimiento de un oscilador con frecuencia
λ0(t) y una expansión en cumulantes a segundo orden encontramos que:
< q(t)q(0) >∝ Re [< eiR t
0λ1(t′)dt′ > eiλ0t] ∼ Re [eiλ0tei
R t
0<λ1(t′)>dt′ . (4.96)
e−R t
0(t−t′)<λ1(t′)λ1(0)>dt′e−
R t
0(t−t′)<λ1(t′)λ1(0)>dt′ ]
Notar que < λ1(t′) > es el desplazamiento ∆λ0, ec. (4.94) . Usando ec. (4.65)
para λ0 se puede expresar la función de dispersión intermedia como:
I(K, t) ∝ Re ei(ω1+∆λ0)te−γt/2e−R t
0(t−t′)<λ1(t′)λ1(0)>dt′ (4.97)
a primer orden en K4 se obtiene que < λ1(t′)λ1(0) >= 0 y el factor de estructura
dinámico es una lorenciana centrada a la frecuencia ω1+∆λ0( y por tanto un

CAPÍTULO 4. RESULTADOS. 158
desplazamiento lineal con la temperatura), con una FWHM igual a la anchura del
oscilador armónico γ y por tanto una anchura independiente de la temperatura).
La corrección de segundo orden en λ0(t) producirá una anchura dependiente con
la temperatura debido a la última exponencial en (4.97). A segundo orden, la
frecuencia instantánea viene dada por:
λ0(t) ∼ λ0 +6K4u
200
λ0[(
N∑
j=0
uj0y(0)j )2 + 8K4(
N∑
j=0
uj0y(0)j )(
N∑
j=0
uj0y(1)j )− (4.98)
18K24u
400
λ30
(
N∑
j=0
uj0y(0)j )4] + o(K3
4 )
donde se ha obtenido la solución perturbativa de primer orden y(1)j , (4.77). Para
la función de correlación, los términos< λ1(t′)λ1(0) >dependientes de y(1)
j se
cancelan y se obtienen términos dependientes únicamente de los promedios sobre
la posición del oscilador armónico. Ya que este es un proceso gaussiano (por
simplicidad analítica se simplifica la anarmonicidad en la función de partición),
se utilizan las identidades ya conocidas:
< q2osc(t) >=< q2
osc(0) >=1
βω20
(4.99)
< q2osc(t)q
2osc(0) >= 2 < qosc(t)qosc(0) >2 + < q2
osc(0) >2 (4.100)
por tanto todo puede ser derivado en términos del promedio < q2osc(0) > y la
función de correlación < qosc(t)qosc(0) > las cuales son conocidas. Entonces
encontramos que:

CAPÍTULO 4. RESULTADOS. 159
< λ1(t′)λ1(0) >= 36K2
4
u400
λ20
< q2osc(0) >2 +72K2
4
u400
λ20
< qosc(t)qosc(0) >2
(4.101)
sustituyendo ahora ec. (4.101) y [viendo ec. (4.95)]
< qosc(t)qosc(0) >=e−γt/2
βω20
[cos(ω1t) +γ
2ω1
sin(ω1t)] (4.102)
se pueden calcular las integrales en el último exponente en ec. (4.97). Además,
se tiene que indicar que, para tiempos largos, el primer término en (4.101) es
dominante y da una función de dispersión gaussiana de la forma:
I(∆K, t) ∝ Re ei(ω1+∆λ0)te−γt/2e−σ2(t2/2) (4.103)
donde
σ =6|K4|βω1ω2
0
=ω2
0
2βV ‡ω1(4.104)
El factor de estructura dinámico es la transformada de Fourier de (4.103) . La
exponencial imaginaria da la posición del pico, ya discutido arriba. La anchura
del pico es determinado por las exponenciales reales. A tiempos cortos, o sea si
βV ‡ 1 [ver ec. (4.104)], el primer exponente domina, por tanto el pico tiene
un perfil lorenciano con la FWHM dada principalmente por γ, como en el caso
del oscilador armónico. A tiempos largos, o si βV ‡ es pequeño, la contribución
gaussiana domina y el pico tiene un perfil gaussiano con un FWHM pricipalmente
dado por σ. En situaciones intermedias ambas contribuciones han sido tomadas en
cuenta, la parte central del modo T siendo cercano a un pefil gaussiano mientras
que las alas son mejor aproximadas por un perfil lorenciano. Esto es análogo
al efecto de estrechamiento dinámico, primeramente discutido en el contexto de
resonancia magnética. Esto es importante para notar que este modelo simple da
una anchura proporcional a σ, y por tanto una anchura lineal dependiente de la
temperatura. En la Fig. 32 se compara la anchura experimental con la numérica,

CAPÍTULO 4. RESULTADOS. 160
y la FWHM obtenida de la transformada de Fourier de la función de dispersión
intermedia de Kubo, ec. (4.97).
Figura 32. FWHM del modo T como una función de la temperatura parael sistema Na/Cu(001). Círculos: resultados experimentales, [16, 17]. Líneacontinua: resultados numéricos obtenidos empleando la ecuación de Langevin conun potencial coseno, (4.67). Los parámetros usados son los mismos que en Fig,30. Línea discontinua: estimación analítica, (4.104). Línea con puntos: predicciónteórica, estimada por la transformada de Fourier de la función de dispersiónintermedia del modelo de Kubo, ec. (4.97).
Este modelo simple es capaz de tener en cuenta el crecimiento lineal de la
anchura con la temperatura observada experimentalmente. Las correcciones de
segundo orden pueden ser calculadas para el desplazamiento en (4.98) , con el
resultado:
∆λ0 =6K4
βω20ω1
− 54K24
β2ω60ω1
(4.105)
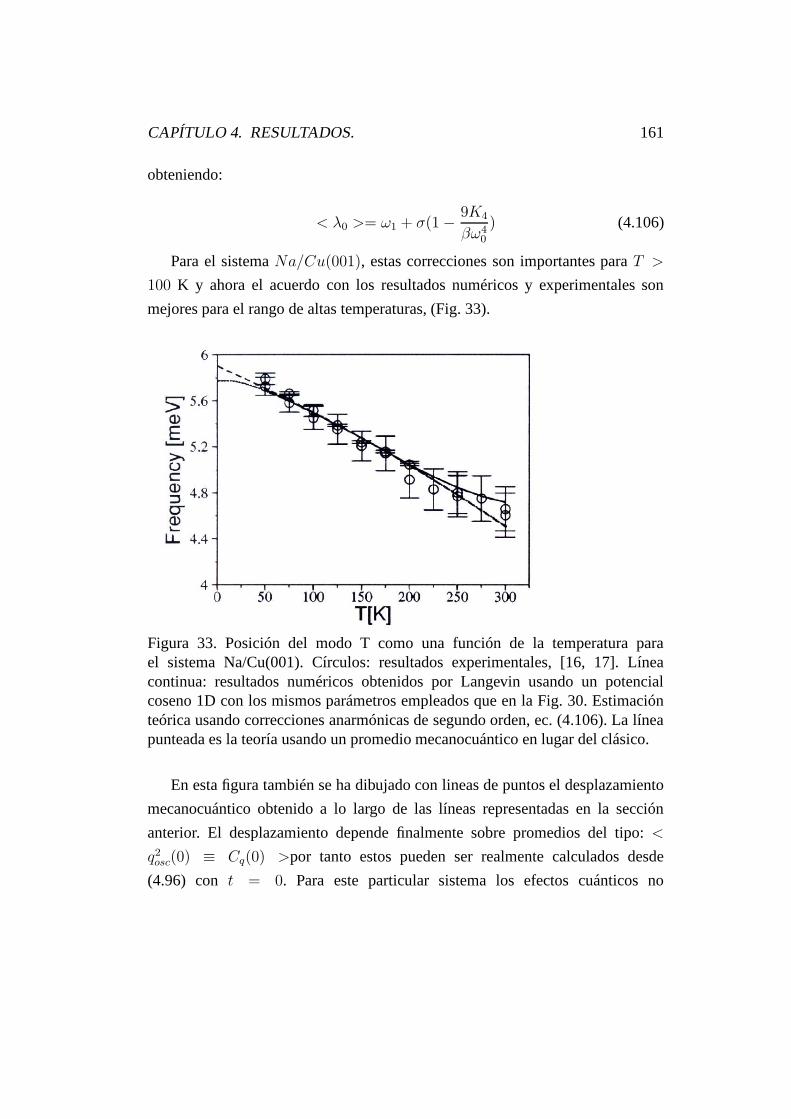
CAPÍTULO 4. RESULTADOS. 161
obteniendo:
< λ0 >= ω1 + σ(1 − 9K4
βω40
) (4.106)
Para el sistema Na/Cu(001), estas correcciones son importantes para T >
100 K y ahora el acuerdo con los resultados numéricos y experimentales son
mejores para el rango de altas temperaturas, (Fig. 33).
Figura 33. Posición del modo T como una función de la temperatura parael sistema Na/Cu(001). Círculos: resultados experimentales, [16, 17]. Líneacontinua: resultados numéricos obtenidos por Langevin usando un potencialcoseno 1D con los mismos parámetros empleados que en la Fig. 30. Estimaciónteórica usando correcciones anarmónicas de segundo orden, ec. (4.106). La líneapunteada es la teoría usando un promedio mecanocuántico en lugar del clásico.
En esta figura también se ha dibujado con lineas de puntos el desplazamiento
mecanocuántico obtenido a lo largo de las líneas representadas en la sección
anterior. El desplazamiento depende finalmente sobre promedios del tipo: <
q2osc(0) ≡ Cq(0) >por tanto estos pueden ser realmente calculados desde
(4.96) con t = 0. Para este particular sistema los efectos cuánticos no

CAPÍTULO 4. RESULTADOS. 162
son experimentalmente observables para el rango de temperaturas abarcadas.
También hay que hacer notar que a bajas temperaturas el corrimiento hacia
el azul mecanocuántico compensa el desplazamiento al rojo inducido por la
anarmonicidad.
4.8. Tratamiento cuántico para explicar la forma de
línea vibracional.
4.8.1. Modelo de un oscilador armónico amortiguado excitado.
En esta sección se muestra un desarrollo teórico que comienza a partir de
un modelo de oscilador armónico amortiguado. Primero, un oscilador armónico
es un buen punto de comienzo para simular el adsorbato, simple, aislado que
experimenta un movimiento de oscilación translacional frustrado (modo-T) sobre
la superficie. Segundo, la superficie metálica se considera como una colección de
osciladores independientes en equilibrio térmico a la temperatura de superficie
(baño térmico), cuyas fluctuaciones térmicas son introducidas en dicho baño
térmico, haciendo que el oscilador sienta un ruido, produciéndose al mismo
tiempo un mecanismo de pérdida de energía del adsorbato con dicho baño
térmico. Como consecuencia de este intercambio de energía entre el sistema
(adsorbato) y el baño térmico (sustrato) diremos que estamos ante un problema
de sistema abierto, ya que el sistema absorbe energía del baño térmico debido a la
presencia de estas fluctuaciones, y este cede energía al baño por un mecanismo
de amortiguamiento. Tercero, para un haz energético a baja energía, el efecto
neto del proceso de dispersión puede ser visualizado por medio de la creación
o aniquilación de sólo un cuanto vibracional a una frecuencia dada, aceptando la
“rotating wave approximation” [163]. El oscilador con frecuencia ω0 se encuentra
acoplado débilmente y de forma bilineal al baño térmico y pierde energía con
éste; del mismo modo, pero a la inversa, las fluctuaciones en el baño térmico se
reacoplan al modo T. Con todos estos ingredientes, el hamiltoniano total puede

CAPÍTULO 4. RESULTADOS. 163
ser escrito como:
H = HA +HR +HAR +HHe−A (4.107)
donde el hamiltoniano para el sistema dinámico es:
HA = ~ω0a†a (4.108)
La red de superficie se expresa como una suma de modos normales independientes
j, no acoplados entre sí :
HR =∑
j
~ωjb†jbj (4.109)
la interacción entre el adsorbato y la red se supone bilineal y bajo la “rotating
wave approximation” se escribe como:
HAR =∑
j
~[kja†bj + k†jab
†j ] (4.110)
donde los coeficientes de acoplamiento kj = cj/(mjωj)1/2 están escritos en
términos de fuerzas de acoplamiento cj y las masas efectivasmj de los osciladores
armónicos que constituyen la red de modos normales. En ecs. (4.108)-(4.110), a†
y a son los operadores de creación y aniquilación que describen el modo-T del
adsorbato y cumplen la relación de conmutación [a, a†]=1, los operadores bj y
b†j satisfacen la relación de conmutación de bosones [bjb†k]=δjk, los coeficientes
kj nos da cuenta de la fuerza del acoplamiento. Entonces, en (4.110) se supone
una interacción efectiva, parcialmente mediada por las excitaciones de fonones.
Diferentes formulaciones teóricas podrían seguir a este modelo, como por ejemplo
la formulación del operador densidad [163, 164] la cual incorpora velocidades
de relajación de coherencias y poblaciones y permite un tratamiento del efecto
de la temperatura. Aquí se ha utilizado una formulación cuántica de Langevin
[163] para interpretar los resultados experimentales en un lenguaje similar al
tratamiento clásico de Langevin. Para ello se ha partido de unas ecuaciones de

CAPÍTULO 4. RESULTADOS. 164
movimiento en la representación de operadores de Heisenberg mecanocuánticos.
Las fluctuaciones sobre el sistema son minimizadas en la escala de tiempo
durante el cual el baño térmico se encuentra correlacionado, pero tienen una gran
influencia sobre la escala de tiempo en la cual se produce el amortiguamiento.
Este procedimiento estándar consiste en resolver las ecuaciones acopladas para
los operadores del adsorbato y del baño térmico. Primero, se resolvieron las
ecuaciones del baño y esta suma sobre los modos de los osciladores armónicos
con términos de amortiguamiento la sustituimos en las ecuaciones de Heisenberg
de movimiento para el adsorbato. Escribiendo la ecuación general del movimiento
de un operador M como:
dM
dt=
1
i~[M,H] (4.111)
el cual puede ser usado para ambos operadores M = a(t) y M = bj(t). Para
derivar sus ecuaciones acopladas partiendo de condiciones iniciales a(0) y bj(0),
y de una distribución térmica para los modos del baño térmico j. Se resuelve
formalmente para los bj(t) y reemplazando esta suma de bj(t) obtenidas en la
ecuación para a, nos queda la siguiente expresión:
da
dt= −iω0a−
∑
j
|kj|2∫ t
0
dt′a(t′) exp[iωj(t′ − t)] + Fr(t) (4.112)
donde Fr(t) es el operador de Langevin para la fuente de ruido que viene
expresada como:
Fr(t) = −i∑
j
kjbj(0) exp(−iωjt) (4.113)
La ec.(4.112) puede ser interpretada como una ecuación cuántica de Langevin
generalizada con una fricción no óhmica del tipo:

CAPÍTULO 4. RESULTADOS. 165
γ(t′ − t) =∑
j
|kj|2 exp[iωj(t′ − t)] (4.114)
Bajo la conocida aproximación de Wigner-Weisskopff para este modelo [163], en
el que supone un tiempo suficientemente largo para asegurar que el sistema se
ha relajado, se obtiene la solución de la ecuación de Heisenberg de movimiento
para el operador a(t) de un oscilador amortiguado con un hamiltoniano dado por
ecs.(4.107)-(4.110) como:
a(t) = a(0) exp[−i(ω0 + ∆ω)t− γt/2]− (4.115)
∑
j
kj exp(−iωjt)[1 − exp(iωj − ω0 − ∆ω)t− γt/2]
ω0 − ωj + ∆ω − iγ/2bj(0).
Aquí, el desplazamiento en frecuencia ∆ω y la velocidad de relajación (coeficiente
de fricción) γ han sido definidos tal que:
−i∑
j
|kj|2(ωj − ω0 − iε)
= −i∫
dωjg(ωj)|k(ωj)|2/(ωj − ω0 − iε) ' γ/2 + i∆ω
(4.116)
donde g(ωj) es la densidad de modos normales del baño térmico a su temperatura
y ε → 0 es un pequeño incremento positivo. Después de la integración, la
velocidad de relajación o coeficiente de fricción viene dado por:
γ = 2πg(ω0)|k(ω0)|2 (4.117)
y cuyo desplazamiento ∆ω está dado por:
∆ω = −PP∫ ∞
−∞
g(ωj)|k(ωj)|2ωj − ω0
dωj (4.118)
donde PP significa el valor principal de Cauchy de la integral. Indicar que

CAPÍTULO 4. RESULTADOS. 166
la integral temporal (4.112) se simplifica para el caso que la disipación sea
instantánea, es decir γ(t′−t) = γδ(t′−t). Para este caso concreto, dicha ecuación
puede ser reescrita como una ecuación no generalizada de Langevin:
da
dt= −iω0a−
γa
2+ Fr(t) (4.119)
pudiendo ver que en el término γa2
aparece la fricción de forma explícita en
el movimiento vibracional. Las propiedades de Fr(t) son obtenidas de forma
sencilla cuando quedan definidos los promedios térmicos sobre la distribución
estadística del baño: con media < Fr(t) >R= 0 y función de correlación
< F †r (t1)Fr(t2) >R= γ(t1−t2)n , y para el caso en el que no tengamos disipación
instantánea, la transformada de Fourier de esta función varía con la frecuencia,
dando lugar a un ruido coloreado, donde n es la distribución de Bose-Einstein
a la temperatura T y energía cuántica vibracional ~ω0. Como una consecuencia,
encontramos en (4.115) que los promedios sobre el baño térmico de a y N = a†aestán dados por:
< a(t) >R=< a(0) >R exp[−i(ω0 + ∆ω)t− γt/2] (4.120)
< N(t) >R=< N(0) >R exp(−γt) (4.121)
4.8.2. Forma de línea para el modo T.
Partiendo de la ec. (3.65), la función de dispersión intermedia se escribe como:
I(∆K, t) =< e−i∆K.R(0)ei∆K.R(t) > (4.122)
La componente RK(t) = u(t) del desplazamiento paralelo al adsorbato a lo
largo de ∆K es la única coordenada que se considera explícitamente. Para
un oscilador armónico amortiguado, este desplazamiento unidimensional puede
escribirse como:

CAPÍTULO 4. RESULTADOS. 167
u(t) = (~
2mω T)1/2 exp(−γt/2) × [a exp(−iωT t) + a† exp(iωT t)] (4.123)
donde ωT = ω0 + ∆ω. Si partimos de la identidad:
exp[−i∆Ku(0)] exp[i∆Ku(t)] = (4.124)
exp1∆
2K2[u(0), u(t)] × exp−i∆K[u(0) − u(t)]
y usando la conocida fórmula [165]
< expQ >= exp(1
2< Q2 >) (4.125)
la función de dispersión intermedia con fricción puede expresarse como:
< exp[−i∆Ku(0)] exp[i∆Ku(t)] >= (4.126)
exp−∆K2[< [u(0)]2 > (1 + exp(−γt)
2)− < u(0)u(t) >]
la función de autocorrelación de desplazamientos viene dada por:
< u(0)u(t) >=~
2mωexp(−γt/2)[(1+ < a†a >) × exp(iωT t)+ (4.127)
< a†a > exp(−iωT t)]
donde la distribución de población de Bose-Einstein depende de la temperatura :
< a†a >=1
exp(~ωTβ) − 1= n (4.128)

CAPÍTULO 4. RESULTADOS. 168
por tanto, la función de dispersión intermedia, usando el parámetro adimensional
λ2 = ~∆K2/(2mω)se escribe como:
I(∆K, t) = exp[−λ2(2n + 1)(1 + exp(−γt)
2)]× (4.129)
expλ2 exp(−γt/2)[(n + 1) exp(iωt) + n exp(iωT t)]
Esta expresión puede ser reescrita como:
I(∆K, t) = exp[−λ2 coth(~ωTβ
2)(
1 + exp(−γt)2
)]× (4.130)
expλ2 1
sinh(~ωT β2
)exp(−γt/2) × [exp(iωT +
~ωTβ
2) + exp(−iωT t−
~ωTβ
2)]
si además, usamos la expansión:
exp[1
2y(x+ x−1)] =
∞∑
n=−∞xnIn(y) (4.131)
donde In es la función de Bessel modificada de primera especie, llegamos a la
expresión compacta:
I(∆K, t) = exp−λ2 coth(~ωTβ
2)(
1 + exp(−γt)2
)× (4.132)
∞∑
n=−∞exp(iωTn +
~ωTβ
2)In[y(t)]
donde
y(t) = λ2 1
sinh(~ωT β2
)exp(−γt/2) (4.133)

CAPÍTULO 4. RESULTADOS. 169
Para terminar con el desarrollo numérico, nos disponemos a calcular el factor de
estructura dinámico S(∆K,ω). Como se puede ver, en esta expresión compacta
para la función de dispersión intermedia I(∆K, t) aparecen tres exponenciales
con argumentos en el que aparecen la fricción γ y la frecuencia ωT . El factor de
Debye-Waller viene entonces dado por:
exp(−2$) ≡ exp[−λ2(n+ 1/2)] (4.134)
Si a continuación se realiza una expansión en serie de Taylor para cada
exponencial, se puede obtener la siguiente expresión para el factor de estructura
dinámico:
S(∆K,ω) = exp(−2$)∞
∑
n,m,l=0
(−1)lλ2(n+m+l)nn(n+ 1)m(n+ 1/2)l
n!m!l!×
(4.135)
(n+m + 2l)γ
[ω − (n−m)ωT ] + [(n+m + 2l)γ/2]2
Como puede verse, la forma de línea del factor de estructura dinámico es una
combinación de infinitas lorencianas. El pico cuasielástico se obtiene de los
eventos inelásticos con un balance neto de energía nulo (n=m) y los picos
inelásticos correspondientes al modo T son el resultado de eventos inelásticos
con n6=m. El desplazamiento y anchura global es el resultado de un número de
desplazamientos y anchuras parciales. La dependencia de este factor de estructura
dinámico con la temperatura proviene de la fricción γ, la frecuencia ω, y el factor
de Debye-Waller.

CAPÍTULO 4. RESULTADOS. 170
4.8.3. Relajación vibracional en los sistemas Na/Cu(001),
CO/Cu(001) y CO/Pt(111).
La dispersión inelástica de átomos de He ha sido usada para estudiar los
movimientos vibracionales a baja frecuencia del Na y CO sobre Cu(001) y CO
sobre Pt(111) a baja concentración, usando un haz monoenergético de He entre
10 meV y 20 meV [152, 20, 22]. En particular, para el sistema Na/Cu(001) los
resultados experimentales para la excitación por colisión del modo T muestran
una forma de línea con un desplazamiento del pico y una anchura que dependen
de la temperatura de superficie, estos datos fueron obtenidos entre 50 y 200
K. Los valores experimentales fueron entonces extrapolados a 0 K, dando una
energía de excitación ~ω0 = 6 meV para la vibración del modo T y una
fricción constante γ = 0,1ω0. Para relacionar este sistema con nuestro modelo,
es fundamental estimar los ordenes de magnitud temporales de interacción de
los átomos de He con la superficie, así como los tiempos de respuesta de la
excitación vibracional, ya sea por mecanismos vía fonones de la red, o par
electrón-hueco. Entonces, dicho esto, se sabe que la duración ∆texc del proceso
de transferencia de energética que conduce a una transición 0→1 para la vibración
del Na puede ser estimada de la relación de incertidumbre ∆E∆t=~, con ∆E=6
meV. Esto da un valor de ∆texc '110 fs. Por otro lado, el tiempo de colisión
∆tcol se estima considerando una distancia RHe−Na ' 4 Å y una velocidad
vHe conocida para el haz monoenergético de He empleado. Por tanto, se tiene
que ∆tcol > RHe−Na/vHe' 425 fs, y del orden de 1 ps. La tercera cantidad
importante, es la velocidad promedio de difusión kdif = 1/τMFPT que proviene
de nuestros cálculos del MFPT [30, 36], el cual nos da un valor de 3427 fs a
T = 200 K.
Primero, puede verse que con estos tiempos es claro que la difusión es
lenta sin la comparamos con el tiempo de colisión de transferencia energética
pudiendo entonces ignorarse. Segundo, se pueden comparar tiempos de colisión
y de excitación con tiempos típicos de relajación, par electrón-hueco, que son del
orden de 20 fs en Cu [166, 167, 168], concluyendo que este tipo de excitación

CAPÍTULO 4. RESULTADOS. 171
juega un papel en el fenómeno de relajación sólo durante un intervalo corto de
tiempo dentro del tiempo de colisión. Una justificación física similar se puede
aplicar a los otros dos sistemas de interés, CO/Cu(001) y CO/Pt(111). Nuestro
modelo teórico se deriva de asumir que la relajación es dominada a tiempos
largos por un mecanismo vía fonones de red pudiendo aplicarse a estos tres
sistemas. Además, se ha mostrado [169, 170] que la contribución par electrón-
hueco es independiente de la temperatura si las vibraciones son armónicas y
el correspondiente acoplamiento es lineal, mientras que en la dependencia con
la temperatura, es el mecanismo fonónico el que juega el papel importante.
Asumiendo pues, un simple modelo de Debye para las vibraciones de red del
Cu y Pt, la densidad de los modos vibraciones por unidad de frecuencia está dada
por:
g0(ω) = gD(ω) = 18πNω2/ω3D (4.136)
con g0(ω) = 0 para ω > ωD, donde N es el número de átomos de red
y ωD es la frecuencia de corte de Debye. Se han usado energías de Debye
εD = ~ωD igual a 27,57 meV para el Cu(001) y 19,82 meV para el Pt(111).
La correspondiente temperatura de Debye TD para el Cu y para el Pt son 320 K
y 230 K, respectivamente. Como bien sabemos, los experimentos se llevaron a
cabo a una temperatura de superficie comprendida entre 50 y 200 K, por debajo
de la TD y por tanto la probabilidad de eventos multifonónicos son bajos. La
transferencia de energía del adsorbato excitado a la red involucra estados iniciales
de red adicionales y una probabilidad de excitación proporcional a la población
inicial n(ω, T ) para cada modo de la red con frecuencia ω. Por tanto, la densidad
g debe ser multiplicada por un factor 1 + n (factor de población térmica), que
cambia esto a:
gT (ω) = g0(ω)(1 + n) = g0(ω)exp[~ω/(kBT )]
exp[~ω/(kBT ) − 1](4.137)
La otra cantidad que aparece en los cálculos es el acoplamiento k(ω) entre la

CAPÍTULO 4. RESULTADOS. 172
vibración del adsorbato y los modos de red a frecuencia ω. Como se indicó arriba,
ésto se considera como un acoplamiento efectivo el cual conserva la energía y
momento y experimenta valores significantes alrededor de la frecuencia ω0 del
adsorbato. Aquí se necesitan sólo frecuencias cercanas a ω0, como se vio en la
aproximación de Wigner-Weisskopff, (4.116) . Incluyendo ahora la dependencia
con la temperatura, se tiene que:
γT (ω) = 2πgT (ω)|k(ω)|2 (4.138)
∆ω(T ) = −PP∫ ωD
0
dωgT (ω)|k(ω)|2ω − ω0
(4.139)
Esta última expresión es el desplazamiento en frecuencias que depende de la
temperatura, mientras que ω0 +∆ω(T = 0) se ve como el valor que queda a 0 K y
podría referirse al desplazamiento residual que está presente a 0 K. El coeficiente
de fricción no es cero en este límite, porque el adsorbato excitado puede todavía
transferir su energía a la red, para inducir excitaciones desde movimientos del
punto cero de los modos de red, y puede también crear excitaciones par electrón-
hueco en la distribución electrónica.
Para este propósito de evaluación de la integral, se ha parametrizado la
cantidad positiva |k(ω)|2, localmente alrededor de ω = ω0 con la siguiente forma
lineal (expansión de la serie de Taylor hasta primer orden) :
|k(ω)|2 = [p+ q(ω − ω0)]/N (4.140)
Los parámetros p y q nos proporcionan información sobre la fuerza de los
acoplamientos adsorbato-red vibracional y sobre las masas de los modos de red,
para las frecuencias cercanas a la frecuencia vibracional del adsorbato. Los valores
de p y q pueden obtenerse dentro de un procedimiento de ajuste al desplazamiento
y anchura experimental del modo T. Primero, se comienza con (4.138) a ω = ω0,
nosotros calculamos p de la anchura experimental a una temperatura determinada,
se eligió 150 K. Ha sido observado que el parámetro p depende muy débilmente

CAPÍTULO 4. RESULTADOS. 173
de la temperatura de superficie para los tres sistemas estudiados. Para la misma
temperatura (150 K) y de (4.139) y el resultado del valor de p, se calcula el
parámetro q. Estos dos valores, p y q se usan para todo el rango de temperaturas.
Este procedimiento se realiza para los tres sistemas estudiados aquí. Se han usado
los siguientes valores para las frecuencias de los adsorbatos: ~ω0(Na/Cu) = 6
meV, ~ω0(CO/Cu) = 3,94 meV, y ~ω0(CO/Pt) = 5,94 meV. El signo del
parámetro q es una mera consecuencia de este procedimiento; es decir, no es
forzado a priori. Como q es proporcional a la primera derivada de ω0, su signo
relaciona la pendientes de k(~ω0)alrededor de ω0 y nos proporciona información
del perfil del acoplamiento vibracional alrededor de la frecuencia producida por
las vibraciones del adsorbato. Con este procedimiento, obtenemos los siguientes
valores de (p, q) para cada uno de los sistemas: (2,31 × 10−7a.u−2, 5,80 ×10−5a.u−1) para el Na/Cu(001); (6,44 × 10−8a.u−2, 1,58 × 10−5a.u−1) para el
CO/Cu(001), y (1,10×10−8a.u−2, 3,98×10−6a.u−1) para el CO/Pt(111). En Fig.
34 se representa la correspondiente anchura γT , y desplazamiento en frecuencia
ωT frente a la temperatura de superficie T para los diferentes sistemas.

CAPÍTULO 4. RESULTADOS. 174
Figura 34. Anchuras (FWHM) γT y frecuencias vibracionales ωT del modo Tinelástico frente a la temperatura de superficie T . Los círculos cerrados conlínea continua muestran los resultados teóricos que se comparan con los círculosabiertos. Arriba se muestra el sistema Na/Cu(001), en el medio el CO/Cu(001) yabajo el CO/Pt(111).
Observando el buen acuerdo entre los resultados experimentales (círculos
abiertos con las barras de error) y los calculados numéricamente (círculos
cerrados). El desplazamiento al rojo y al azul observados en los sistemas Na y CO
respectivamente son bien reproducidos con el cambio de signo en el valor de q.
Una interesante medida adicional de esta teoría es que nos proporciona una ley que
se puede extrapolar a T = 0. Las correspondientes anchuras y desplazamientos
provenientes de dicha extrapolación en nuestro modelo difiere sólo ligeramente de
los valores extrapolados experimentalmente. A altas temperaturas, los efectos de
anarmonicidad podrían jugar un papel más y más importante, pudiendo extender

CAPÍTULO 4. RESULTADOS. 175
nuestro modelo a tales efectos.
Por otro lado, para el sistema Na/Cu(001) , y usando los valores de p y
q obtenidos previamente, se ha calculado la función de dispersión intermedia
I(∆K, t) así como la forma de línea del factor de estructura dinámico S(∆K,ω).
El papel dominante lo supone una simple función lorenciana. La Fig. 35 nos
muestra la I(K, t) frente a ambos parámetros, de tiempo y de fricción, para una
transferencia de momento paralelo de 1,43 Å−1 y T = 150 K, con la dirección de
K a lo largo de [100] , una diagonal de la superficie.
Figura 35. Aquí se representa la función de dispersión intermedia I(∆K, t) frenteal tiempo t y frente al coeficiente de fricción γ reescalados por la frecuencianatural ω0, calculada teóricamente a un ∆K = 1,43 Å−1y T = 150 K a lo largode la dirección diagonal [100].
Sólo los tres términos n=-1,0,1 , en (4.132) han sido incluidos, ya que son los
necesarios para obtener una buena convergencia. La frecuencia de oscilación está
dada por ωT . Puede observarse que cuando la fricción incrementa, el decaimiento

CAPÍTULO 4. RESULTADOS. 176
de la función de dispersión intermedia viene a ser más y más pronunciada. En
la Fig. 36 se muestran los correspondientes valores numéricos del factor de
estructura dinámico frente a la frecuencia y fricción, apareciendo los dos picos
inelásticos de transferencia energéticas para la excitación y desexcitación de un
cuanto vibracional del adsorbato.
Figura 36. Factor de estructura dinámico S(∆K,ω) calculado numéricamente enfunción de la frecuencia ω y el coeficiente de fricción γ. Se muestran los dospicos inelásticos correspondiente a los modos T de los procesos de excitación ydesescitación de un fonón. Para mayor claridad, se ha suprimido la intensidad delpico cuasielástico, correspondiente al proceso n = m.
Para mayor claridad, el gran pico cuasielástico, proviene de la contribución
(m=n), y no ha sido incluido en la Fig. 36. Como se esperaba, las formas de
líneas van decreciendo en altura y van ensanchándose a medida que la fricción
va incrementándose. Esto sigue de (4.135), que los perfiles de líneas parciales
contribuyen a la forma de línea total, desplazándose (n-m) ωT y ensanchándose
(n+m+2l)γT/2. Si sólo el primer armónico del modo T es excitado, entonces

CAPÍTULO 4. RESULTADOS. 177
n = ±1,m = 0, y l = 0, siempre y cuando contribuciones debido a n −m = 1
para los demás n y m no son considerados, el desplazamiento está dado por ωTen (4.139), y la correspondiente anchura está dada por γT en (4.138).
4.9. Formas de línea para el pico cuasielástico y el
modo T.
4.9.1. Forma de línea de difusión.
En este modelo se va a proponer una forma de línea que tenga en cuenta ambos
procesos de difusión y vibración, y compararla así con el factor de estructura
dinámico que se mide experimentalmente por QHAS. Como ya comentamos
anteriormente, este modelo se apoya en un aproximación gaussiana para la función
de dispersión intermedia, (3.77). Por tanto, si partimos en general de (3.81)
combinada con:
τr ≡1
< v2∆K >
∫ ∞
0
< v∆K(t)v∆K(0) > (4.141)
nos lleva a la relación de Green-Kubo para el coeficiente de difusión (3.83) y
por tanto, bajo la aproximación gaussiana, la función de dispersión intermedia
contiene el coeficiente exacto de difusión contenido en el parámetro χ, (3.80).
Este parámetro gobernará la coherencia dinámica del proceso de difusión y por
tanto la forma de línea del pico Q. En el límite χ→ ∞, o equivalente, τr → ∞ o
l→ ∞, se obtiene un perfil gaussiano (aproximación a tiempos cortos, t τr):
I(∆K, t) ∝ e−D|∆K|2t2/2τr (4.142)
y el correspondiente factor de estructura dinámico presenta un perfil gaussiano:
S(∆K, ω) ∝ 1
|∆K|v0exp[−ω2/2|∆K|2v2
0] (4.143)

CAPÍTULO 4. RESULTADOS. 178
con la velocidad media isotrópica v0 =√
< v2∆K > =
√
D/τr. La correspon-
diente anchura energética depende linealmente de la transferencia del momento
paralelo y v0, Γ ∝v0|∆K|. El significado físico de este resultado es claro. La
correspondiente función de van Hove es de la forma:
G(R, t) ∝ 1
(v0t)2exp[−R2/(t2v2
0)] (4.144)
la cual es interpretada como la probabilidad que la partícula se desplace una
distancia R en un tiempo t asumiendo una velocidad constante R/t y una
distribución inicial de Maxwell de velocidades, con v0 =√
kBT/m. Eso significa
que para tiempos mucho más cortos que el tiempo medio de colisión la partícula se
comporta como casi libre y la coherencia dinámica domina, esto es, las partículas
mantienen memoria de sus velocidades y las variaciones de probabilidad toman
importancia sobre distancias más cortas que el recorrido libre medio. Este
comportamiento ha sido encontrado en experimentos con QHAS de Xe adsorbidos
sobre Pt(111) [15], proporcionando evidencias de la completa movilidad del gas.
Para el caso opuesto, χ 1, nosotros tenemos una aproximación a tiempos largos
(t τr),
I(∆K, t) ∝ e−∆K2Dt (4.145)
y el espectro tiene la ya conocida forma lorenciana, (3.70).
Aquí las correlaciones entre velocidades a diferentes tiempos son práctica-
mente nulas y por tanto el proceso es únicamente difusivo (caminante aleatorio).
La FWHM es Γ = 2D|∆K|2. La transformada de Fourier exacta de (3.79) pa-
ra todos los rangos de tiempos pueden ser expresadas en términos de la función
gamma completa e incompleta:
S(∆K, ω) =eχ
2
τrπ
χ−2χ2
Re χ−i2ωτr [Γ(χ2+iωτr)−Γ(χ2+iωτr ,χ2)] = (4.146)

CAPÍTULO 4. RESULTADOS. 179
eχ2
2π
∞∑
n=0
(−1)nχ2n
n!
2[(χ2 + n)/τr]
ω2 + [(χ2 + n)/τr]2
Como el valor del parámetro χ decrece, el factor de estructura dinámico va desde
una casi gaussiana a una forma de linea lorenciana llegando el espectro a ser más
y más estrecho. Este efecto general es llamado estrechamiento dinámico [171],
(Fig. 37).
Figura 37. En esta figura se representa el factor de estructura dinámico teórico,ec.(4.146) (curva continua) y el numérico (círculos abiertos) sin la presenciadel potencial adiabático, a T = 200 K y diferentes transferencias de momentoparalelo. En la Fig. de arriba se utilizó un ∆K =0.11 Å−1 y en la Fig. de abajoun ∆K =1.23 Å−1. Los valores de χ =0.33 y 3.67 muestran una forma de línealorenciana y gaussiana respectivamente. En la ventana se representa la función dedispersión intermedia numérica.
Aquí se representa el factor de estructura dinámico para una partícula sin
interacción potencial adiabática (círculos abiertos) y las predicciones teóricas
(línea continua, (4.146) a T = 200 K a dos valores diferentes de ∆K, 0.11 Å−1

CAPÍTULO 4. RESULTADOS. 180
y 1.23 Å−1. El parámetro χ toma los valores de 0.33 y 3.67, respectivamente,
mostrando una clara transición entre el perfil lorenciano y gaussiano. En
la ventana, se han representado las correspondientes funciones de dispersión
intermedia. Un importante punto es determinar el rango de valores de ∆K en el
que la aproximación gaussiana es válida para la función de dispersión intermedia.
Sabemos que, a largas transferencias de momento paralelo, el adsorbato comienza
a sentir la presencia del potencial adiabático, y por tanto la FWHM no es
cuadrática en ∆K como en (3.70), y como consecuencia, la aproximación
gaussiana comienza a fallar, por lo que podemos decir, que este proceso comienza
a perder el comportamiento gaussiano. Ahora bien, el modelo más simple que
incluye la periodicidad de la superficie es el dado por Chudley-Elliot, que como
bien sabemos proponen una ecuación maestra para la función de van Hove,
asumiendo saltos discretos instantáneos en una red de Bravais. Esto da de nuevo
una exponencial para la función de dispersión intermedia:
I(∆K, t) = I(∆K, 0)e−t/τ1(∆K) (4.147)
donde el tiempo de correlación τ1(∆K) presenta una dependencia periódica en
∆K de la forma:
τ−11 (∆K) = ν
∑
j
Pj[1 − cos(j.∆K)] (4.148)
siendo ν la frecuencia total de saltos fuera de un sitio de adsorción y Pj
la probabilidad relativa para un salto con un desplazamiento vectorial j. Para
pequeños valores de ∆K, y considerando saltos no correlacionados en la dirección
paralela, nosotros vemos que:
τ1(∆Kx,y) ∼ν
2
∑
j
Pjj2∆K2
x,y =ν
2< j2 > ∆K2
x,y (4.149)
donde Pj es ahora la probabilidad de saltar sobre j sitios de red en un salto simple
a través de la dirección x, o y, e ∆Kx,y la transferencia del vector de onda paralelo

CAPÍTULO 4. RESULTADOS. 181
en esta dirección. La expresión para el coeficiente de difusión con el modelo de
difusión de salto [139]:
D =ν
2< j2 > (4.150)
recuperando la aproximación lorenciana en el límite a tiempos largos, (3.70). En
orden a ilustrar la discusión precedente , se elige un prototipo de modelo para la
difusión átomo-superficie: átomos de Na a baja concentración en una superficie
simétrica de Cu(100). Este modelo ha sido investigado de forma experimental
[152, 172, 15, 16] y de forma teórica [152, 172, 173, 14, 27, 113, 33] usando
diferentes aproximaciones. A partir de la simulaciones numéricas y extensivos
ajustes a los experimentos, se propuso una superficie de energía potencial
bidimensional [16, 17]. Los parámetros importantes PES para la discusión del
sistema son la altura de la barrera de difusión, V = 75 meV a través de las
direcciones x e y, y la posición del modo T extrapolado a temperatura 0 K,
ω0 = 6 meV. El coeficiente de fricción ha sido estimado de la extrapolación de
la anchura del pico T y comparación con los experimentos y las simulaciones
por Langevin, dando un γ = 0,1ω0. Se toman esta PES y estos parámetros
como nuestro punto de inicio para las simulaciones numéricas, resolviendo la
ec. de Langevin por un algoritmo de Verlet de tercer orden [116]. Las funciones
de correlación importantes son determinadas por promedios sobre apropiados
colectivos de trayectorias. Como primera comparación, en Fig. 38 se muestra la
FWHM predicha por (3.70) y la anchura obtenida del ajuste lorenciano al factor
de estructura numérico a T=150 K.

CAPÍTULO 4. RESULTADOS. 182
Figura 38. FWHM del pico Q a T = 150 K como una función de la transferenciade momento paralelo, simbolizado en esta figura como K para el sistemaNa/Cu(001). Los círculos llenos muestran la FWHM extraída de la lorenciana(3.70) que se ajusta al pico Q obtenido de la simulación numérica de Langevin.Con línea discontinua se muestra los resultados obtenidos con el modelo deChudley-Elliot en dos dimensiones, (4.149). La curva continua nos muestrala FWHM de la aproximación gaussiana, (4.146), utilizando el coeficiente dedifusión obtenido numéricamente.
Primero, indicar que para el rango de temperaturas experimental (50 < T <
350 K) los valores del parámetro χ son tales que la lorenciana es una buena
aproximación. Segundo, sólo para ∆K < 0,15Å−1 la aproximación gaussiana va
bien. Por otro lado, el modelo de Chudley-Elliot da un buen ajuste de la anchura
angular del pico Q como función de ∆K, para casi toda la región de ∆K excepto
para los valores cercanos a la primera zona de Brillouin , donde el solapamiento
del proceso de vibración con la difusión comienza a ser considerable [30]. En la
práctica este modelo presenta dos fuentes de error:

CAPÍTULO 4. RESULTADOS. 183
Primera: la figura de saltos instantáneos es una buena aproximación para
barreras V ‡/(kBT )≥ 3 [150, 30]; Segunda, y más importante: la FWHM no
es realmente una función periódica de ∆K cuyo periodo es el vector de la red
recíproca [152, 16, 15]. Esto se debe al hecho de que a largos valores de la
transferencia de momento paralelo la contribución del modo vibracional T al
pico cuasielástico es considerable, y los procesos de difusión y vibración no
pueden separarse. Hemos visto [30] que en efecto, las frecuencias de salto y sus
distribuciones pueden ser calculadas analíticamente en muy buena aproximación
(barreras donde V ‡/(kBT ) ≥ 3) usando la teoría del movimiento de Kramers
generaliza a potenciales periódicos [158]. La aproximación de Kramers tiene la
ventaja que la FWHM dentro del modelo de Chudley-Elliot puede ser estimada
usando únicamente dos parámetros físicos: una frecuencia de salto efectiva y la
pérdida de energía δ que la partícula experimenta con el baño térmico cuando pasa
de una barrera de potencial a la siguiente. Estos dos parámetros determinaran el
completo proceso de difusión si la difusión es activada térmicamente y no hay
fuerte acoplamiento con la vibración. Además, estos dos parámetros se encuentran
fácilmente relacionados con la curvatura de energía potencial en la barrera, y
con el coeficiente de fricción, y por tanto se puede estimar alturas de barreras
y constantes de fricción de un ajuste al experimento por medio de un modelo de
Kramers generalizado [30].
4.9.2. Forma de línea de vibración.
La aproximación gaussiana para la función de dispersión intermedia, puede
ser también el punto inicial para una investigación analítica del modo T. Ahora, la
importancia en la dinámica del adsorbato tiene lugar cerca del mínimo del pozo
de potencial y este puede ser expandido en una serie de Taylor en la variable
R. Reteniendo solo el primer término se obtiene un potencial parabólico y la ec.
(3.77) es exacta, ya que el proceso es gaussiano. La función de autocorrelación
de velocidades para el oscilador armónico tiene la forma de ec.(4.72) y el factor
de estructura dinámico puede ser expresado analíticamente ahora por (4.75)

CAPÍTULO 4. RESULTADOS. 184
donde se obtiene que el perfil del modo T no es afectado por la transferencia
de momento paralelo (modo sin dispersión) como ya se ha observado en el
experimento. Experimentalmente, el valor del coeficiente de fricción se estima
usualmente por extrapolación con la anchura del pico T a baja temperatura, un
procedimiento el cual es justificado por (4.75). Cuando la temperatura aumenta,
usualmente se observa un desplazamiento y una anchura en el modo T, y una
de las posibles interpretaciones es la atribución a la anarmonicidad del potencial,
otra interpretación se atribuye a una desplazamiento armónico con ruido coloreado
como ya vimos también en la sección anterior. Recientemente, se han propuesto
fórmulas analíticas para el desplazamiento y la anchura del modo T, así como su
forma de línea, interpretado de forma anarmónica. En esta vía, el desplazamiento
y anchura experimental se puede relacionar fácilmente a la curvatura del potencial
adiabático cerca del mínimo del pozo de potencial. Un tratamiento de primer
orden de esta curvatura, produce un desplazamiento lineal con la temperatura,
mientras que para obtener la dependencia lineal con la temperatura de la anchura
hay que irse a una aproximación de segundo orden [173]. La aproximación
gaussiana es también buena para el modo T, opuesto al caso del pico Q. De
forma interesante, el perfil de línea para el modo T puede también mostrar
un efecto de estrechamiento dinámico similar al predicho para el pico Q en
la aproximación gaussiana. Primero, nosotros expandimos la segunda identidad
(3.76) en momentos, en lugar de cumulantes, y mantenemos el segundo orden (el
cual es equivalente a la expansión de Taylor en segundo orden de (3.77) alrededor
de ∆K = 0). Entonces, la función de dispersión intermedia es ahora proporcional
a la función de autocorrelación de posiciones. La función de dispersión puede ser
calculada asumiendo el modelo del oscilador de Kubo [74] en la cual la frecuencia
harmónica es perturbada por una frecuencia aleatoria incluyendo el efecto de
anarmonicidad. Entonces, se puede mostrar [173] que, a tiempos largos, la función
de dispersión intermedia I(∆K, t) viene dada por (4.103). Cuando se realiza
la transformada de Fourier en tiempos, la primera exponencial en (4.103) da la
posición del pico, con el desplazamiento ∆ω1 para el primer orden en K4[173].
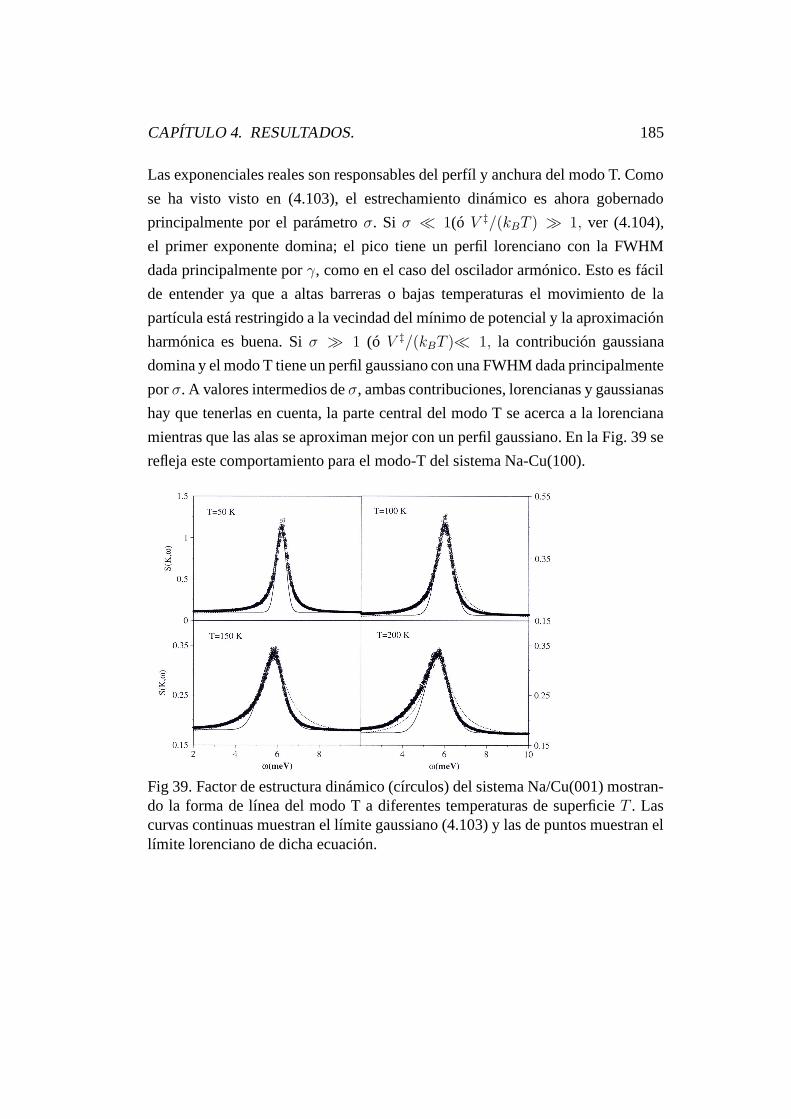
CAPÍTULO 4. RESULTADOS. 185
Las exponenciales reales son responsables del perfíl y anchura del modo T. Como
se ha visto visto en (4.103), el estrechamiento dinámico es ahora gobernado
principalmente por el parámetro σ. Si σ 1(ó V ‡/(kBT ) 1, ver (4.104),
el primer exponente domina; el pico tiene un perfil lorenciano con la FWHM
dada principalmente por γ, como en el caso del oscilador armónico. Esto es fácil
de entender ya que a altas barreras o bajas temperaturas el movimiento de la
partícula está restringido a la vecindad del mínimo de potencial y la aproximación
harmónica es buena. Si σ 1 (ó V ‡/(kBT ) 1, la contribución gaussiana
domina y el modo T tiene un perfil gaussiano con una FWHM dada principalmente
por σ. A valores intermedios de σ, ambas contribuciones, lorencianas y gaussianas
hay que tenerlas en cuenta, la parte central del modo T se acerca a la lorenciana
mientras que las alas se aproximan mejor con un perfil gaussiano. En la Fig. 39 se
refleja este comportamiento para el modo-T del sistema Na-Cu(100).
Fig 39. Factor de estructura dinámico (círculos) del sistema Na/Cu(001) mostran-do la forma de línea del modo T a diferentes temperaturas de superficie T . Lascurvas continuas muestran el límite gaussiano (4.103) y las de puntos muestran ellímite lorenciano de dicha ecuación.

CAPÍTULO 4. RESULTADOS. 186
La transformada de Fourier de la función de dispersión intermedia completa,
(4.103), no mostrada aquí, reproduce numéricamente el perfil del modo T
mostrado con círculos abiertos. La curva punteada es una aproximación lorenciana
y la curva continua muestra la gaussiana. A altas temperaturas la parte derecha del
pico (no afectada por el solapamiento con el pico Q) se aproxima mejor al perfil
gaussiano, pero a bajas temperaturas el perfil lorenciano es el que domina.
4.10. Difusión y vibración a baja frecuencia.
Un procedimiento para entender mejor el acoplamiento vibración-difusión en
la dinámica de adsorbatos consiste en asumir un modelo simple para la función de
autocorrelación de velocidades dada por (3.107). A altas temperaturas, la difusión
es predominante y τ2 es aproximadamente el coeficiente de difusión dividido por
la temperatura escalada kBT . A bajas temperaturas (para el sistema estudiado
aquí, con una barrera de difusión V = 75 meV, y a T < 50 K, los saltos a través
de sitios de adsorción son eventos poco frecuentes y la dinámica vibracional es
la única que aparece. Entonces, τ−12 ∼ γ/2 , y a baja temperatura, se tiene que
ω2 ∼ ω1, ver (4.72). La fase ϕ debería ser ajustada a cada temperatura. En fig. 40
se muestra la velocidad de autocorrelación numérica para el sistema Na/Cu(110)
a dos diferentes temperaturas: T= 50 K (curva de puntos) y T=150 K (curva
continua).

CAPÍTULO 4. RESULTADOS. 187
Figura 40. En esta figura se representa la función de autocorrelación numérica parael Na/Cu(001) a dos temperaturas diferentes. Curva de puntos: T=50 K. Curvacontinua: T=150 K. La línea discontinua nos muestra el mejor ajuste realizadocon la ec. (3.107) a T=150 K.
El desplazamiento del modo T así como el rápido decaimiento con la
temperatura debido a la aparición del proceso de difusión puede verse claramente.
El mejor ajuste a la ecuación analítica (3.107) a T=150K es dibujada como curva
discontinua, mostrando un buen acuerdo. Los parámetros tomados del ajuste son
ω2 = 2,13,10−4a.u−1 y τ−12 = 2,21,10−5a.u−1. Estos últimos valores comparan
bien con el coeficiente de fricción γ = 0,1ω0 = 2,2,10−5a.u−1, mostrando que
un decaimiento de la función de correlación como un modelo de partícula libre
es una buena aproximación para esta temperatura. Ahora, si (3.107) se incluye en
(3.77) (aproximación gaussiana) y la integración es llevada a cabo analíticamente,
la función de dispersión intermedia puede ser escrita como:

CAPÍTULO 4. RESULTADOS. 188
I(∆K, t) = exp[−χ2f(ω2, t)]e−χ2A1−χ2A2t
∞∑
n,m
(−1)n(−1)m
n!m!χ2(n+m)An3A
m4 ×
(4.151)
e−i(m−n)ϕe−(m+n)t/τ2e−i(m−n)ω2t
y la función f(ω2, t) es definida como:
f(ω2, t) = A1 + A2t+ A3eiϕe−(τ−1
2−iω2)t + A4e
−iϕe−(τ−1
2+iω2)t (4.152)
donde los coeficientes son expresados como:
A1 =τ−22 [2τ−1
2 ω2 sinϕ+ (ω22 − τ−2
2 ) cosϕ]
(τ−22 + ω2
2)2
(4.153)
A2 =τ−22 [2τ−1
2 ω2 cosϕ− (ω2 sinϕ]
τ−22 + ω2
2
(4.154)
A3 =1
2τ 22 (τ−1
2 − iω2)2(4.155)
A4 =1
2τ 22 (τ−1
2 + iω2)2(4.156)
Finalmente, el factor de estructura dinámico queda como:
S(∆K, ω) =e−χ
2A1
π
∞∑
n,m=0
(−1)n+mχ2(n+m)An3Am4
n!m!e−i(m−n)ϕ× (4.157)

CAPÍTULO 4. RESULTADOS. 189
[χ2A2 + (n+m)τ−12 ]
[ω − (n−m)ω2]2 + [χ2A2 + (n+m)τ−12 ]2
Ahora se tiene una doble suma sobre perfiles lorencianos. El pico Q resulta de
los términos m = n y el pico-T viene de las diferentes combinaciones de los
n y m índices correspondiente a los procesos de creación y aniquilación del
modo T. Pero otra vez, dependiendo de los valores de χ, tales sumas pueden
globalmente contribuir a los dos casos extremos, yendo de una pura gaussiana a un
perfil lorenciano , o incluso perfiles intermedios. De uno puede ver claramente la
contribución del modo T y del pico Q y viceversa. Contrario a lo visto a veces en
la literatura, la forma total de línea no es una simple suma de dos contribuciones.
En la Fig. 41 se representa el factor de estructura dinámico numéricamente para el
sistema Na/Cu(001) a T=150 K (círculos) comparado con la predicción analítica
(4.157) para ∆K = 0,11Å−1
.
Figura 41. Factor de estructura dinámico numérico para el Na/Cu(001) (círculos) aT=150 K y ∆K = 0,11Å−1. La línea continua nos muestra la predicción analítica,(4.157). En la ventana de muestra el pico T ampliado a un ∆K = 0,88 Å−1, a lamisma temperatura.

CAPÍTULO 4. RESULTADOS. 190
Se ha observado que para el rango de valores de ∆K dentro del intervalo
[0, 0,11] Å−1,la aproximación gaussiana es válida, esto es, (4.157) reproduce
muy bien la forma de línea de los picos Q y T. A grandes transferencias de
momento paralelo (ventana), se observa un solapamiento apreciable entre los
dos picos pudiendo ver cómo la aproximación gaussiana falla para el pico Q.
Sin embargo, la forma de línea permanece con muy buena aproximación para
el modo T. Esto se debe a la naturaleza sin dispersión del modo T. Hay que
remarcar que la aproximación gaussiana para vibraciones es precisa no sólo para
todos los valores de ∆K, sino también para un amplio rango de temperaturas. En
este punto de la discusión, se examina que el alcance de (4.157) podría ser usada
como una formula de trabajo para determinar, en la zona de Brillouin , qué perfil
(gaussiano-o-lorenciano) domina en el pico Q, bien sea en el cálculo numérico
o en el experimento. Este es un importante asunto debido al hecho que muchas
veces en el procedimiento de convolución el perfil es asumido de antemano. Ya
que (4.157) es obtenida con la aproximación gaussiana, el correspondiente valor
de χ extraído de un ajuste no es en general el mismo como el nominal obtenido
de (3.80), una vez que τr es reemplazado por τ2. En Fig. 42 se ha representado
el valor de χ frente a la transferencia de momento paralelo extraída del ajuste a
(4.157) en la simulación numérica a T=150 K.

CAPÍTULO 4. RESULTADOS. 191
Figura 42. En esta figura se representa χ en función de la transferencia demomento paralelo, obtenido ajustando (4.157) al pico cuasielástico numérico aT=150 K. En la ventana se muestra la calidad del ajuste para ∆K=1.23 Å−1.
En la ventana, se muestra la calidad del ajuste a ∆K = 1,23Å−1
(punto negro)
una vez que el pico-T es multiplicado por un factor constante (En efecto, los
coeficientes A3 y A4 en (4.152). La información es obtenida del análisis del perfil
del pico Q. A muy bajos y altos valores de ∆K el pico Q se aproxima a una
lorenciana, y un valor intermedio es predicho a χ∼ 1. Por último mencionar que
el perfil de una lorenciana efectiva asumido en el procedimiento de deconvolución
[152, 172, 16] a altas transferencias de momento paralelo es, en nuestra opinión,
un punto débil del método de trabajo empleado por los experimentalistas.

Capítulo 5
CONCLUSIONES.
En este último capítulo se va a proceder a mostrar las conclusiones a las que
se ha llegado después de haber realizado esta memoria de investigación con el
objetivo de estudiar la dinámica disipativa de los procesos de difusión y vibración
de adsorbatos sobre superficies metálicas.
Conclusión 1. Se ha estudiado en detalle la dinámica caótica y las propiedades
de transporte de adsorbatos de Na sobre la superficie de Cu(001) usando un
potencial semiempírico no separable. Se ha visto que para el rango de energías
comprendido entre la barrera de difusión (74.6 meV) y el máximo de potencial,
el espacio de fases muestra una coexistencia de regiones caóticas con islas
de estabilidad producidas por los movimientos periódicos más simples. Los
movimientos periódicos encontrados son de dos clases: los localizados dentro del
pozo de potencial, no originando difusión, y los que originan largos saltos a lo
largo de las direcciones paralelas x e y , y las diagonales. Hay que señalar que
los movimientos localizados son estables para energías mucho más altas que la
correspondiente al punto silla de la barrera de potencial (barrera de difusión).
Las órbitas correspondientes a las traslaciones paralelas frustradas sufren una
bifurcación abrupta cuando las barreras energéticas se abren, originando una
estructura de islas alrededor de islas que originan movimientos de arrastre de
todo tipo de periodos posibles. Las traslaciones diagonales son inestables pero
192

CAPÍTULO 5. CONCLUSIONES. 193
experimentan la misma bifurcación cuando se alcanza una energía determinada.
Dándose la interesante situación en que la órbita diagonal llega a estabilizarse por
las pequeñas depresiones situadas en los máximo de potencial, donde se sitúan
los átomos de Cu, peculiaridad que nos lleva a una posible explicación para la
inusual alta frecuencia de trayectorias que migran a lo largo de esta dirección
detectada experimentalmente. En este sistema Na/Cu(001) se ha encontrado
difusión anómala para un rango de energías en el que ambos movimientos
coexisten y son estables. Por otro lado, a altas energías (desde 95 hasta ∼120
meV), sólo se ha observado difusión normal. Se han calculado coeficientes
de difusión microcanónicos en este rango energético usando simulaciones de
dinámica molecular y dos expresiones estadísticas aproximadas, verificando
ambos tratamientos numérico y teórico la influencia de saltos largos debido a la
estabilización del movimiento de difusión libre. La influencia de vuelos largos
correlacionados ha sido observado experimentalmente en otros sistemas, pero
no así difusión anómala. Esto nos da una idea de la importancia del tipo de
potencial empleado en este sistema, pues el que sea no separable, genera una
dinámica determinista compleja, mostrando diferentes grados de integrabilidad
y caos dependiente de la energía que se utilice, y esta dinámica caótica es la
que induce a diferentes comportamientos difusivos. También se ha observado que
si en esta dinámica determinista, se introduce el efecto de una fricción, y una
temperatura simulando la fuerza estocástica, estos elementos alteran la dinámica
determinista haciendo que el adsorbato alcance el equilibrio térmico, y obteniendo
así difusión normal, debido a que la fricción elimina todo tipo de difusión anómala
que pudiera generar el potencial adiabático no separable. Por otro lado se ha
visto que este tiempo de relajación al equilibrio es por definición la inversa del
coeficiente de fricción, por tanto si la constante de fricción es pequeña, es posible
que el tiempo necesario para que el adsorbato alcance el equilibrio térmico sea
muy superior al tiempo necesario para que se equilibre la dinámica caótica, y por
tanto, bajo esas condiciones poder observarse una difusión anómala pasajera antes
de que el sistema alcanzara el equilibrio térmico. No ha sido este el caso, ya que

CAPÍTULO 5. CONCLUSIONES. 194
el coeficiente de fricción γ medido experimentalmente fue de alrededor de 1 ps−1,
impidiendo la detección de la difusión anómala pasajera generada por el potencial
no separable.
Conclusión 2. Como ya es bien sabido el efecto de la temperatura hace
que la partícula encerrada dentro de un pozo de potencial puede ganar energía
térmica para remontar la barrera de potencial y difundirse por la superficie. Este
idea intuitiva y simple, básica en la teoría de Kramers sobre superficies, se ha
aplicado por primera vez en esta memoria para explicar los procesos de difusión
activados térmicamente de adsorbatos sobre superficies. Esta teoría debería ser
usada como una herramienta estándar para ajustar e interpretar los resultados
experimentales para difusión de adsorbatos sobre superficie a baja concentración.
Con esta teoría de Kramers hemos podido reproducir la dependencia obtenida
experimentalmente de la FWHM del pico cuasielástico frente a la transferencia
de momento paralelo haciendo uso únicamente de dos parámetros, la velocidad de
difusión espacial y la pérdida de energía cuando la partícula atraviesa un pozo de
potencial. Esta aproximación de Kramers nos proporciona expresiones analíticas
faciles de implementar a la simulación numérica, y de ajustar al experimento,
debido a que se evita el tiempo que se tarda en la simulación numérica. Además,
se ha demostrado que esta teoría de Kramers da una muy buen estimación de
las velocidades, coeficientes de difusión y distribuciones de saltos de partículas
adsorbidas sobre superficies cuando las barreras de difusión son V ‡/(kBT ) > 4
, teniendo en cuenta correcciones de barrera finita, correcciones que por primera
vez se han empleado en esta teoría de Kramers aplicada a superficies. Recalcar
que una restricción en la teoría es la necesidad de que el acoplamiento potencial
sea débil, de forma que la dinámica clásica alrededor de la energía de la
barrera sea regular, para poder tener una buena estimación del valor por pérdida
energética. También se ha visto que si nos encontramos ante un proceso de
difusión unidimensional y la altura de la barrera de potencial V ‡/(kBT ) > 3
esta teoría puede reemplazar a la simulación numérica de Langevin. Ahora bien,
si el proceso de difusión es multidimensional, esto no es posible. Y en este caso,

CAPÍTULO 5. CONCLUSIONES. 195
uno necesita entender la dinámica clásica determinista, debiendo estar seguro que
las medidas experimentales de interés se encuentran en el rango en el que la teoría
de Kramers es aplicable. Y por supuesto, siempre y cuando nos encontremos bajo
una condición de baja concentración, donde la interacción adsorbato-adsorbato
no afecte a la difusión. También decir que en esta teoría se ha asumido que los
saltos en la dirección x no están correlacionados con los saltos en la dirección y,
esto no es siempre cierto, pues como se ha visto precisamente en este sistema,
la frecuencia de saltos máxima en la dirección [100] no es dos veces los saltos
medidos en la dirección [110]. Todo esto implica que esta teoría del cambio de
Kramers aplicada a la difusión activada térmicamente sobre superficies puede
deparar algunas sorpresas en el futuro.
Conclusión 3. Se ha desarrollado una teoría analítica clásica hamiltoniana para
explicar la forma de línea, desplazamiento y anchura del modo T basado en dos
suposiciones: la primera que la dinámica de los adsorbatos aislados está bien
reproducida por una ecuación de Langevin generalizada y la segunda suposición
es que la primera corrección anarmónica de la interacción potencial es suficiente
para tener en cuenta la dependencia con la temperatura. La validez de la primera
suposición se ha demostrado para diferentes sistemas átomo-superficie a bajo
recubrimiento, donde los resultados obtenidos por las simulaciones numéricas
han sido contrastados con los resultados experimentales de QHAS. También se
ha demostrado que la segunda suposición es correcta para sistemas incluso a
regímenes a baja fricción si βV ‡ 1. La forma de línea obtenida es lorenciana
sólo en la aproximación armónica, ya que las correcciones anarmónicas de primer
orden producen un desplazamiento dependiente de la temperatura y asimetría en el
pico. El acuerdo es bueno con simulaciones numéricas para βV ‡ ∼ 30 en un rango
de baja fricción (γ = 0,1ω0), y βV ‡ ∼ 8 en el régimen de fricción intermedia
(γ = 0,5ω0). Para el desplazamiento y anchura, se ha empleado una expansión
perturbativa en la frecuencia instantánea del sistema. Las correcciones de primer
orden son suficientes para tener en cuenta el desplazamiento linear dependiente
de la temperatura. En cambio, para obtener la dependencia con la temperatura de

CAPÍTULO 5. CONCLUSIONES. 196
la anchura, se han tenido que incluir correcciones a segundo orden empleando
para ello un modelo de oscilador de Kubo. Estas expresiones reproducen
cualitativamente el desplazamiento y anchura numérico y experimental del modo
T, mostrando también un efecto de movimiento de estrechamiento entre el perfil
lorenciano y gaussiano cuando la altura de la barrera (βV ‡) decrece. Comparando
estos resultados con el experimento se muestra que la teoría predice correctamente
los efectos con la temperatura observados en el modo T.
En definitiva, se ha visto que el formalismo hamiltoniano empleado en esta
memoria reproduce las formas de línea de los picos Q y T medidos por QHAS.
En el primer caso se tiene que considerar la dinámica energética cercana a la
barrera de difusión donde la frecuencia del modo normal del sistema en la barrera
es la cantidad física importante obtenida, la cual es imaginaria y da el prefactor
de Kramers, por tanto juega un papel importante en el intento de frecuencia para
que la partícula escape del pozo de potencial. Y en el segundo caso, hay que
tener en cuenta la dinámica en el pozo, donde la frecuencia del modo normal del
sistema resulta ser compleja. La parte real da la posición del pico a temperatura
cero, incluyendo el desplazamiento en la frecuencia de oscilación del adsorbato,
debido al acoplamiento con el baño térmico. Y la parte imaginaria da la anchura
a temperatura cero, la cual corresponde con el coeficiente de fricción que mide la
velocidad de transferencia de energía del oscilador armónico (adsorbato) al baño
térmico (sustrato).
Conclusión 4. Al mismo tiempo, se ha realizado una interpretación cuántica
de la dependencia de la temperatura del modo T bajo un formalismo de
funciones de correlación temporal por colisiones incluyendo efectos disipativos
dentro de un modelo de oscilador armónico amortiguado, acoplado a un baño
térmico (superficie) simulado por una colección de osciladores con bosones
como excitación. Se ha proporcionado una interpretación para dicha dependencia
con la temperatura del modo translacional frustrado (modo T) basado en la
transferencia de energía de un adsorbato excitado a la superficie, incluso a T = 0
K. El acuerdo encontrado con los resultados experimentales en el rango de

CAPÍTULO 5. CONCLUSIONES. 197
temperaturas medido es muy bueno cuando el parámetro de fricción depende de
la frecuencia del adsorbato y de la temperatura de la superficie, siempre y cuando
se asuma un modelo de Debye para las vibraciones del sustrato. La importancia
de este modelo es que no se necesita ningún potencial efectivo adsorbato-
sustrato, sino que sólo tiene en cuenta la fuerza del acoplamiento vibracional
adsorbato-sustrato, donde dos parámetros p y q nos proporcionan la información
de dicho acoplamiento. Además, el signo de q relaciona la pendiente de dicha
constante de acoplamiento κ(ω) alrededor de ω0 y nos proporciona información
sobre el perfil del acoplamiento vibracional alrededor de la frecuencia natural
del adsorbato. Además, proporciona una ley de extrapolación a 0K para el
cálculo intrínseco del desplazamiento y anchura (denominada residual) el modo
T. También, la consideración cualitativa realizada sobre los tiempos de colisión
y relajación sugieren que los mecanismos de relajación del fonón y del par
electrón-hueco no son paralelos pero si secuenciales, con una excitación del par
electrón-hueco seguido de excitaciones del fonón, por tanto la relajación total
γtot no es una simple suma de velocidades γph y γe−h, y en su lugar decimos
que a largos tiempos domina la velocidad de relajación dada por γph. Esta
nueva interpretación de considerar una aproximación armónica en la excitación
vibracional correspondiente al modo T del adsorbato supone la inclusión de un
ruido coloreado en lugar de un ruido blanco para describir el mecanismo de
disipación, suponiendo una velocidad no instantánea en la transferencia de energía
del adsorbato con el sustrato como consecuencia del acoplamiento.
Conclusión 5. Como última conclusión, y pensamos la más importante,
es que con esta memoria de investigación se ha cumplido con el objetivo
que nos propusimos años atrás de tratar de dar una visión unificada de los
movimientos de difusión y de vibración de adsorbatos sobre superficies analizadas
experimentalmente por QHAS dentro de un formalismo teórico estocástico. El
desarrollo de una nueva teoría que tenga en cuenta ambos procesos físicos
reproduciendo de forma satisfactoria las formas de líneas para el pico Q y modo T
medidas por QHAS. Se ha observado también por primera vez en estos sistemas

CAPÍTULO 5. CONCLUSIONES. 198
un efecto de movimiento por estrechamiento bajo ciertas condiciones para el
pico Q y T. Por último decir que el formalismo teórico estocástico propuesto en
esta memoria sirve para explicar el acoplamiento entre la difusión y la vibración
que aparecen conjuntas bajo ciertas condiciones experimentales, llegando a la
conclusión que el espectro de tiempos de vuelo obtenido experimentalmente no
se puede reproducir como una simple suma de factores de estructuras dinámicos
para el pico Q y el modo T como propusieron en su día algunos autores, sino que
se ha mostrado que es algo más complejo.

Apéndice A
Procesos estocásticos.
A.1. Procesos estacionarios.
Se intuye la idea de un proceso estacionario, un proceso en el que no
evoluciona en el tiempo. Por tanto en este proceso la variable aleatoria X(t)
obedece a una distribución que no cambia frente a cualquier traslación temporal.
Tomemos X = X(t) : −∞ < t <∞ que obedece a un proceso estacionario.
La función de autocovarianza, o también denominada función de autocorrela-
ción C viene dada por:
cov[X(t)X(t′)] = C(t, t′)
y por tanto la condición que requiere un proceso estacionario es que la función
de correlación no dependa de la traslación temporal t, t′, sino que dependa de la
diferencia de tiempos (t′ − t) = τ :
C(t, t′) = C(0, t′ − t) = C(0, τ)
Siendo τ el tiempo de correlación, tiempo en el que se encuentran correlacionadas
las variables. Además otra característica de un proceso estacionario es que el valor
199

APÉNDICE A. PROCESOS ESTOCÁSTICOS. 200
medio < X(t) > de la distribución estacionaria no depende del tiempo:
< X(t) >=< X(0) >=< X >
A.2. Proceso Gaussiano.
Un procesoX(t) continuo y real es gaussiano si cada vector de dimensiones fi-
nitas X[X(t1), X(t2), ..., X(tn)] obedece a una distribución normal multivariada
(distribución multinormal), escrita N(µ,V) del tipo:
f(x) =1
√
(2π)n|V |exp[−1
2](x − µ)V−1(x − µ−1)t
por tanto si X es N(µ, V )entonces:
1) < X >= µ, lo cual significa que < Xi >= µi para todo i.
2)V = (vij) se denomina matriz de covarianza, ya que vij= cov(Xi,, Xj).
Es importante recalcar que un proceso gaussiano no tiene por qué ser
necesariamente estacionario.
Decimos pues, que un proceso gaussiano es estacionario si y solo si:
1) < X(t) > es constante para todo t, o dicho de otro modo, no depende del
tiempo.
2) La matriz de covarianza V(t) satisface la condición V(t)=V(t + h) para
todo t y h >0.
A.3. Proceso markoviano.
Un proceso continuo X(t) es llamado proceso markoviano si se cumple que:
P [X(tn) ≤ x|X(t1) = x1, ..., X(tn−1) = xn−1] = P [X(tn) ≤ x|X(tn−1) = xn−1]
para todo x, x1, x2, ..., xn−1, y todos los tiempos t1 < t2 < ... < tn.

APÉNDICE A. PROCESOS ESTOCÁSTICOS. 201
Por otro lado, un proceso gaussiano X es un proceso de markov si y solo si:
< X(tn) ≤ x|X(t1) = x1, ..., X(tn−1) = xn−1 >=< X(tn) ≤ x|X(tn−1) = xn−1 >,
para todo x, x1, x2, ..., xn−1, y todos los tiempos t1 < t2 < ... < tn.

Bibliografía.
[1] R. Gomer, Rep. Prog. Phys. 53, 917 (1990).
[2] S. J. Lombardo y A. Bell, Surf. Sci. Rep. 13, 1 (1991).
[3] J. W. M. Frenken y B. J. Hinch, Helium Atom Scattering from Surfaces,
volume 27, Springer, New York, 1992.
[4] J. V. Barth, Surf. Sci. Rep. 40 (2000).
[5] R. Brown, Philosofical Magazine 4, 161 (1828).
[6] A. Einstein, Ann. Physik 17, 549 (1905).
[7] H. A. Kramers, Physica. 7, 284 (1940).
[8] P. Hänggi, P. Talkner y M. Borkovec, Rev. Mod. Phys. 62, 251 (1990).
[9] V. Melnikov, Phys. Rep. 209, 1 (1991).
[10] D. Ruelle, Thermodynamic formalism, Addison-Wesley, 1978.
[11] P. Gaspard, Chaos, Scattering ans Statistical Mechanics, Cambridge:
Cambridge University Press, 1998.
[12] J. R. Dorfman, An Introduction to Chaos in Non-Equilibrium Statistical
Mechanics, Cambridge: Cambridge University Press, 1999.
[13] J. D. Meiss, Rev. Mod. Phys. 64, 795 (1992).
202

BIBLIOGRAFÍA. 203
[14] R. Guantes, J. L. Vega y S. Miret-Artés, Phys. Rev. B 64, 245415 (2001).
[15] J. Ellis y J. P. Toennies, Phys. Rev. Lett. 70, 2118 (1993).
[16] A. P. Graham, F. Hofmann, J. P. Toennies, L. Y. Chen y S. C. Ying, Phys.
Rev. Lett. 78, 3900 (1997).
[17] A. P. Graham, F. Hofmann, J. P. Toennies, L. Y. Chen y S. C. Ying, Phys.
Rev. B. 56, 10567 (1997).
[18] J. Ellis, A. P. Graham, F. Hofmann y J. P. Toennies, Phys. Rev. B. 63,
195408 (2001).
[19] A. P. Jardine, J. Ellis y W. Allison, Phys.: Condens. Matter. 14, 6173 (2002).
[20] A. Graham, F. Hofmann y J. P. Toennies, J. Chem. Phys. 104, 5311 (1996).
[21] F. Hofmann y J. P. Toennies, Chem. Rev. 96, 1307 (1996).
[22] A. P. Graham y J. P. Toennies, Europhys. Lett. 42, 449 (1998).
[23] M. Bertino, J. Ellis, F. Hofmann y J. P. Toennies, Phys. Rev. Lett. 73, 605
(1994).
[24] W. L. Silvestri, Theory y Experiments on surface diffusion, PhD thesis,
Göttingen, 1998.
[25] L. V. Hove, Phys. Rev. 95, 249 (1954).
[26] C. T. Chudley y R. Elliot, Proc. Phys. Soc. 77, 353 (1961).
[27] L. Y. Chen y S. C. Ying, Phys. Rev. Lett. 26, 4361 (1993).
[28] L. Y. Chen y S. C. Ying, Phys. Rev. B 49, 13838 (1994).
[29] A. Cuchetti y S. C. Ying, Phys. Rev. B 60, 11110 (1999).

BIBLIOGRAFÍA. 204
[30] R. Guantes, J. L. Vega, S. Miret-Artés y E. Pollak, J. Chem. Phys. 119,
2780 (2003).
[31] T. Alla-Nissila y S. C. ying, Prog. Surf. Sci. 39, 227 (1992).
[32] R. Ferrando, R. Spadacini y G. E. Tommei, Phys. Rev. E 48, 2437 (1993).
[33] L. Y. Chen, M. Baldan y S. C. Ying, Phys. Rev. B 54, 8856 (1996).
[34] E. Pollak, J. Bader, B. J. Berne y P. Talkner, Phys. Rev. Lett. 70, 3299
(1993).
[35] J. L. Vega, R. Guantes y S. Miret-Artés, J. Phys.: Condens. Matter. 14,
6193 (2002).
[36] J. L. Vega, R. Guantes y S. Miret-Artés, Phys. Chem. Chem. Phys. 4, 4985
(2002).
[37] J. L. Vega, R. Guantes, S. Miret-Artés y D. A. Micha, J. Chem. Phys. 121,
8580 (2004).
[38] J. L. Vega, R. Guantes y S. Miret-Artés, J. Phys.: Condens. Matter 16,
S2879 (2004).
[39] T. Engel y K. H. Rieder, Structural Studies of Surfaces with Atomic y
Molecular Beam Diffraction, Springer-Verlag, 1982.
[40] N. Esbjerg y J. K. Norskov, Phys. Rev. Lett 45, 807 (1980).
[41] E. W. Müller, Field Ion Micrscopy, Principles y Applications, 1969.
[42] T. R. Linderoth, S. Horch, J. S. E. Laegsgaard y F. Besenbacher, Phys. Rev.
Lett 78, 4978 (1997).
[43] N. G. V. Kampen, Stochastic Processes in physics y chemistry, North-
Holland Physics Publishing, The Netherlands, fourth edition, 1985.

BIBLIOGRAFÍA. 205
[44] J. A. Strossio y D. M. Eigler, Science , 1319 (254).
[45] Y. M. Mo, Science , 886 (1993).
[46] e. a. T. C. Shen, Science , 1590 (1995).
[47] G. L. Kellogg, Surf. Sci. Rep. 21, 1 (1994).
[48] A. G. Naumovets y Y. S. Vedula, Surf. Sci. Rep 4, 365 (1985).
[49] J. W. Gadzuk y A. C. Luntz, Surf. Sci. 144, 429 (1984).
[50] J. C. Tully, Annu. Rev. Phys. Chem 51, 153 (2000).
[51] N. N. Persson y E. Zaremba, Phys. Rev. B , 1863 (1985).
[52] M. Bee, Quasielastic Neutron Scattering, Bristol, 1988.
[53] W. Marshall y S. W. Lovesey, Theory of Thermal Neutron Scattering,
Oxford University Press, New York, 1971.
[54] H. Ibach y D. L. Mills, Eletron Energy Loss Spectroscopy y Surface
Vibrations, Academic, New York, 1982.
[55] Y. J. Chabal, Surf. Sci. Rep 8, 211 (1988).
[56] J. P. Toennies y K. J. Winkelmann, J. Chem. Phys 66, 3965 (1977).
[57] G. Brusdeylins, R. B. Doak y J. P. Toennies, Phys. Rev. B 27, 3662 (1983).
[58] J. P. Toennies, J. Phys. Condens. Matter 5, A25 (1993).
[59] G. Comsa, Surf. Sci 299/300, 77 (1994).
[60] D. Farias y K. H. Rieder, Rep. Prog. Phys 61, 664 (1998).
[61] A. Lahee, J. P. Toennies y C. Wöll, Surf. Sci 177, 371 (1986).

BIBLIOGRAFÍA. 206
[62] J. P. Toennies, Solvay Conference on Surface Science, volume 14, Springer
Series in Surface Sciences, 1998.
[63] F. Hofmann y J. P. Toennies, J. Chem. Phys 101, 10155 (1994).
[64] A. P. Graham, W. Silvestri y J. P. Toennies, Elementary processes of surface
diffusion studied by quasielastic helium atom scattering, pages 1–15, 1996.
[65] T. Ala-Nissila, R. Ferrando y S. Ying, Adv. in Phys 51, 949 (2002).
[66] J. P. Toennies, volume 21 of Springer, Springer Series in Surface Sciences,
Berlin.
[67] J. ellis, G. witte y J. P. Toennies, J. chem. Phys. 102, 5059 (1995).
[68] H. Steininger, S. Lehwald y H. Ibach, Surf. Sci 123 (1982).
[69] B. Poelsema, S. T. Wart y G. Comsa, Phys. Rev. Lett. 49, 578 (1982).
[70] G. E. Uhlenbeck y L. S. Ornstein, Phys. Rev. 36, 823 (1930).
[71] P. Langevin, Comptes Rendus 146, 530 (1908).
[72] S. Chandrasekhar, Rev. Mod. Phys. 15, 1 (1943).
[73] L. Onsager, Phys. Rev. 37, 405 (1931).
[74] R. Kubo y N. Hashitsume, Statistical Physics II, Springer-Verlag, Berlin,
1991.
[75] D. Chandler, Introduction to Modern Statistical Mechanics, Oxford
University Press, New York, 1987.
[76] N. G. V. Kampen, Stochstic Processes in Physics y Chemistry, North-
Holland, Amsterdam, 1996.
[77] P. W. Paul y J. Baschnagel, Stochastic processes from Physics to Finance,
springer, Berlin, 1999.

BIBLIOGRAFÍA. 207
[78] J. L. Doob, Stochastic Processes, Wiley, New York, 1953.
[79] A. D. Fokker, Ann. Physik. 43, 810 (1914).
[80] M. Planck, Sitzber. Preub. Akad. Wiss , 324 (1917).
[81] M. Weissbluth, Photon-Atom interactions, Academic Press, New York,
1989.
[82] A. Kolmogorov, Grundbegriffe der Wahrscheinlichkeitsrechnung, Springer,
Berlin, 1933.
[83] H. Risken, The Fokker-Planck Equation, Springer, Berlin, 1989.
[84] M. V. Smoluchowski, Ann. Physik. 48, 1103 (1915).
[85] C. W. Gardiner, Handbook of Stochastic Methods for Physics, Chemistry y
the Natural Sciences, Springer, Berlin, 1998.
[86] L. V. Hove, Phys. Rev. 95, 249 (1954).
[87] J. P. Hanssen y I. R. McDonald, Theory of Simple Liquids, Academic Press,
New York, 1976.
[88] P. A. Egelstaff, An Introduction to the Liquid State, Academic Press, New
York, 1967.
[89] D. A. McQuaerrie, Statistical Mechanics, Harper y Row, New York, 1976.
[90] T. Ala-Nissila y S. Ying, Prog. Surf. Sci. 39, 227 (1992).
[91] R. S. R. Ferrando y G. E. Tommei, Phys. Rev. E 51, 126 (1995).
[92] D. Forster, Hydrodynamic Fluctuations, Broken Symmetry y correlation
Functions, Addison.Wesley, 1990.
[93] L. F. Richardson, Proc. R. Soc. 110, 709 (1926).

BIBLIOGRAFÍA. 208
[94] R. Metzler y J. Klafter, Phys. Rep. 339, 1 (2000).
[95] T. Geisel, A. Zacherl y G. Radons, Phys. Rev. Lett. 59, 2503 (1987).
[96] G. Radons, T. Geisel y A. Zacherl, Z. Phys. B 71, 117 (1988).
[97] J. Klafter y G. Zumofen, Phys. Rev. E 49, 4873 (1994).
[98] W. Feller, An Introduction to Probability Theory ans its Applications,
Wiley, New York, 1971.
[99] M. F. Shlesinger, G. M. Zaslavsky y U. Frisch, Lévy Flights y related Topics
in Physics, springer, Berlin, 1995.
[100] M. F. Shlesinger, G. M. Zaslavsky y J. Klafter, Nature 363, 31 (1993).
[101] M. F. Shlesinger y J. K. B. J. West, Phys. Rev. Lett. 58, 1100 (1987).
[102] M. F. Shlesinger y J. Klafter, Phys. Rev. Lett. 54, 2551 (1985).
[103] I. M. Sokolov, Phys. Rev. E 63, 11104 (2000).
[104] J. Klafter y G. Zumofen, Phys. Rev. E 49, 4873 (1994).
[105] J. Klafter, A. Blumen y M. F. Shlesinger, Phys. Rev. A 35, 3081 (1987).
[106] J. Klafter, M. F. Shlesinger y G. Zumofen, Phys. Today 49, 33 (1996).
[107] G. Zumofen y J. Klafter, Phys. Rev. E 47, 851 (1993).
[108] T. Geisel, J. Niertwetberg y A. Zacherl, Phys. Rev. Lett 54, 616 (1985).
[109] H. Scher y E. W. Montroll, Phys. Rev. B 12, 2455 (1975).
[110] J. B. W. Paul, Stochastic Processes, Springer-Verlag, Berlin, 1999.
[111] G. H. Herzberg, Molecular Spectra y Molecular Structure, volume 1, Van
Nostrand Reinhold, New York, 1950.

BIBLIOGRAFÍA. 209
[112] H. Dürr y A. P. Baddorf, J. Electron. Spectrosc. Relat. Phenom 64/65, 691
(1993).
[113] L. Y. Chen y S. C. Ying, Phys. Rev. B. 60, 16965 (1999).
[114] R. Ferrando y G. Tréglia, Phys. Rev. B 50, 12104 (1994).
[115] G. Caratti, R. Ferrando, R. Spadacini y G. E. Tommei, Phys. Rev. E 54,
4708 (1996).
[116] M. P. Allen y D. J. Tildesley, Computer Simulations of Liquids, Clarendon,
Oxford, 1987.
[117] S. A. Lindgren, C. Svensson y L. Wallden, Phys. Rev. B. 42, 1467 (1990).
[118] L. Y. Chen, M. R. Baldan y S. C. Ying, Phys. Rev. B. 54, 8856 (1996).
[119] A. Weinstein, Invent. Math. 20, 47 (1973).
[120] W. H. Press, B. P. Flannery, S. A. Teukolsky y W. T. Vetterling, Numerical
Recipes, Cambrige: Cambridge University Press, 1992.
[121] R. Seydel, Practical Bifurcation y Stability Analysis, Springer, New York,
1994.
[122] K. R. Meyer, Trans. Am. Math. Soc. 149, 95 (1970).
[123] T. Geisel, A. Zacherl y G. Radons, Phys. Rev. Lett. 59, 2503 (1987).
[124] T. Geisel, A. Zacherl y G. Radons, Z. Phys. B. 71, 117 (1988).
[125] E. W. Montroll y M. F. Shlesinger, Nonequilibrium PhenomenaII: From
Stochastic to Hydrodynamics, Elsevier Science Publishers BV, 1984.
[126] D. S. Sholl y R. T. Skodje, Physica D. 71, 168 (1994).
[127] S. J. Lombardo y A. T. Bell, Surf. Sci. Rep. 13, 1 (1991).

BIBLIOGRAFÍA. 210
[128] H. Risken, The Fokker-Planck Equation, Springer, Berlin, 1984.
[129] P. Riemann, Chem. Phys. 268, 337 (2001).
[130] S. I. Denisov y W. Horsthemke, Phys. Rev. E. 62, 7729 (2000).
[131] J. Masoliver y K. G. Wang, Phys. Rev. E. 51, 2987 (1995).
[132] K. G. Wang y M. Tokuyama, Physica A. 265, 341 (1999).
[133] M. A. Lieberman y K. Y. Tsang, Phys. Rev. Lett. 55, 908 (1985).
[134] J. P. Crutchfield, J. D. Farmer y B. A. Huberman, Phys. Rep. 92, 45 (1982).
[135] P. Landa y P. V. E. McClintock, Phys. Rep. 323, 1 (2000).
[136] Y. Georgievskii y E. Pollak, J. Chem. Phys. 102, 6908 (1995).
[137] Y. Georgievskii, M. A. Kozhushner y E. Pollak, J. Chem. Phys. 102, 6908
(1995).
[138] E. Pollak, H. Grabert y P. Hänggi, J. Chem. Phys. 91, 4073 (1989).
[139] G. Wanshtröm, Interactions of Atoms and Molecules with Solid Surfaces,
Plenum, New York.
[140] E. Pollak y E. Hershkovitz, Chem. Phys. 180, 191 (1994).
[141] E. Hershkovitz y E. Pollak, J. Chem. Phys. 106, 7678 (1997).
[142] A. M. Berezhkovskii, E. Pollak y V. Y. Zitserman, J. Chem. Phys. 97, 2422
(1992).
[143] P. Talkner y E. Pollak, Phys. Rev. E. 47, R21 (1993).
[144] E. Pollak y P. Talkner, Phys. Rev. E. 47, 922 (1993).
[145] G. Caratti, R. Ferrando y G. E. Tommei, Chem. Phys. 235, 157 (1998).

BIBLIOGRAFÍA. 211
[146] S. Lifson y J. L. Jackson, J. Chem. Phys. 36, 2410 (1962).
[147] M. Borromeo y F. Marchesoni, Surf. Sci. 465, L771 (2000).
[148] P. Reimann, C. V. den Brock, H. Linke, P. Hänggi, J. M. Rubi y A. Pérez-
Madrid, Phys. Rev. Lett. 87, 10602 (2001).
[149] Y. Georgievskii y E. Pollak, Surf. Sci. 355, L366 (1996).
[150] R. Ferrando, R. Spadacini y G. E. Tommei, Phys. Rev. A 46, R699 (1992).
[151] A. P. Graham y J. P. Toennies, Surf. Sci. 427-428, 1 (1999).
[152] A. P. Graham, F. Hofmann, L. Y. C. J. P. Toennies y S. C. Ying, Phys. Rev.
B. 56, 10567 (1997).
[153] A. O. Caldeira y A. J. Leggett, Ann. Phys. 149, 374 (1983).
[154] E. Cortés, B. J. West y K. Lindenberg, J. Chem. Phys. 82, 2708 (1985).
[155] E. Pollak, J. Chem. Phys. 85, 865 (1986).
[156] E. Pollak, Phys. Rev. A 33, 4244 (1986).
[157] A. M. Levine, M. Shapiro y E. Pollak, J. Chem. Phys. 88, 1959 (1988).
[158] E. Pollak, Dynamics of Molecules and chemical Reactions, Dekker, New
York, 1996.
[159] S. W. Lovesey, Theory of Neutron Scattering from Condensed Matter,
volume 1, Clarendon, Oxford, 1984.
[160] J. R. Manson y V. Celli, Phys. Rev. B. 39, 3605 (1989).
[161] R. Kubo, J. Math. Phys. 4, 174 (1963).
[162] R. G. Gordon, Adv. Magn. Reson. 3, 1 (1968).

BIBLIOGRAFÍA. 212
[163] W. H. Louisell, Quantum Statistical Properties of Radiation, Wiley, New
York, 1973.
[164] K. Blum, Density Matrix Theory and Applications, Plenum, New York,
1981.
[165] W. Marshall y S. W. Lovesey, Theory of Thermal Neutron Scattring, Oxford
University Press, New York, 1971.
[166] J. A. Prybyla, H. W. K. Tom y G. D. Aumiller, Phys. Rev. Lett. 68, 3737
(1998).
[167] D. A. Micha, A. Santana y A. Salam, J. Chem. Phys. 116, 5173 (2002).
[168] E. Knoesel, A. Hotzel y M. Wolf, Phys. Rev. B 57, 12812 (1998).
[169] J. C. Tully, Annu. Rev. Phys. Chem. 51, 153 (2000).
[170] B. N. J. Persson y J. W. Gadzuk, Surf. Sci. 410, L779 (1998).
[171] J. H. V. Vleck, Phys. Rev. 74, 1168 (1948).
[172] F. H. J. P. T. L. Y. C. J. Ellis, A. P. Graham y S. C. Ying, Phys. Rev. B 63,
195408 (2001).
[173] R. Guantes, J. L. Vega, S. Miret-Artés y E. Pollak, J. Chem. Phys. 120,
10768 (2004).