
Transferencia de Energía Triplete-Triplete · singlete-triplete y triplete-singlete, prohibidas...
81
CAPÍTULO 6 T T r r a a n n s s f f e e r r e e n n c c i i a a d d e e E E n n e e r r g g í í a a T T r r i i p p l l e e t t e e - - T T r r i i p p l l e e t t e e 6.1 Introducción 237 6.1.1 Consideraciones sobre la TET 237 6.1.2 Tratamientos Clásicos para la TET 242 6.1.3 Tratamientos Cuánticos para la TET 246 6.1.4 TET Vertical vs. No-Vertical en Fase Condensada 248 6.2 Aplicación de la Teoría del Estado de Transición para Procesos Diabáticos a la TET 249 6.2.1 La Superficie de Energía Potencial del Proceso de Transferencia de Energía Triplete 250 6.2.2 La Constante de Velocidad de Transferencia de Energía 254 6.3 Aproximación de Primer Orden 256 6.3.1 Cálculo de la Energía del Complejo Activado 256 6.3.2 TET No-Vertical en el COT 264 6.3.3 Métodos 268 6.3.4 Conclusiones 269 6.4 Estudio de la TET con SEPs Completas: Algoritmo TET-ACC 270 6.4.1 Algoritmo TET-ACC 270 6.4.1.1 Cruce entre Superficies de Energía Potencial Completas 271 6.4.1.2 Descripción del Algoritmo 277 6.4.1.3 Análisis y Topología de las curves TET-ACC 281 6.4.1.4 Transferencia de Energía Triplete Exotérmica. La Región Invertida 286 6.4.2 Funciones Termodinámicas de Activación a partir de las Curvas TET-ACC Calculadas 289 6.4.3 TET No-Vertical en los Estilbenos 292 6.4.3.1 Funciones de Partición y Constante de Velocidad 308 6.4.4 Limitaciones del Algoritmo e Interpretación de Resultados 311 6.4.5 Métodos 314 6.5 Conclusiones 315 -235-
-
Upload
nguyencong -
Category
Documents
-
view
221 -
download
0
Transcript of Transferencia de Energía Triplete-Triplete · singlete-triplete y triplete-singlete, prohibidas...

CAPÍTULO
6
TTrraannssffeerreenncciiaa ddee EEnneerrggííaa TTrriipplleettee--TTrriipplleettee
6.1 Introducción 237 6.1.1 Consideraciones sobre la TET 237 6.1.2 Tratamientos Clásicos para la TET 242 6.1.3 Tratamientos Cuánticos para la TET 246 6.1.4 TET Vertical vs. No-Vertical en Fase Condensada 248
6.2 Aplicación de la Teoría del Estado de Transición para Procesos Diabáticos a la TET 249
6.2.1 La Superficie de Energía Potencial del Proceso de Transferencia de Energía Triplete 250
6.2.2 La Constante de Velocidad de Transferencia de Energía 254 6.3 Aproximación de Primer Orden 256 6.3.1 Cálculo de la Energía del Complejo Activado 256 6.3.2 TET No-Vertical en el COT 264 6.3.3 Métodos 268 6.3.4 Conclusiones 269 6.4 Estudio de la TET con SEPs Completas: Algoritmo TET-ACC 270 6.4.1 Algoritmo TET-ACC 270 6.4.1.1 Cruce entre Superficies de Energía
Potencial Completas 271 6.4.1.2 Descripción del Algoritmo 277 6.4.1.3 Análisis y Topología de las curves TET-ACC 281 6.4.1.4 Transferencia de Energía Triplete Exotérmica.
La Región Invertida 286 6.4.2 Funciones Termodinámicas de Activación a partir de las Curvas TET-ACC Calculadas 289 6.4.3 TET No-Vertical en los Estilbenos 292 6.4.3.1 Funciones de Partición y Constante de Velocidad 308 6.4.4 Limitaciones del Algoritmo e Interpretación de Resultados 311 6.4.5 Métodos 314 6.5 Conclusiones 315
-235-

6.1 Introducción 237
66..11 IInnttrroodduucccciióónn
En este capítulo se estudia uno de los procesos más comunes e importantes de
transferencia de energía en fotoquímica orgánica (Turro, 1966), la transferencia de
energía triplete-triplete o simplemente transferencia de energía triplete (TET). Además
se desarrollan diversas aproximaciones teóricas al problema, que abarcan tanto la
transferencia de energía vertical como la no-vertical. Estas aproximaciones teóricas
pretenden establecer una relación cuantitativa entre las propiedades de las superficies de
energía potencial de los estados electrónicos involucrados en el proceso de transferencia
y las constantes cinéticas de velocidad del mismo. De la misma manera, se pretende
ahondar en las características estructurales y termodinámicas que controlan el proceso.
El modelo teórico usado en todas ellas está basado principalmente en el cálculo de SEP
precisas así como en la aplicación de la teoría del estado de transición (TST) para
procesos diabáticos.
Esta sección (6.1) se organiza de la siguiente manera. En primer lugar se presentarán
brevemente una serie de consideraciones generales sobre la TET: el mecanismo de
intercambio electrónico en la TET, la evaluación de la energía de acoplamiento y la
dependencia de la velocidad del proceso con la densidad de estados resonantes entre
otras. En el segundo punto (6.1.2) se desarrollarán las ideas básicas concernientes a los
modelos clásicos para los procesos de TET (verticales y no-verticales), continuando con
los cuánticos en el siguiente punto (6.1.3). Por último, a la luz de lo expuesto en los
puntos anteriores, se discutirá la situación actual en lo que concierne a la TET vertical y
no-vertical.
6.1.1 Consideraciones generales sobre la TET
La transferencia de energía triplete-triplete (TET) fue observada por primera vez en
1956 (Terenin y Ermolaev, 1956). En este experimento, se observó que la presencia de
una molécula que absorbía luz a una cierta longitud de onda era capaz de inducir la
emisión de fosforescencia de otra molécula que se encontraba en la misma disolución y
que de otra manera no se observaba A este fenómeno se de denominó

238 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
fotosensibilización triplete§. En este tipo de reacciones, como ya apuntaron los autores
desde el comienzo (Terenin y Ermolaev, 1956), existe una transferencia de energía
electrónica desde una molécula excitada electrónicamente 3D, a otra molécula en estado
singlete, 1A (Esquema 6.1):
exp3 1 1 3enkD A D A+ ⎯⎯→ +
Esquema 6.1. Transferencia de Energía Triplete
El proceso de TET transcurre mediante un mecanismo de intercambio electrónico
(Lamola, 1969; Förster, 1948; Dexter, 1953; Naqvi y C. Steel, 1970) en el que ambas
moléculas intercambian un electrón conservándose durante el proceso la multiplicidad
de espín (más adelante se discutirá -ver por ejemplo el Esquema 6.2- la analogía con las
reacciones de transferencia de carga), y la velocidad de reacción es del orden de los
picosegundos (Anderson et al. , 1974; Saltiel et al., 1974; Saltiel y Atwater, 1988).
Existen dos magnitudes que determinan la constante de velocidad del proceso de
transferencia de energía y que han sido ampliamente discutidas en estos procesos, éstas
son, la energía de interacción electrostática entre las dos moléculas implicadas en el
proceso, y la densidad de estados resonantes entre ambas moléculas. A continuación se
resumen algunas propiedades importantes de estas dos magnitudes.
1. Evaluación de la energía de interacción.
Las funciones de onda del sistema antes y después de la transferencia de energía son:
(6.1) [ ] ( ) ( )** ··· D AD A iψ ψΨ =℘ j
j (6.2) [ ] ( ) ( )*···* D AD A iψ ψΨ =℘
donde el asterisco denota excitación electrónica y el operador ℘ actúa antisimetrizando
el producto de las dos funciones de onda moleculares sobre los respectivos electrones
“i” y “j” de dador y aceptor (Naqvi y C. Steel, 1970). La interacción que existe entre las
dos moléculas es de naturaleza electrostática, por lo que la energía de interacción entre
ambas se puede hallar calculando la integral:
( ) ( ) ( ) ( )*if D A D A*H i j i jψ ψ ψ ψ′= ℘ ℘ (6.3)
que puede descomponerse como suma de dos términos, uno primero culómbico (Hc), y
un segundo debido al intercambio electrónico entre las dos moléculas (He): § Del término inglés triplet photosensitization.

6.1 Introducción 239
( ) ( ) ( ) ( ) ( ) ( ) ( ) ( )
* * *if D A D A D A D A
if c e
H i j i j i j iH H H
* jψ ψ ψ ψ ψ ψ ψ ψ′ ′= + ℘
= +
(6.4)
El primer término (culómbico) es nulo si la transición electrónica de dador o aceptor
está prohibida, ya sea porque la integral ( ) ( ) ( ) ( )* *D A D Ai j i jψ ψ µ ψ ψ sea nula
(transición electrónica prohibida por mecanismo de dipolo eléctrico) o porque no
conserve la simetría de espín en la transición, de tal manera que si dador o aceptor
cambian de multiplicidad de espín en la transición electrónica este término se hace igual
a cero. Esto ocurre por ejemplo en la TET, en la cual están involucradas transiciones
singlete-triplete y triplete-singlete, prohibidas por el principio de conservación del
momento de espín. Por lo tanto, en este caso, la energía de interacción vendrá dada por
el único término que no se anula:
( ) ( ) ( ) ( )*e D A D AH i j i * jψ ψ ψ ψ′= ℘ (6.5)
Como se demostrará cualitativamente en la próxima sección (Sección 6.2), el valor de
esta integral, tal y como se predijo para procesos de TET en sistemas simples (Dexter,
1953; Ermolaev, 1967), decae exponencialmente al aumentar la distancia entre dador y
aceptor. Esta dependencia exponencial es debida a la necesidad de que exista un
solapamiento de las nubes de carga de ambas moléculas para que haya un intercambio
electrónico entre dador y aceptor. En el caso de átomos el solapamiento está bien
descrito mediante una dependencia exponencial con la distancia interatómica a través de
un “radio orbital efectivo” (Dexter, 1953) que depende de la pareja de átomos entre los
que ocurre el proceso de TET. Esta dependencia exponencial se complica notablemente
en la TET intermolecular (la que es principal objeto de estudio en este capítulo) debido
a los efectos de la orientación espacial relativa de ambas moléculas dentro del complejo
de encuentro. De hecho, se ha demostrado la gran dependencia que existe entre la
constante de velocidad del proceso de transferencia de energía y la orientación de los
grupos cromóforos de dador y aceptor a través del término de acoplamiento electrónico
(para un review sobre el tema ver: Speiser, 1996).
En cualquier caso, el valor de la integral (ecuación 6.5), decrece rápidamente a medida
que ambas moléculas se alejan, siendo la distancia máxima de separación para que se
verifique el proceso de TET de aproximadamente 10 Angstroms.
El proceso de TET se puede analizar usando la regla de oro de Fermi (ecuación 3.31) ya
que se trata de un proceso en el que aparece una perturbación dependiente del tiempo,
esta es, la interacción electrostática entre ambas moléculas:

240 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
22ifk Hπ ρ= (6.6)
En esta ecuación se observa cómo la constante de velocidad depende de la integral de
intercambio al cuadrado. Además depende también de la densidad de estados, es decir,
del número de estados resonantes entre ambas moléculas por unidad de energía. Este es
el segundo factor que controla la velocidad de transferencia de energía, el cual se trata a
continuación.
2. Densidad de estados resonantes en TET intermolecular.
La transferencia de energía triplete (TET) intramolecular ocurre cuando dos moléculas
con sendos grupos cromóforos intercambian energía electrónica según el esquema de
reacción 6.1. La TET intramolecular añade un factor de complejidad con respecto a
dicho proceso entre átomos. Para estos últimos, los estados energéticos entre los que
puede existir resonancia energética y por lo tanto transferencia de energía vienen dados
exclusivamente por los espectros electrónicos de ambos átomos. De esta manera, y
puesto que las funciones de partición electrónica no dependen de la temperatura, la TET
entre átomos tampoco depende de ésta, y solamente depende de las propiedades del
espectro electrónico.§
Para moléculas esta situación es distinta, ya que en este caso los niveles energéticos de
la molécula dependen en gran medida de la temperatura a través de las funciones de
partición rotacionales y vibracionales, las cuales son específicas de cada molécula.
Además, cuando el proceso ocurre en disolución, el disolvente puede provocar
perturbaciones en dichas funciones de partición.
Este comportamiento en principio complejo, se revela asombrosamente simple en
muchos casos a la luz de los resultados experimentales. Cuando se mide la constante de
velocidad de TET entre un aceptor y diversos dadores con distintas energías triplete,¤ se
observa cómo la constante de velocidad de transferencia de energía decae
exponencialmente cuando la energía triplete del dador disminuye respecto de la del
aceptor (como se verá con más detalle en el punto 6.1.2). Sin embargo, para algunas
moléculas aceptoras esta dependencia es más compleja. De hecho, las moléculas
§ Realmente el átomo en una red cristalina también posee energía vibracional, y sus propiedades electrónicas se ven modificadas con la temperatura. No se profundiza en ello ya que el objetivo de este estudio es la TET intermolecular. ¤ Se usará el término TE para denotar la energía vertical de excitación (triplete) , mientras que el término
0S → 1T00TE se usará para denotar la energía triplete correspondiente a la transición vibracional 0-0.

6.1 Introducción 241
aceptoras que no presentan este comportamiento toman valores de la constante de
velocidad mayores de los que toman las primeras.
Fue en 1963 (Saltiel y Hammond, 1963; Hammond y Saltiel, 1963) cuando se observó
por primera vez este comportamiento en los Estilbenos. La constante de velocidad del
proceso de TET entre distintos dadores con una energía triplete bien definida, y el cis- y
trans-estilbeno, tomaba valores mayores a los “normales” (que como se verá
posteriormente siguen una cinética dada por la ecuación 6.8) que exhiben la
dependencia exponencial antes mencionada de la constante de TET. Este
comportamiento se denominó TET no-vertical, ya que en él parecía haber un gran
cambio estructural en el proceso de excitación de la molécula aceptora (estilbenos) y
por lo tanto no era posible observar la banda 0-0.
A partir de este descubrimiento se han propuesto distintas teorías para explicar y
predecir esta desviación de la constante de velocidad con respecto al comportamiento
“normal”. Asimismo, se ha estudiado dicho comportamiento en otros compuestos como
el 1,3,5,7-Ciclooctatetraeno (Forward et al., 1993; Das y Priyadarsini, 1994) o
compuestos relacionados estructuralmente con los estilbenos.
Son distintos los aspectos que se han estudiado con respecto a las propiedades
características de los procesos de TET no-vertical, destacando los aspectos estructurales
y energéticos, es decir, las deformaciones que sufre la molécula aceptora en el proceso,
así como su relación con los factores energéticos que lo controlan.
Si bien en los primeros trabajos (Saltiel y Hammond, 1963; Hammond y Saltiel, 1963)
se postuló un cambio geométrico concurrente con la excitación del aceptor,
posteriormente se recurrió a una explicación más sencilla, esta es, la activación térmica
de uno o varios modos normales de vibración (bandas calientes) (Lamola, 1969; Bylina,
1968; Yamauchi y Azumi, 1973; Sandros, 1973; Ramamustly y Liu, 1976; Scaiano y
Wubbels, 1981; Saltiel et al., 1984) que permiten el descenso del la diferencia de
energía singlete-triplete. Algunos estudios que consisten en la aplicación de estos
conceptos a SEP multidimensionales (Caldwell et al., 1992; Catalán y Saltiel, 2001) se
ha realizado en los últimos años. Incluso se ha llegado a asociar una distorsión
geométrica a la constante de velocidad del proceso de TET (Lalevée et al., 2002)
postulando una relación clara entre la deformación geométrica en el estado fundamental
del aceptor y la disminución de la energía triplete necesaria del dador para que ocurra la
transferencia.

242 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
En los tres próximos puntos se ampliarán algunos de estos tratamientos, desarrollando
en primer lugar los modelos clásicos y posteriormente cuánticos que se han desarrollado
para explicar los procesos de TET. Por último se tratará el problema del
comportamiento vertical y no-vertical en fase condensada.
6.1.2 Tratamientos Clásicos de la TET
1. Tratamiento de Sandros
El tratamiento que desarrolló Sandros en 1964 (Sandros, 1964) es el primero realizado
para la determinación de la constante cinética en los procesos de transferencia de
energía. Las reacciones de transferencia de energía en disolución ocurren cuando
aceptor y dador, en el complejo de encuentro, mediante colisiones efectivas
intercambian la energía. El modelo desarrollado por Sandros tiene únicamente dos
premisas. La primera consiste en suponer que no hay una relajación vibracional
significativa en el proceso de excitación (S0 → T1 en la molécula aceptora) y
decaimiento (S0 ← T1 para la molécula dadora) lo cual es equivalente a que la principal
transición es la 0-0 en ambos casos. En segundo lugar, las dos moléculas en el complejo
de encuentro se suponen en equilibrio térmico, siguiendo una distribución de
Boltzmann. Así, la relación de poblaciones [*D···A] frente a [D···A*] debe ser:
[ ]
[ ]
[ ]
[ ] [ ]
exp* ··· 1* ··· exp exp
ATD
T T
E RTA DD A E RT E RT
−= =
− −∆ (6.7)
donde es la energía triplete del aceptor (dador). De esta manera, cuando las dos
moléculas tienen igual probabilidad de estar excitadas, lo cual ocurre cuando
( )A DTE
[exp 1TE RT−∆ =] , o lo que es lo mismo, cuando: 0TE∆ = , la velocidad de
transferencia debe ser igual a la mitad de la velocidad de difusión (ya que la mitad de
los complejos difunden con D excitada y la otra mitad con A, y por lo tanto solamente
la mitad de ellos son efectivos en la transferencia de D a A), por lo que la constante de
velocidad debe ser:
[ ]
exp
1 expd
enT
kkE RT
=+ −∆
(6.8)
Esta ecuación predice para valores positivos de D AT TE E ET∆ = − un rápido acercamiento
de a , mientras que a medida que la reacción se hace más expenk dk ( exp
en dk k= )

6.1 Introducción 243
“endotérmica”, esto es, , la constante cinética de velocidad decrece
exponencialmente.
0D AT T TE E E∆ = − <
La ecuación de Sandros es de gran utilidad cuando las condiciones expuestas antes se
cumplen. Como se verá más adelante, a veces no existe equilibrio entre los dos estados
electrónicos del complejo en el encuentro, por lo que dicha ecuación deja de ser válida.
Por otra parte, la otra de las condiciones o supuesto del modelo en general no se
cumple, sin embargo, para moléculas que no presenten una gran deformación
geométrica en el proceso de excitación o decaimiento puede ser considerada válida.
Como se verá, esta última condición tampoco se cumple en algunos casos, por lo que la
ecuación de Sandros para el cálculo de deja de ser correcta. expenk
2. Modelo de Balzani. Relación con los procesos de transferencia electrónica.
Balzani y colaboradores (Balzani y Bolleta, 1978; Balzani et al., 1980) fueron los
primeros en hacer notar la similitud entre los procesos de transferencia electrónica y los
de transferencia de energía triplete-triplete. Mientras que para los primeros un electrón
es cedido de una molécula a otra, para los segundos, un segundo electrón es captado,
por lo que no hay cambio neto en la carga electrónica de ninguna de las dos moléculas,
aunque sí lo hay de energía (Esquema 6.2).
D A D+ A−+ + 3 D 3 A+ +1 A 1 D
Esquema 6.2. Saltos electrónicos implicados en el proceso de transferencia electrónica (izquierda) y de transferencia de energía triplete mediante mecanismo de intercambio electrónico (derecha).
En base a esta similitud entre los procesos de transferencia electrónica y de intercambio
electrónico, Balzani y colaboradores recurrieron a las relaciones de energía libre que
existían para los procesos de transferencia electrónica para aplicarlos a la TET.
Fue R. A. Marcus quien desarrolló en base a las propiedades de las SEP que intervienen
en el proceso de transferencia electrónica la descripción a nivel molecular de los
mismos. Dentro de esta teoría se enmarca la expresión teórica desarrollada por Marcus

244 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
que relaciona la energía libre de activación con la energía libre del proceso (Marcus,
1960; Marcus, 1964):
( )( )
( )2
0 14 0
GG GG∆
∆ = ∆ +∆
‡ ‡‡
(6.9)
Posteriores variantes de esta ecuación han sido propuestas. Cabe destacar las relaciones
de Rehm y Weller (Rehm y Weller, 1969; Rehm y Weller, 1970):
( ) ( )( )1 22
202 2G GG G∆ ∆⎡ ⎤∆ = + + ∆⎢ ⎥⎣ ⎦
‡ ‡ (6.10)
Así como la obtenida por Agmon y Levine (Agmon, 1978; Agmon y Levine, 1977):
( )
( )( )0 lln 1 exp
ln 2 0G GG G
G∆ ∆⎡ ⎤∆ = ∆ + + −
⎣ ⎦∆
‡‡
‡
n 2 (6.11)
En todas ellas es la denominada barrera intrínseca de reorganización, que
corresponde a la energía libre de activación para un proceso con
( )0G∆ ‡
0G∆ = .
Estas relaciones de energía libre pueden ser aplicadas a través del correspondiente
modelo cinético (ver detalles en el punto 6.3.2) al problema de la TET:
( )exp
1 1
en d
d e
en d
kk k kk k− −
−
=+ + n
(6.12)
Donde es la constante de difusión de formación (destrucción) del complejo
[A···D], y es la constante del paso de transferencia de energía desde el dador al
aceptor (y viceversa) en el complejo de encuentro. Siendo
( )d dk −
( )en enk −
0 G RTen enk k e−∆=
‡ , y G RT
en enk k e−∆− = , para dar (aplicando el modelo del Agmon y Levine al cálculo de la
energía libre de activación):
( )
( )( )
exp
0
0 ln 1 expln 2 01 exp
en d
G RT d
en
kkG GGk Ge
k RT∆ −
=∆ ∆⎧ ⎫ln 2⎡ ⎤∆ + + −⎣ ⎦⎪ ⎪∆+ + ⎨ ⎬
⎪ ⎪⎩ ⎭
‡
‡
(6.13)
La constante cinética de velocidad queda expresada en función de la energía libre del
proceso y del parámetro de reorganización ( )0G∆ ‡ , así como de las constantes: ,
y , las cuales deben ser prácticamente iguales para una serie de experimentos de
TET en los que un mismo aceptor sea desactivador (quencher) de una serie de dadores
con energías triplete distintas, pero con una similitud estructural y electrónica (
0enk dk
dk−
Balzani y
Bolleta, 1978; Balzani et al., 1980).

6.1 Introducción 245
Ahora el problema se limita a evaluar los parámetros y constantes de la ecuación (6.13).
Por un lado se toma como: G∆
(6.14) ( ) (00 00* , * ,G E D D E A A∆ − + )
Esta simplificación que hacen los autores en el cálculo de la energía libre del proceso se
basa en varios hechos. En primer lugar, si no hay un gran cambio en las funciones de
partición (vibracional y rotacional) de las moléculas en un estado electrónico y otro, la
entalpía a temperatura ambiente se puede tomar como la diferencia de las energías
correspondientes a las transiciones 0-0 de ambas moléculas. En segundo lugar, el
término entrópico de la energía libre puede ser considerado nulo por dos motivos
principales:
-Los cambios en el momento dipolar de las moléculas dadora y aceptora deben ser
pequeños, ya que ninguna de ellas sufre ganancia ni pérdida de carga. Tampoco es de
esperar que haya una redistribución importante de la carga electrónica interna en las
transiciones S0 → T1 de la molécula aceptora y S0 ← T1 de la molécula dadora, por lo
tanto la variación de energía de solvatación debe ser pequeña y por lo tanto su
contribución a la energía libre prácticamente nula.
-Por otro lado, al no formarse ni romperse enlaces químicos no se ven afectados los
grados de libertad del sistema, y este factor no contribuye al término de variación de
entropía, y por lo tanto tampoco a la energía libre del proceso.
El resto de parámetros de la ecuación (6.13), o bien son evaluados a partir de datos
experimentales conocidos, o bien ajustados a partir de una serie de experimentos en los
que se usa un mismo aceptor con varios dadores con distintas energías triplete pero con
características estructurales comunes.
En resumen, a partir de este tratamiento clásico, es posible calcular las constantes de
velocidad de TET recurriendo a relaciones de energía libre aplicadas en procesos de
transferencia electrónica.
3. Factores Estructurales
Desde el comienzo del estudio de los procesos de TET, especialmente los no-verticales,
se han usado criterios estructurales concernientes a los cambios que sufre la molécula
aceptora en el proceso de TET no-vertical para racionalizar este tipo de procesos desde
un punto de vista cualitativo. Esto es debido a que hay una clara relación entre este
comportamiento y los grandes cambios estructurales que sufre la molécula.

246 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
Aunque desde que se observó en el cis-Estilbeno el comportamiento no-vertical, se
postuló un cambio importante en la estructura de la molécula en el proceso de
transferencia de energía triplete, no ha sido hasta hace pocos años cuando se ha podido
estudiar con detalle estos procesos desde un punto de vista teórico, debido al creciente
desarrollo de los medios para calcular teóricamente las propiedades de las SEP con
métodos ab initio. Se han estudiado de diversas maneras las trayectorias sobre dichas
superficies que dan información sobre los aspectos estructurales y energéticos que
pueden controlar el proceso de transferencia no-vertical en moléculas aceptoras.
Cabe destacar dentro de este tipo de estudios, el de Catalán y Saltiel (Catalán y Saltiel,
2001) sobre las deformaciones geométricas que sufren los estilbenos y su relación con
la energética del proceso. Por otro lado, una serie de trabajos realizados por Allonas y
colaboradores (Lalevée et al., 2002, 2002b) han abordado la TET no-vertical (ver
siguiente punto) también desde esta perspectiva, en distintas oximas. En concreto, estos
autores aplican el modelo de Agmon-Levine-Balzani (ver más arriba en este mismo
punto) con el objetivo de estudiar la relación que tienen las deformaciones geométricas
que ocurren durante el proceso de TET con la constante cinética de velocidad.
6.1.3 Tratamientos Cuánticos de la TET
Ulstrup y Jortner (Ulstrup y Jortner, 1975) desarrollaron un modelo cuántico para la
transferencia electrónica en disolución. Éste ha sido la base de posteriores modelos
cuánticos análogos para la transferencia de energía triplete-triplete. De hecho, Orlandi y
colaboradores lo aplicaron al estudio del comportamiento no-vertical del cis- y trans-
Estilbeno (Orlandi et al., 1980) tal y como se resume a continuación:
El modelo se basa en la aplicación de la regla de oro de Fermi (ver sección 3.2). La TET
pueden ser considerada como un proceso no radiante en el que una supermolécula
(entendiendo por tal al conjunto del aceptor, el dador y el disolvente) pasa de un estado
electrónico [*D···A] a otro [D···A*]. La constante de velocidad del proceso es el
producto de dos términos, uno electrónico y otro nuclear:
k (6.15) EN=
donde el término electrónico (E), es la energía de interacción entre los dos estados
electrónicos al cuadrado, es decir, 2ifE H= .

6.1 Introducción 247
El término nuclear de la ecuación (6.15) se corresponde con la integral de solapamiento
(J) entre los espectros de emisión del dador y absorción del aceptor según la ecuación:
22ek Hif Jπ= (6.16)
La integral de solapamiento depende de la temperatura, ya que a su vez depende de la
distribución de población de las moléculas en los distintos niveles energéticos
(traslacionales, rotacionales y vibracionales).
Sin embargo, la integral de solapamiento J es difícil de evaluar en el proceso de
transferencia de energía triplete, ya que los espectros de emisión y absorción
corresponden a transiciones prohibidas y por lo tanto la intensidad de las mismas es
prácticamente nula.
En base a las propiedades de la superficie de energía potencial de las dos moléculas es
posible formular teóricamente el término nuclear (N), que dentro del tratamiento de
Ulstrup y Jortner es igual a:
( ) ( ) ( )[ ]
( )( ) ( )
1 21
2
2
exp
exp 4
; ;
D A D Ai i f f
D AD A i i
S
D D A AS i f i f S
D D A AD i f A i f
N q qE
E E E
S S
ε ε ε ε
πβ β ε ε
β ε ε ε ε
ε ε ε ε
−= −
× − ∆ − + − + −⎡⎣×
∑∑∑∑ +
⎤⎦ (6.17)
donde 1 Bk Tβ = , es la energía de reorganización de los modos normales de
vibración clásicos, esto es, los que cumplen que
SE
Bh k Tν < . Dq y son las funciones
de partición de dador y aceptor,
AqDiε , D
fε , Aiε y A
fε son los niveles de energía vibracional
de dador y aceptor en sus estados inicial (i) y final (f). E∆ es la diferencia de energía
entre las energías espectroscópicas 0-0 de dador y aceptor, y DS y son los términos
de solape vibracional (FC) entre los estados inicial y final de dador y aceptor
respectivamente.
AS
La determinación de este término nuclear (N) requiere el conocimiento de todas las SEP
involucradas en el proceso, además de las funciones de onda vibracionales (incluyendo
correcciones anarmónicas), por lo que esta formulación teórica debido a la complejidad
en la determinación de todos los términos, ya sea teórica como experimentalmente, la
hacen poco útil. Sin embargo, es posible simplificar esta ecuación. Para ello, es posible
simular el espectro electrónico como debido únicamente a uno o unos pocos modos
normales vibracionales con frecuencias bajas que son las que dan lugar a una progresión
mayor de bandas de vibración en el espectro (de absorción o emisión), e incluyendo el

248 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
resto de modos normales (con frecuencias mayores) como un solo modo normal. Ambas
frecuencias pueden ser tomadas en principio del espectro de absorción y emisión de
aceptor y dador respectivamente. Sin embargo, como ya se ha apuntado para los
procesos de TET las transiciones son prohibidas por la conservación de la multiplicidad
de espín, por lo que estos espectros suelen ser muy débiles y difícilmente detectables.
En hidrocarburos insaturados la naturaleza de los estados electrónicos T1 y S1 suele ser
similar, por lo que los espectros en los que están involucrados ambos estados y S0,
suelen tener una forma parecida. A partir de los espectros S0 → S1 y S1 → S0 se pueden
obtener por lo tanto los parámetros de la ecuación (6.17) correspondientes a los
espectros S0 → T1 y T1 → S0.
6.1.4 TET Vertical vs. No-Vertical en fase condensada
Hasta ahora se han presentado las más destacadas teorías desarrolladas para los procesos
de transferencia de energía triplete-triplete vertical y no-vertical. En este punto se
comparan y detallan las principales diferencias entre ambos tipos de TET.
Como ya se ha dicho anteriormente, el adjetivo no-vertical se aplica a procesos de TET
en los que existe una desviación respecto de una cinética de Sandros. En general se dice
que un quencher o aceptor presenta un comportamiento no-vertical cuando para una
serie de procesos de TET frente a distintos dadores (los cuales pueden ser elegidos de
tal manera que no presenten grandes deformaciones geométricas en el proceso de
transferencia) las constantes de velocidad de TET son mayores que las predichas por la
ecuación de Sandros (6.8).
Existen dos aspectos fundamentales en la explicación del proceso. La primera de ellas
es la relacionada con las deformaciones geométricas que deben ocurrir en la molécula
aceptora en el proceso. En todos los casos estudiados de TET no-vertical parece existir
un gran cambio conformacional de la molécula aceptora en el estado excitado (triplete).
Sin embargo, en hidrocarburos insaturados distintos autores atribuyen este
comportamiento a distintas deformaciones de la molécula aceptora. Así por ejemplo, en
el caso de compuestos con enlaces dobles C=C conjugados (con distinto grado de
deslocalización) se ha propuesto tanto que la rotación entorno al enlace sencillo
(Gorman et al., 1991; Caldwell et al., 1992; Gorman et al., 1993; Davies et al., 1995)

6.2 Aplicación de la Teoría del Estado de Transición 249
como entorno al enlace doble (Catalán y Saltiel, 2001) son las responsables de este tipo
de comportamiento.
Desde un punto de vista experimental éste es un tema difícil de abordar, ya que la única
manera de estudiar el problema (usada hasta ahora) es sintetizando compuestos con
distintas restricciones estructurales y midiendo la constante de TET en cada caso, sin
embargo, en muchos casos (como se verá más adelante) no está claro en qué medida se
bloquean estas coordenadas moleculares, lo que impide una clara interpretación de los
resultados. Por otro lado, desde un punto de vista teórico, si bien se puede disponer de
SEP ab initio con un alto grado de precisión, surge la cuestión de la elección de las
coordenadas que contribuyen definitivamente al comportamiento no-vertical, elección
que parece en algunos casos subjetiva (como también se discutirá más adelante en una
crítica a estos trabajos teóricos).
En segundo lugar, está el aspecto energético, el cual evidentemente está relacionado con
las deformaciones estructurales antes discutidas. Con respecto a los factores energéticos
que controlan la TET no-vertical existen dos interpretaciones claramente enfrentadas.
Por un lado, distintos autores sostienen que es la activación térmica de el/los modo/s
normales en el estado fundamental del aceptor la que incrementa la velocidad de
transferencia de energía a través del término entálpico de activación en los procesos no-
verticales frente a la TET “normal”. Por otro lado, una segunda interpretación basada
principalmente en un trabajo de Saltiel y colaboradores (Saltiel et al., 1984) atribuye al
factor entrópico de activación el control del proceso de TET no-vertical.
En los próximos apartados se desarrollará un modelo teórico en sus distintas
aproximaciones que permite estudiar los factores geométricos y termodinámicos que
modulan las reacciones de TET verticales y no-verticales.
66..22 AApplliiccaacciióónn ddee llaa TTeeoorrííaa ddeell EEssttaaddoo ddee TTrraannssiicciióónn ppaarraa pprroocceessooss
ddiiaabbááttiiccooss aa llaa TTEETT
La transferencia de energía triplete-triplete (TET) es un proceso diabático, ya que
implica un cruce entre dos superficies de energía potencial en el espacio de coordenadas
internas del sistema molecular, y gracias a un acoplamiento débil (Speiser, 1996) debido
al solape de las nubes electrónicas de ambas moléculas (Dexter, 1953; Naqvi y C. Steel,

250 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
1970), se produce la transferencia de energía. Debido a esto, es posible tratar el estudio
de la constante de velocidad del proceso con la teoría del estado de transición para
procesos diabáticos (Laidler, 1987). Para poder aplicar esta teoría es necesario conocer
la topología de las SEP que intervienen en el proceso con un alto grado de precisión. En
este primer punto se van a estudiar en primer lugar las características generales y
comunes de las SEP que intervienen en el proceso de TET, así como el papel de las
coordenadas internas del complejo dador-aceptor [D···A] en el acoplamiento electrónico
y en la energía de activación. Para ello se dividirán las coordenadas internas del
complejo en dos conjuntos bien diferenciados. Una vez realizada esta descripción
general de las SEP, se pasará a la obtención de una expresión para la constante cinética
en base a las propiedades ya estudiadas.
6.2.1 La Superficie de Energía Potencial del Proceso de Transferencia
de Energía Triplete
El esquema de reacción de transferencia de energía se puede expandir (Balzani y
Bolleta, 1978; Balzani et al., 1980; Orlandi et al., 1980) separando los procesos de
difusión (kd, k-d,) y de transferencia de energía (ke, k-e) como (Esquema 6.3):
3D + 1A d
d
k
k−⎯⎯→←⎯⎯ [ 3D···1A ] e
e
k
k−⎯⎯→←⎯⎯ [1D···3A] d
d
k
k
−⎯⎯→←⎯⎯ 1D + 3A
Esquema 6.3
En el proceso de transferencia de energía están implicadas solamente las SEP del
complejo de encuentro reactivo [3D···1A] y del producto [1D···3A], los cuales pueden
considerarse como una misma supermolécula en dos estados electrónicos distintos (Fig.
6.1a) (de ahora en adelante se usarán por simplicidad los símbolos [*D···A] [D···*A]). El
proceso de transferencia de energía consiste en el paso del sistema desde el primero de
los estados al último. Asumiremos que no se forman exciplejos estables en la reacción
de TET en disolución (Scaiano y Wubbels, 1981), ya que la interacción dador-aceptor
es muy débil. Si esta interacción es despreciable, la energía relativa de cada complejo
viene dada por: E[*D···A] = E(*D) + E(A) y E[D···*A] = E(D) + E(*A). Sin embargo,
cuando las dos SEP toman valores de energía cercanos, debe existir un acoplamiento

6.2 Aplicación de la Teoría del Estado de Transición 251
entre ambos estados para que se pueda verificar el paso de un estado a otro. Por lo tanto,
se debe introducir una corrección a la energía debida a este efecto.
Las SEP de ambos complejos de colisión se pueden expandir como función únicamente
de las coordenadas internas del mismo, con F grados de libertad, dados por F = 3 (ND +
NA) - 6, donde ND(A) es el número de átomos del dador (aceptor), cada uno de ellos
asociado a una coordenada. El número total de coordenadas, Q, se puede separar en dos
grupos, { }, ,r t vQ Q Q= , donde define la posición relativa entre dador y aceptor (tres
coordenadas de traslación y tres de rotación) y corresponde a todas las vibraciones
moleculares [3(N
,r tQ
vQ
D + NA) – 12]. En el paso de transferencia de energía, la simetría
espacial y de espín de la función de onda total se conserva, dando lugar a un cruce
evitado§ en la región de aproximación de ambas SEP (Neumann y Wigner, 1929) (Fig.
6.1b).
[ ]* ···D A
[ ]···*D A
Energy
G∆
2 ifH
ATE
DTE
vQ
†0E
,r tQ
Energy
GD
vQ
)a )b
2 ifHEC
Energía
Energía
Fig. 6.1. Superficies de energía potencial correspondientes al cruce entre los dos estados del complejo de encuentro [D···A] para la reacción de transferencia de energía. a) SEP en función de una coordenada de vibración (Qv). D
TE y ATE son las energías ópticas de
excitación al estado triplete para dador y aceptor respectivamente; †0E es la energía de cruce
de las SEP en ausencia de acoplamiento electrónico; Hif representa la magnitud (exagerada) de la energía de acoplamiento, y ∆G es la energía libre del proceso. b) Otra perspectiva de las SEP, esta vez en función de la posición relativa de ambas moléculas, dada por las coordenadas . Las flechas denominadas EC y GD muestran la dirección de los vectores de acoplamiento electrónico y diferencia de gradientes respectivamente.
,r tQ
§ Del término inglés “Avoided Crossing”

252 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
Las dos dimensiones especiales donde la degeneración de energía desaparece están
definidas por los vectores Acoplamiento Electrónico (vEC) y Diferencia de Gradientes
(vGD). El primero de ellos (EC) indica la máxima magnitud del acoplamiento, mientras
que el segundo (GD) da la diferencia entre los vectores gradiente de la energía
correspondientes a las dos SEP de los dos complejos para una configuración nuclear
determinada: [*D···A] y [D···*A]. A continuación, a cada vector antes nombrado se le
asigna un subespacio de coordenadas bien definido.
La magnitud de la energía de intercambio electrónico entre el dador y el aceptor viene
dada por el término matricial de acoplamiento [ ] [ ]* ··· ··· *ˆ
if D A D AH H ′= Ψ Ψ (i=inicial,
f=final) donde ,
1ˆi j ij
Hr
′ = ∑ es el hamiltoniano del acoplamiento electrónico, y los índices
i,j denotan los electrones de dador y aceptor respectivamente. La función de onda puede
ser tomada como [ ] ** ··· D AD A ψ ψΨ =℘ , y [ ] *···* D AD A ψ ψΨ =℘ , donde ψ representa la
función de onda electrónica de las moléculas aisladas, y ℘ es un operador de
permutación que actúa antisimetrizando el producto de las funciones de onda sobre las
permutaciones entre los electrones i y j (Naqvi y C. Steel, 1970).
La función de onda del complejo [A···D], en una primera aproximación, se puede tomar
como aquella compuesta por el HOMO y el LUMO de ambos estados, triplete y
singlete, de las moléculas que intervienen en el proceso (Dexter, 1953). De acuerdo con
esto, ifH es solamente función de la posición relativa de dador y aceptor, dada por las
coordenadas , ya que los cambios en el solape orbital debido a vibraciones internas
es de esperar que sean mucho menos importantes. Así pues, el vector de acoplamiento
electrónico
,r tQ
( )ECv debe depender únicamente de la posición relativa de dador y aceptor,
esto es, de las coordenadas . Por lo tanto, para una configuración nuclear dada el
vector de acoplamiento electrónico viene dado por:
,r tQ Q
( )6
,
1
if r tEC
H Qqµ
µ µ=
⎛ ⎞∂= ⎜ ⎟⎜ ⎟∂⎝ ⎠∑
Q
v u ,r tq Qµ ∈ . (6.18)
donde µu es el µ-ésimo vector unitario y ifH la energía de acoplamiento entre los
estados inicial y final.
Puede ser posible también añadir un término adicional de acoplamiento vibrónico en el
Hamiltoniano, debido a la degeneración energética de las SEP de los dos estados

6.2 Aplicación de la Teoría del Estado de Transición 253
electrónicos. Sin embargo, la contribución de la energía de acoplamiento vibrónico debe
ser solamente una pequeña fracción de la debida al acoplamiento electrónico. Esto se
puede demostrar de manera cualitativa:
Según la teoría de perturbaciones, el término de acoplamiento vibrónico puede ser
formulado como:
[ ] [ ] [ ]( )
[ ]
0 0
* ··· ··· * * ··· ··· *, , , ,
D Aif D A D A D A D A
i r t v i r t vi i
H H HW
Q Q= =
′∂ + +∂= Ψ Ψ ≈ Ψ Ψ
∂ ∂∑ ∑ (6.19)
donde 0,A DH es el hamiltoniano electrónico de la molécula aislada, y H ′ es el
acoplamiento electrónico anteriormente definido. Si la constante de velocidad de
conversión interna para cada una de las moléculas aisladas es despreciable (de otra
manera el proceso de transferencia de energía no tendría lugar), los dos primeros
términos en la suma de la ecuación (6.19) se anulan, es decir,
[ ]( )
[ ]
0,
* ··· ··· *, ,
0D AD A D A
i r t v i
HQ=
∂Ψ Ψ
∂∑ = , y la expresión resultante es:
[ ] [ ]
1
* ··· ··· *, ,
ijif D A D A
i j r t
rW
Q
−∂≈ Ψ Ψ
∂∑ (6.20)
Tomando como constante (la distancia media entre electrones) la ecuación
(6.20) se puede simplificar más para dar:
ijr R=
[ ] [ ] [ ] [ ] (1
2,* ··· ··· * * ··· ··· *
,if if r tD A D A D A D A
r t
RW RQ
−−∂
≈ Ψ Ψ = Ψ Ψ =∂
)2R S Q− (6.21)
donde representa el solapamiento orbital electrónico en función de las coordenadas
de posición relativa entre dador y aceptor: . Puesto que en la presente aproximación
la dependencia del acoplamiento electrónico con la separación entre electrones de una
molécula y otra (R) viene dada por , la contribución del término vibrónico
a la energía total debe ser muy pequeña para una misma separación electrónica en
comparación con
ifS
,r tQ
1if ifH R S−≈
ifW
ifH . En resumen, el acoplamiento entre los estados definidos por las
SEP de reactivos y productos se debe principalmente al intercambio electrónico, con
una pequeña contribución de acoplamiento vibrónico. Las componentes del vector
asociado a esta interacción ( se extienden únicamente sobre las coordenadas )ECv

254 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
traslacionales y rotacionales que describen la proximidad y orientación entre
dador y aceptor dentro del complejo de colisión.
( ,r tQ )
Por otro lado, el vector gradiente de la energía para los dos estados del complejo de
encuentro, y por lo tanto, el vector diferencia de gradientes, depende únicamente de las
coordenadas internas (vibracionales), ya que la interacción dador-aceptor en el proceso,
es tan pequeña que no se forman nuevos enlaces (que puedan acumular energía). De
acuerdo con esto, la diferencia de gradientes de energía no depende significativamente
de la posición relativa entre el dador y el aceptor: [ ] [ ]( )* ··· ··· *0
D A D AE E
qµ
⎛ ⎞∂ −⎜ ⎟ ≈⎜ ⎟∂⎝ ⎠Q
,
{ },r tq Qµ ∈ , para cualquier configuración Q .
Por lo tanto, el vector diferencia de gradientes para una configuración Q se puede
definir como:
[ ] [ ]( )* ··· ··· *
1
N D A D AGD
E E
qµµ µ=
⎛ ⎞∂ −⎜ ⎟
⎟=
⎜ ∂⎝ ⎠
∑Q
v u vQµ q ∈ (6.22)
En conclusion, el vector está asociado al subespacio de coordenadas , mientras
que el vector lo está al subespacio de coordenadas .
ECv ,r tQ
GDv vQ
6.2.2 La Constante de Velocidad de Transferencia de Energía
La constante de velocidad para el cruce entre las superficies de energía potencial de
reactivos y productos, de acuerdo con la teoría del estado de transición (Laidler, 1987)
se puede expresar como:
0
1 2···B
Vk TB
ek Tqk
q q hχ
≠ −
= e (6.23)
donde es la función de partición del complejo activado y qq≠i las funciones de
partición de reactivos y productos, V0 es la energía del complejo activado, χ el
coeficiente de transmisión, y el resto de símbolos tienen el significado habitual.

6.2 Aplicación de la Teoría del Estado de Transición 255
La etapa de cruce puede ser descrita como una transición diabática en un sistema que
presenta un acoplamiento débil.§ En este caso, el valor de χ viene dado por (Landau,
1932; Zener, 1933):
24
vif
f i
Hs sπ
χ−
(6.24)
donde v es la velocidad relativa con la que el sistema pasa el punto de mayor
acercamiento entre superficies y f is s− es la magnitud absoluta de la diferencia entre
vectores gradiente de las dos superficies que se cruzan. El cruce diabático débilmente
acoplado lleva consigo un descenso de la barrera de energía en comparación con aquella
de cruce entre superficies en las que no se tiene en cuenta dicho acoplamiento ( )†0E ,
esto es: (Fig. 6.1b). De acuerdo con esto, la constante de velocidad para
un cruce diabático se puede expresar como:
†0 0 ifV E H= −
†02
1 2
4···v
if
B B
H Eif k T k T
ef i
H q kTkq q hs s
π ≠ −
=−
e e (6.25)
donde †0E es la energía del complejo activado que se puede obtener a partir de las
superficies de energía potencial adiabáticas individuales de cada molécula (como se
verá más adelante en la Sección 6.3). Como se ha hecho notar más arriba, el valor †0E
no depende de la separación entre dador y aceptor dentro del complejo de colisión,
mientras que ifH cambia muy rápidamente (exponencialmente) con dicha separación.
Es más, debido a que if BH k T< , el término exponencial cumple 1if
B
Hk Te ≈ , y la constante
de velocidad dada por la ecuación (6.25) muestra una dependencia exponencial con la
distancia dador-aceptor, observada en sistemas simples (Dexter, 1953; Ermolaev, 1967).
§ El término transición diabática se usa aquí en el sentido usual, esto es, para indicar la transición que implica dos (o más) SEPs acopladas (interaccionantes).

256 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
66..33 AApprrooxxiimmaacciióónn ddee pprriimmeerr oorrddeenn
En esta sección se desarrollará la aproximación de primer orden para el cálculo de la
energía de activación del proceso de TET (factor exponencial en la constante cinética).
Básicamente, dicha aproximación consiste en expandir la función diferencia de energía
potencial entre los dos estados electrónicos del complejo hasta el primer orden:
( ) ( ) tA Dv T TE Q E E∆ ≈ − + x g (6.26)
donde son las coordenadas internas del complejo, es el vector de coordenadas
nucleares transpuesto, y
vQ tx
g es el vector gradiente de la energía.
A partir de esta aproximación se calculará la energía correspondiente al complejo
activado, y se obtendrá la constante cinética del proceso.
6.3.1 Cálculo de la energía del complejo activado
En esta sección se deducirá el valor de la energía del complejo activado en función de la
energía triplete del dador. El término de acoplamiento ifH (ecuación 6.5) depende
únicamente del solape orbital y, por lo tanto, es independiente de la energía del dador.
De acuerdo con esto, solamente se necesita determinar la dependencia de †0E (la energía
de activación sin considerar el acoplamiento electrónico) en función de la energía
triplete del dador. Para temperaturas moderadas, la energía potencial del complejo en su
estado inicial, [*D···A], definida como ( ) ( )0 * ···D A vE Q E Q≡ , se puede representar
clásicamente como función de las energías electrónicas ,*A DelecE , las coordenadas
vibracionales ( ),,
A Dqµ ν , y las constantes de fuerza armónicas ,*A Dµνκ , como:
( ) ( )0 1 2 3 6 1 2 3 6
3 6 3 6 3 6 3 6* *
, ,... , , ,...
1 1 ...2 2
A D
A A D D
A A A D D Do N N
N N N NA D A A A D D Delec elec
E Q E q q q q q q
E E q q q qµν µ ν µν µ νµ ν µ ν
κ κ
− −
− − − −
= =
= + + + +∑ ∑ ∑ ∑ (6.27)

6.3 Aproximación de Primer Orden 257
donde 2
,*, ,
A D oD A D A
Eq qµνµ ν
κ⎛ ⎞∂
= ⎜⎜ ∂ ∂⎝ ⎠e
⎟⎟ . El subíndice “e” se usa aquí para identificar la
configuración de equilibrio del complejo [*D···A]. Como se ha visto antes, debido al
débil acoplamiento entre estados, no existe acumulación de energía en el otro
subespacio de coordenadas . De manera similar, la superficie de energía potencial
del complejo en su estado final, [D···A*], definida como
,r tQ
( ) (1 ··· * )D A vE Q E Q≡ , se puede
expresar por:
( ) ( ) *
1 1 1 2 3 6 1 2 3 6
3 6 3 6 3 6 3 6 3 6 3 6* *
1 1
, ,... , , ,...
1 1 ...2 2
A D
A D A A D D
A A A D D D A DN N elec elec
N N N N N NA D A A A D D D
E Q E q q q q q q E E
q q q q q qµ µ µ µ µν µ ν µν µ νµ µ µ ν µ ν
α α κ κ
− −
− − − − − −
= =
= = +
+ + +∑ ∑ ∑ ∑ ∑ ∑
+
+ (6.28)
donde * 1AA
Eqµµ
α⎛ ⎞∂
= ⎜ ⎟⎜ ⎟∂⎝ ⎠e
, 1DD
Eqµµ
α⎛ ⎞∂
= ⎜ ⎟⎜ ⎟∂⎝ ⎠e
y 2
* , 1* , * ,
A DA D A D
Eq qµνµ ν
κ⎛ ⎞∂
= ⎜ ⎟⎜ ⎟∂ ∂⎝ ⎠e
.
la ecuación (6.28) corresponde a la expansión de la superficie de energía potencial de
[D···A*] hasta el término cuadrático, usando el mismo origen y conjunto de
coordenadas usadas en la ecuación (6.27).
Para el espacio de cruce y, por lo tanto, de las ecuaciones (6.27) y (6.28)
tenemos:
0 1( ) ( )E Q E Q=
( )
( )
3 6 3 6 3 6 3 6* *
1 1 1 1
3 6 3 6*
1 1
12
1 ...2
A D A A
D D
N N N ND A A A D D A AT T
N ND D D D
A AE E q q
q q
q qµ µ µ µ µν µν µ νµ µ µ ν
µν µν µ νµ ν
α α κ κ
κ κ
− − − −
= = = =
− −
= =
− = + + −
+ − +
∑ ∑ ∑ ∑
∑ ∑ (6.29)
donde y ( )*A A AT elec elecE E E≡ − ( )*D D D
T elec elecE E E≡ − .
en aquellos casos donde el primer término es el que presenta la mayor contribución a la
diferencia de energía, la ecuación (6.29) puede ser simplificada para dar:
3 6 3 6
*
1 1
A DN ND A A AT TE E q qD D
µ µµ µ
α− −
= =
− ≈ +∑ ∑ µ µα (6.30)
esta expresión muestra que la diferencia de energía triplete entre el dador y el aceptor en
la región de cruce de las superficies energía potencial se corresponde con la activación
térmica de los grados de libertad de los dos reactivos. Es evidente que la ecuación (6.30)
será más precisa cuanto mayor sea el valor de los coeficientes * ,,A D
µ να . De hecho, estos

258 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
coeficientes son las componentes del vector gradiente de la energía en el estado final del
complejo después de la excitación vertical; un alto valor del gradiente (esto es, grandes
valores de los coeficientes α ) indican un importante cambio en la conformación de D,
A o de ambos en el proceso de transferencia. Esta es precisamente una de las
características de los procesos no-verticales, como se ha discutido antes, en los cuales la
desviación del comportamiento normal en la transferencia de energía triplete-triplete se
asoció a una estructura flexible para el aceptor (por ejemplo para el COT ver: Gorman
et al., 1991). Por otra parte, se debe a hacer notar que un gran cambio conformacional
no implica necesariamente un valor del gradiente de la energía alto.
Otra propiedad distintiva del estado singlete del aceptor en los procesos de transferencia
de energía no-vertical, es que pequeños cambios en las coordenadas vibracionales ( ) ,
llevan consigo un gran cambio en la diferencia de energías
iq
A DT TE E− , lo cual también
contribuye a la validez de la aproximación en la ecuación (6.30).
Los procesos de transferencia de energía triplete de interés en la presente discusión son
aquellos en los cuales están implicadas moléculas con una estructura flexible (como por
ejemplo el cis-stilbene y el COT). Por lo tanto, la ecuación (6.30), puede ser
simplificada para aquellos casos en los que la contribución de la deformación del dador
al cambio en la energía de excitación sea despreciable, obteniéndose:
3 6
*
1
AND AT TE E qA A
µ µµ
α−
=
− = ∑ (6.31)
Finalmente, la ecuación (6.31) se puede reformular en una nueva base ortonormal, en la
cual el vector gradiente para la excitación vertical de *A, es uno de los vectores de la
base, para dar:
1A D
T TE E Aξ− = g (6.32)
donde ( ) ( )1
23 6 2*1
1
ANAE Q µ
µ
α−
=
⎧ ⎫= ∇ =⎡ ⎤ ⎨⎣ ⎦
⎩ ⎭∑g ⎬ y 1
Aξ es la coordenada a lo largo del vector
gradiente.
En la expresión de arriba |g| (≡ g) es la norma del vector diferencia de gradientes (vGD)
entre los gradientes en las superficies T1 y S0, es decir ( ) ( )1T S= −g g g 0 . Puesto que la
excitación desde el estado S0 del aceptor tiene lugar desde una configuración cercana a
la posición de equilibrio donde el gradiente es nulo, el valor de g corresponde

6.3 Aproximación de Primer Orden 259
básicamente al gradiente en la superficie del estado triplete solamente .
De acuerdo con esto, la energía de excitación del aceptor en esta nueva base depende
únicamente de una sola coordenada
( )( )1T ≡ 1g g g
1Aξ , que es aquella correspondiente al vector
gradiente del aceptor en el estado triplete:
1 1A D
T TE E g Aξ− = (6.33)
Ésta puede ser considerada como una generalización del concepto de “hot-band”
discutido antes, ya que como muestra la ecuación (6.33), el déficit de energía puede ser
reducido por una distorsión geométrica del aceptor en su estado fundamental a lo largo
de una coordenada vibracional ( )1Aξ , la cual corresponde al mayor cambio de energía
en la superficie de energía potencial triplete. Las distorsiones en otras direcciones,
corresponden a vectores de desplazamiento ortogonales al vector gradiente g, y por lo
tanto no contribuyen, dentro de este formalismo, a la expansión de energía dada por la
ecuación (6.33). Además, queda claro que el conjunto completo de componentes del
gradiente (elongaciones de enlace, torsiones y flexiones) determinan la energía de
excitación del aceptor.
La energía vibracional ( del complejo inicial [*D···A] puede ser obtenida de la
ecuación (6.27) restando a la energía total la energía electrónica, para dar:
)0,vibE
3 6 3 6 3 6 3 6
*0,
1 1 ...2 2
A A D DN N N NA A A D D D
vibE q qµν µ ν µν µ νµ ν µ ν
κ κ− − − −
= +∑ ∑ ∑ ∑ q q + (6.34)
Esta expresión puede ser también redefinida en una nueva base, en la cual las
coordenadas son 1Aξ y aquellas, para el aceptor solamente, que diagonalizan la matriz
hesiana de la energía en el subespacio de coordenadas ortogonales a 1Aξ , esto es, A
iξ
con . Si las coordenadas del dador 2,...,3 6Ai N= − Diq se mantienen, tenemos:
( ) ( )
3 6 3 6 3 6 3 6*
0,
3 6 3 6 3 6 3 62 2 *11 1 1 1
2 2
1 12 2
1 1 12 2 2
A A D D
A A D D
N N N NA A A D D D
vib ij i ji j
N N N NA A A A A A A D D D
i i ii ii i
E q q
q q
µν µ νµ ν
µν µ νµ ν
κ ξ ξ κ
κ ξ κ ξ ξ κ ξ κ
− − − −
− − − −
= =
= +
= + + +
∑ ∑ ∑ ∑
∑ ∑ ∑ ∑ (6.35)
donde 2
0Aij A A
i j
Eκξ ξ
⎛ ⎞∂= ⎜ ⎟⎜ ⎟∂ ∂⎝ ⎠0
.

260 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
La energía vibracional del aceptor, , correspondiente a la configuración de mínima
energía en subespacio de cruce (es decir, la energía de activación sin considerar el
término
†0,vibE
ifH ) consistente con la ecuación (6.33), se puede obtener definiendo la función
Lagrangiana como: ( )L
( )0, 1A D A
vib T TL E E E gλ ξ= + − − (6.36)
Derivando con respecto a 1Aξ y A
nξ se obtiene:
3 6
11 1 121
11 1 1
0
0
ANA A A A
i iAi
AA A A A A Ann nn n nA A
n n
L g
L
κ ξ κ ξ λξ
κκ ξ κ ξ ξ ξξ κ
−
=
⎧ ∂= + + =⎪∂⎪
⎨∂⎪ = + = ⇒ = −⎪∂⎩
∑
n
(6.37)
Finalmente, reemplazando en la ecuación (6.35) el valor obtenido por las ecuaciones
(6.33) y (6.37), se obtiene la siguiente expresión:
( )( ) (
23 6
111
22† 10, 2 2
1 12 2
AANiA
Ai A D Aii
vib T T T TE E Eg g
κκ
κ κ
−
=
−= − ≡
∑)2DE E− (6.38)
Se debe recalcar que la energía mínima en el punto de cruce es independiente en de las
coordenadas del dador, ya que el cambio de energía (descenso) del sistema depende
solamente de la coordenada del aceptor 1Aξ (ecuación 6.33).
Esta relación (ecuación 6.38) da el valor de la barrera energética para la reacción de
transferencia de energía directa en función de las constantes de fuerza en el estado
fundamental para el aceptor, el gradiente de la energía en la superficie de energía
potencial triplete para la molécula aceptora (g) y la diferencia entre las energías de
excitación ópticas del aceptor y el dador a los estados triplete. Puesto que las superficies
energía potencial definidas aquí no incluyen ninguna contribución debida al efecto de
disolvente, la barrera de energía corresponde a un entorno libre del disolvente, o
también a un entorno en el cual el disolvente permanece sin perturbar el sistema. Esto
es, por supuesto, una simplificación pero es de esperar que no lleve consigo un gran
error en el cálculo de las energías de activación, puesto que la transferencia de energía
triplete no implica una gran redistribución de cargas. Por otro lado, el valor del factor
preexponencial en la ecuación (6.25) debe ser sensible a las propiedades del disolvente,
ya que estas influyen en el término de velocidad v.
( )1κ

6.3 Aproximación de Primer Orden 261
Sustituyendo la ecuación (6.38) en la expresión de la constante de velocidad, tenemos:
( )
†20, 1
212 2
2
1 2 1 2
4 4··· ···
ifvib if A DT T
B B BB
HE HE Ek T k Tif if k Tk T gB B
ef i f i
H Hk T k Tq qk e e eq q h q q hv s s v s s
κπ π⎛ ⎞⎜ ⎟− −≠ ≠ − −⎜ ⎟⎝ ⎠= =
− − (6.39)
Esta expresión se aplica a una reacción de transferencia de energía triplete-triplete en la
cual la conformación del aceptor en el estado fundamental cambia sustancialmente en el
proceso de transferencia, comparada con la que sufre la molécula dadora. El factor pre-
exponencial en la ecuación (6.39) puede ser ahora analizado en detalle en base a las
propiedades de la superficie de energía potencial discutidas antes. Así, la velocidad
relativa v con la cual el complejo cruza el espacio de acoplamiento debe estar
determinada por la frecuencia de vibración de los modos contenidos en . Puesto que
en el modelo discutido aquí el vector GD depende únicamente de las propiedades de la
SEP del aceptor (o en otras palabras, el descenso en la energía de excitación es debido
únicamente a deformaciones del aceptor), el valor de v debe depender de las frecuencias
vibracionales del aceptor, del efecto de los modos vibracionales del disolvente
acoplados a aquellos de la supermolécula, así como de la temperatura.
1κ
Por otro lado, el parámetro f is s− es justamente el módulo del vector GD en el espacio
de cruce. En una serie de experimentos donde se usa la misma molécula aceptora, este
término permanece constante ya que, como se discutió antes, el gradiente del estado
excitado es mucho mayor que el estado fundamental. Por lo tanto,
constantef i fs s s− ≈ = .
El término de acoplamiento electrónico ifH , es una integral bielectrónica de dos centros
que depende fundamentalmente de la estructura del HOMO y del LUMO de ambas
moléculas (dador y aceptor). Sin embargo en una serie de experimentos de TET en los
cuales existe un aceptor común y una serie de dadores con las mismas características
electrónicas ( *,π π , por ejemplo) en los cuales la distribución espacial de los orbitales
moleculares se puede tomar como prácticamente constante, el valor de ifH no se debe
alejar sustancialmente de un valor medio característico de la serie de dadores. Un
razonamiento similar se puede aplicar en las funciones de partición en la ecuación
(6.39), haciendo notar que estas funciones pueden dependen de los grados de libertad
del dador y aceptor respectivamente. Por lo tanto, si la conformación de las moléculas
dadoras seleccionadas no cambia de manera importante tras la transición T1→ S0, la

262 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
expresión para la constante de velocidad de transferencia de energía a un aceptor común
puede ser aproximada por:
( )( )
( )( )2 21 1
2 21 1
2 220
A D A DifT T T T
B B
H E E E Ek T k Teg gkT
e ifk H k T e e k T eκ κ
− − − −
= (6.40)
En resumen, la expresión para la constante de velocidad, como se ha deducido arriba,
para una reacción de transferencia de energía triplete-triplete entre una serie de dadores
triplete rígidos y un aceptor flexible, contiene:
i) un factor pre-exponencial k0e asociado con el solape orbital entre dador y aceptor a
través del término 2if
B
Hk T
ifH e así como otros factores, los cuales cabe esperar que no
dependan de la diferencia de energías triplete, como ya se ha propuesto antes (Balzani y
Bolleta, 1978; Balzani et al., 1980; Orlandi et al., 1980; Saltiel et al., 1984; Abu-
Hasanayn y Herkstroeter, 2001), y:
ii) un término exponencial el cual muestra una dependencia cuadrática con la diferencia
de energía entre el dador y el aceptor en sus estados triplete.
De la ecuación (6.40) podemos definir un parámetro de distorsión geométrica:
( 1/ 2212gγ κ= ) (6.41)
Este parámetro determina el grado de comportamiento no-vertical. Para moléculas
aceptoras con un alto valor del gradiente de la energía en la superficie triplete, la fácil
activación de los modos vibracionales del estado fundamental (bajas constantes de
fuerza ) a lo largo de la dirección del gradiente deben ser muy efectivas reduciendo la
energía de excitación triplete (alto valor de γ ), resultando en valores altos de la
constante de velocidad incluso para reacciones “endotérmicas”. Sin embargo, el hecho
de que ocurra un cambio conformacional significativo puede ser completamente
inefectivo a este respecto, a no ser que al mismo tiempo ocurra un gran cambio en la
energía del estado triplete del aceptor (gradiente en T
1κ
1 alto). Es más, puesto que los
valores de γ y ATE están fijados para una molécula aceptora específica, la ecuación
(6.40) predice una región invertida, donde un incremento de la energía disponible del
dador para la excitación conlleva una disminución en la constante de velocidad (Sigman
y Closs, 1991; Parola et al.,1996; Serpa et al., 2003), en común con la teoría de Marcus
para la transferencia electrónica (Marcus, 1956 ; Marcus, 1960; Marcus, 1964). Sin
embargo, este efecto debe ser difícil de observar en las reacciones de transferencia de

6.3 Aproximación de Primer Orden 263
energía triplete-triplete, debido a la presencia de estados triplete de más alta energía y
de canales alternativos de reacción (Formosinho et al., 1998; Bodesheim et al., 1994).
La expresión para la constante de velocidad (ecuación 6.40) es sensible a pruebas
experimentales, ya que el valor de γ de muchas moléculas de interés se puede calcular
con un una alta precisión mediante métodos químico-cuánticos, como se muestra más
abajo para el COT. Es importante remarcar que el cálculo de las superficies energía
potencial completas para los estados singlete y triplete no es necesario en esta
aproximación, y solamente una parte de las superficies 3A y 1A en las proximidades de
la configuración equilibrio de 1A es necesaria (Fig. 6.2).
Fig. 6.2. Regiones de las SEP singlete y triplete del 1,3,5,7-Ciclooctatetraeno (COT) relevantes en el proceso de transferencia de energía, calculadas según está descrito en el texto. Las SEP están representadas en función del ángulo diedro, α, de cuatro átomos de carbono consecutivos (por ejemplo 1-2-3-4), que da el grado de planaridad de la molécula, así como en función de las distancias de enlace
C C C Cd dπ − =∆ = − , donde dC-C y dC=C son la suma de los enlaces simples y dobles (por ejemplo 2-3 y 1-2), respectivamente. Esta última variable crece inversamente con la deslocalización de la carga π. La flecha indica la dirección de relajación inicial (-vGD), que tiene una gran componente en ambas componentes de deslocalización de carga π y aplanamiento de la molécula. La energía 0-0 del 3COT (D8h) ha sido tomada de (Wenthold et al., 1996).

264 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
Finalmente puede ser instructivo comparar la constante de velocidad calculada con la
TST que viene dada por la ecuación (6.40), con aquella obtenida mediante la regla de
oro de Fermi y siguiendo el modelo de Orlando (Orlandi et al., 1980) (ecuación 6.16).
En ambos casos el factor exponencial depende del cuadrado del acoplamiento
electrónico ( 2if )H entre los estados electrónicos que intervienen. Además, se puede
demostrar (ver Apéndice F) que el término exponencial de la ecuación (6.40) es, de
hecho, la integral de solapamiento J entre los espectros de absorción del aceptor: T1←S0
y emisión del dador: T1 →S0 si ambos se aproximan por una única función Gaussiana
con anchuras muy distintas, esto es, 2 2A Dσ σ .
6.3.2 TET No-Vertical en el COT
En este punto se aplicará el modelo desarrollado en los puntos anteriores a la reacción
de transferencia de energía triplete-triplete del 1,3,5,7-Ciclooctatetraeno (COT), para el
cual están disponibles las superficies de energía potencial precisas (ver Sección 5.3).
Este polieno cíclico no aromático es muy flexible, y presenta una primera banda débil
de absorción a 282 nm en disolución de hexano (Perkampus, 1992), y la transición
más baja en el rango de energías de 65-70 kcal/mol, según datos de
espectroscopia de impacto electrónico (Frueholz
1T S← 0
y Kuppermann, 1978). Investigaciones
más recientes (Forward et al., 1993) de la energía triplete del COT en disolución dan un
valor de 59 kcal/mol. Por el contrario un valor mucho más bajo (41 kcal/mol) ha sido
descrito basándose en estudios cinéticos en los que se incluye un paso de transferencia
reversible de energía desde el COT (Das y Priyadarsini, 1994). En este último trabajo,
se le asigna al COT un tiempo de vida media de 100µs en disolución de ciclohexano, lo
cual parece difícil de conciliar con la baja energía del estado triplete (~22 kcal/mol
sobre la estructura D del estado fundamental (Wenthold et al., 1996)) y la ya citada
flexibilidad del mismo.
3
2d
El COT es usado como un eliminador universal de tripletes en láseres con colorantes
orgánicos, ya que puede mejorar la operatividad de dichos colorantes con energías tan
bajas como 40 kcal/mol (Das y Priyadarsini, 1994). Por lo tanto, no es inesperado el
comportamiento del COT como aceptor frente a una serie de dadores, observándose en

6.3 Aproximación de Primer Orden 265
dicho comportamiento (Forward et al., 1993) una fuerte desviación respecto a una
cinética de Sandros (ecuación 6.8). Esta desviación excepcional ha sido explicada en
base a los movimientos de torsión entorno a los enlaces C-C sencillos, los cuales deben
ser responsables de la reducción de la energía necesaria para poblar el estado triplete
(Forward et al., 1993).
La dependencia de ke con la energía de excitación dada por la ecuación (6.40) se puede
comparar con las obtenidas mediante la medición de las constantes de velocidad del
proceso de transferencia de energía para el COT (Forward et al., 1993; Das y
Priyadarsini, 1994), mediante la expansión en términos cinéticos mostrada en el
esquema 6.2. Si todas las moléculas de 3COT (una vez ocurrida la transferencia de
energía) decaen antes de ser interceptadas por una molécula dadora singlete (reacción
irreversible), la constante de velocidad experimental del proceso de transferencia de
energía triplete-triplete, , puede expresarse (Balzani y Bolleta, 1978; Balzani et al.,
1980; Orlandi et al., 1980) como:
expenk
exp
1
en d
d e
e e
kk k kk k− −
=+ +
(6.42)
La probabilidad de la transferencia inversa (k-e) desde el COT triplete dentro del
complejo de colisión es también despreciable (Nickel, 1992), debido a que debe darse
una relajación muy rápida de la energía triplete (ver arriba). Por lo tanto, si el cociente
1e ek k− , la ecuación (6.42) se reducen a:
exp
1
en de d
d
e
kk kk
kη−
= =+
(6.43)
donde ηe = ke /(ke + k-d ) es la eficiencia del proceso de transferencia para el complejo de
encuentro (Saltiel et al., 1974; Saltiel y Atwater, 1988). Por lo tanto, cuando la
constante de velocidad de transferencia de energía es mayor que la de disociación del
complejo (k-d), la reacción queda controlada por difusión (Saltiel et al., 1974; Saltiel y
Atwater, 1988). Se puede obtener una expresión directamente comparable a la
representación logarítmica de las constantes de velocidad experimentales de
transferencia de energía para el COT en función de la energía del dador sustituyendo ke
en la ecuación (6.43) por su valor dado por ecuación (6.40), para dar:
266 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
2
exp 0
1log log log 1 expA D
en d T Td
e
k E Ek kk RT γ−
⎡ ⎤⎡ ⎤⎛ ⎞−⎢= − + ⎥⎢ ⎥⎜ ⎟⎢ ⎥⎢ ⎥⎝ ⎠⎣ ⎦⎣ ⎦
(6.44)
Los valores calculados (ver métodos en el punto 6.3.3) del parámetro de distorsión
geométrica del COT, ( )1 213.5 kcal/molγ = , y de la energía triplete tras la excitación
vertical, , pueden ser sustituidos en la ecuación (6.44) dando una
expresión para
65.0 kcal/molATE =
( )explog enk en función de la energía de excitación triplete del dador, DTE :
2
exp 0
1 65.0log log log 1 exp13.5
Den d T
de
k Ek kk RT−
⎡ ⎤⎡ ⎤⎛ ⎞−⎢= − + ⎥⎢ ⎥⎜ ⎟⎢ ⎥⎢ ⎥⎝ ⎠⎣ ⎦⎣ ⎦
(6.45)
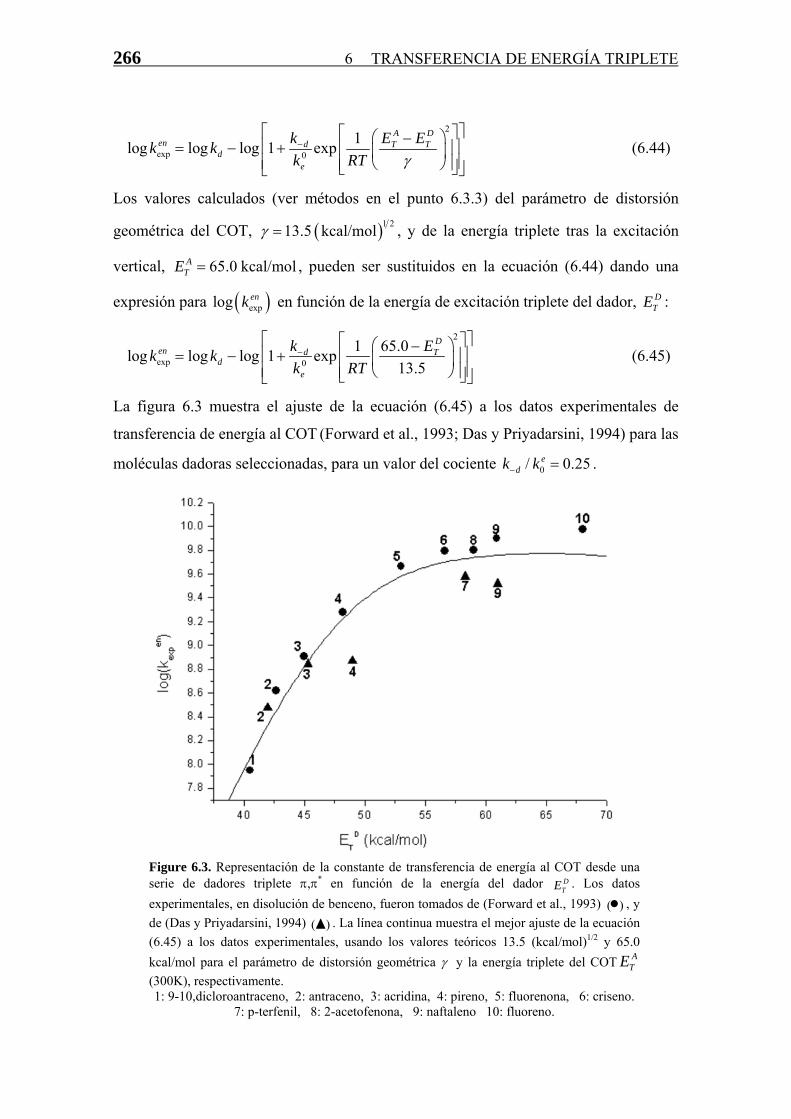
La figura 6.3 muestra el ajuste de la ecuación (6.45) a los datos experimentales de
transferencia de energía al COT (Forward et al., 1993; Das y Priyadarsini, 1994) para las
moléculas dadoras seleccionadas, para un valor del cociente k k . 0/ 0.2ed 5− =
Figure 6.3. Representación de la constante de transferencia de energía al COT desde una serie de dadores triplete π,π* en función de la energía del dador D
TE . Los datos experimentales, en disolución de benceno, fueron tomados de (Forward et al., 1993) , y de (Das y Priyadarsini, 1994) ( . La línea continua muestra el mejor ajuste de la ecuación (6.45) a los datos experimentales, usando los valores teóricos 13.5 (kcal/mol)
( )•)▲
1/2 y 65.0 kcal/mol para el parámetro de distorsión geométrica γ y la energía triplete del COT (300K), respectivamente.
ATE
1: 9-10,dicloroantraceno, 2: antraceno, 3: acridina, 4: pireno, 5: fluorenona, 6: criseno. 7: p-terfenil, 8: 2-acetofenona, 9: naftaleno 10: fluoreno.

6.3 Aproximación de Primer Orden 267
Como se ha mencionado antes, esta serie de moléculas dadoras tienen en común la
naturaleza electrónica de los orbitales frontera ( )*,π π así como una estructura
molecular rígida.
El análisis de las componentes del vector gradiente nos conduce a una descripción
detallada de las distorsiones geométricas que facilitan la excitación al estado triplete
para el COT. En este caso (Fig. 6.4), la mayor contribución proviene de los modos
vibracionales de “stretching” de los dobles enlaces C=C, los cuales hacen disminuir el
orden de enlace e incrementan la densidad electrónica π en los enlaces sencillos C−C.
La segunda contribución importante viene dada por los modos vibracionales de torsión
alrededor de los enlaces sencillos C−C de la molécula. En contraste, la contribución al
vector gradiente de los modos de torsión alrededor de los dobles enlaces es nula.
Como resultado de este movimiento acoplado, la conformación de bote (D2d) del estado
fundamental para el COT se deforma, favoreciendo una geometría más plana.
Curiosamente, cálculos de alto nivel (ver Sección 5.3) del COT así como un estudio
experimental de espectroscopia de estado de transición en este compuesto (Wenthold et
al., 1996) predicen una geometría plana octogonal D8h para el COT en su estado triplete.
Fig. 6.4. Geometría relajada en el estado fundamental del COT (simetría D2d) ilustrando la dirección (flechas) de las deformaciones geométricas que hacen decrecer la energía de excitación al estado triplete.

268 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
El error (desviación típica) del ajuste en la figura 6.3 presenta un único mínimo (no
mostrado) para un valor de 0.25 del cociente , el cual es el único parámetro de
ajuste, ya que el valor de k
0/ edk k−
d esta relativamente bien definido por un plató en los datos
experimentales. Debido a que este cociente es desconocido a priori, ha sido tomado
como idéntico para todas las moléculas dadoras usadas en el experimento (ver arriba) y,
por lo tanto, la consistencia del ajuste depende de la medida en que el valor calculado de
dicho cociente sea próximo al valor correcto. Los valores estimados para la constante de
velocidad de disociación del complejo de encuentro (k-d) en disolventes líquidos, tanto
desde argumentos basados en paso aleatorio (Wagner y Kochevar, 1968), así como en
argumentos más elaborados sobre difusión (Balzani y Bolleta, 1978; Balzani et al.,
1980; Orlandi et al., 1980; Nickel, 1992), caen en el rango de (un valor de
10
10 11 4 ·10 s−−11 s-1 se obtuvo de consideraciones macroscópicas de la entropía (Saltiel et al., 1974;
Saltiel y Atwater, 1988)). Así pues, del ajuste del valor de se puede estimar el
factor pre-exponencial del COT como k
0/ edk k−
e0 ≈ 1011s-1, el cual está en el mismo rango que
aquellos determinados directamente (Anderson et al. , 1974), por medio de medidas de
ke espectroscópicas de pico-segundo para diversos pares dador/aceptor aromáticos no
relacionados estructuralmente con el COT.
6.3.3 Métodos
El parámetro de distorsión geométrica γ fue determinado calculando el gradiente
numérico en la superficie de energía potencial T1 así como las constantes de fuerza
vibracionales numéricas en S0 (Fig. 6.2) usando la teoría de perturbaciones del segundo
orden de Møller-Plesset sobre una geometría optimizada al nivel CASSCF con un
espacio activo (8,8) en una base 6-31g* (CASMP2(8,8)/6-31g*) tal y como está
implementado en el paquete de programas Gaussian 98 (Gaussian98, 1998).; el espacio
activo en todos los cálculos CAS fue compuesto por los orbitales de simetría π (los
cuatro orbitales ocupados más altos energía y los cuatro más bajos desocupados). La
geometría de equilibrio para el estado S0 (D2d) fue optimizada usando gradientes
analíticos a nivel CASSCF(8,8)/6-31g*. El valor ATE fue determinado teóricamente (ver

6.3 Aproximación de Primer Orden 269
Capítulo 5), debido a los limitados datos experimentales, usando el método Complete
Active Space Second Order Perturbation Theory (CASPT2), con una función de
referencia CASSCF(8,8)/ ANO C[4s3p2d]/H[2s1p] tal y como está implementado en el
programa MOLCAS 5.0 (Molcas v.5, 1999; Roos et al., 1995; Roos et al., 1996;
Merchán et al., 1999).
6.3.4 Conclusiones sobre la aproximación de primer orden
El modelo teórico desarrollado para las reacciones de transferencia de energía triplete-
triplete obtenido aquí, permite la identificación de los factores físicos así como de los
cambios moleculares implicados en la excitación triplete no-vertical anómala de
aceptores flexibles, y proporciona una interpretación detallada de los movimientos
moleculares implicados en el proceso. La presente aproximación puede ser también de
utilidad a la hora de predecir si la constante de velocidad de transferencia energía de un
par específico dador/aceptor debiera mostrar o no una dependencia no-vertical de la
constante de velocidad con la energía triplete del dador. La aplicación de esta teoría a
los datos experimentales de transferencia energía para el COT en disolución
proporciona una visión más profunda del origen de la gran desviación de la constante de
transferencia energía respecto de la ecuación de Sandros. Además, los cambios
conformacionales en el estado fundamental responsables del descenso de la energía de
excitación triplete pueden ser identificados satisfactoriamente.

270 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
66..44 TTEETT ccoonn SSuuppeerrffiicciieess ddee EEnneerrggííaa PPootteenncciiaall ccoommpplleettaass:: AAllggoorriittmmoo
TTEETT--AACCCC
Para la aproximación lineal en la expansión de la diferencia de energías triplete
desarrollada en la sección precedente (6.3), una de las limitaciones intrínsecas, que ya se
advirtió en su momento, es que el gradiente en T1 para la geometría FC debe ser
suficientemente grande como para que dicha aproximación sea válida. Es posible
encontrar casos en los que al aplicar esta aproximación se cometa un cierto error que
pueda ser incluso cualitativo. Para subsanar esta carencia, en esta sección se desarrollará
un algoritmo que generaliza en cierta medida el tratamiento de la sección 6.3, ya que
haciendo uso de SEP completas (sin expansiones aproximadas de las SEP) permite
calcular las energías de cruce entre las superficies. A este algoritmo lo denominaremos
“Coordenada del Complejo Activado de Transferencia de Energía Triplete” (TET-
ACC)§.
En esta sección, en un primer punto (6.4.1) se tratará a fondo el planteamiento
matemático del algoritmo, así como la interpretación física de los resultados obtenidos.
En un segundo punto (6.4.2) se abordará el estudio de las funciones termodinámicas de
activación en base a los datos obtenidos de la aplicación del algoritmo.
En un tercer punto (6.4.3) se aplicará el algoritmo desarrollado al estudio del cis- y
trans-estilbeno, compuestos paradigmáticos en el estudio de la TET no-vertical.
Además, en este punto se compararán los distintos modelos teóricos existentes así como
las interpretaciones hechas por diversos autores de dicho comportamiento no-vertical de
la TET. Por último (6.4.4), se discutirán las limitaciones del algoritmo.
6.4.1 Algoritmo TET-ACC
En este punto se desarrollará el algoritmo (cuyo código fortran se encuentra en el
Apéndice G) que permite el cálculo de la estructura óptima (de menor energía) del
complejo activado en el proceso de TET. En primer lugar se presenta el problema del
cruce entre superficies no acopladas de forma general, sin aproximaciones de ningún § Las siglas provienen del nombre en inglés: “Triplet Energy Transfer Activate Complex Coordinate” (TET-ACC).

6.4 TET con Superficies de Energía Potencial Completas 271
tipo en la expansión de las SEP, así como las posibles maneras que hay de resolver
dicho problema matemáticamente. Posteriormente se presenta el algoritmo que recoge
una de las posibles soluciones matemáticas que a su vez es tal vez la de menor coste
computacional.
En el siguiente punto se analizan las curvas obtenidas con el algoritmo (topología y
relación con magnitudes físicas), y por último se añade un estudio sobre la región
invertida (exotérmica) que predice tanto la teoría presentada en 6.3 como la presentada
en esta sección.
6.4.1.1 Cruce entre Superficies de Energía Potencial Completas
Como ya se ha visto en los puntos anteriores, para el cálculo de la constante de
velocidad del proceso de transferencia de energía triplete-triplete es fundamental
conocer con detalle las SEPs de los estados involucrados en el proceso, con el fin de
determinar la estructura del complejo activado así como su energía, y de esta manera
poder calcular el factor exponencial de la constante de velocidad. En esta sección, se
presentan los detalles de un algoritmo que hace uso de toda la superficie de energía
potencial para el cálculo de la energía del complejo activado en función de la energía
triplete de la molécula aceptora: DTE .
A partir de ahora usaremos las SEP sin considerar el acoplamiento electrónico entre
ellas, discutiéndose más adelante la inclusión del mismo (ver punto 6.4.4). La superficie
de energía potencial adiabática (sin considerar acoplamiento entre estados, y por lo
tanto independiente del subespacio de coordenadas: ) del complejo antes de la
transferencia de energía [*D···A] se puede expresar en función únicamente de las
coordenadas internas ( ) de ambas moléculas
,r tQ
vQ ( ) [ ] ( )0 * ··· vD AE Q E Q≡ como:
( ) ( )
( ) ( )0 0 1 2 3 6 1 2 3
* *
, ,... , , ,...A
A A A D D DN
A D A A D Delec elec v v v v
E Q E q q q q q q
E E E Q E Q
−=
= + + +
6DN − (6.46)
donde AelecE y *D
elecE son las energías electrónicas de ambas moléculas para la geometría
de equilibrio del complejo [*D···A], ( )1 2 3 6, ,...A
A A A Av NQ q q q −= y ( )1 2 3 6, ,...
D
D D D Dv NQ q q q −=

272 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
son las 3ND,A-6 coordenadas internas de cada molécula, y ( )A Av vE Q y ( )*D D
v vE Q la
energía potencial de la molécula aceptora y dadora en el estado fundamental y excitado
respectivamente.
Por otro lado, de la misma manera la superficie de energía potencial adiabática del
complejo después de la transferencia de energía [D···A*] se puede expresar como
función únicamente de las coordenadas internas ( ) de ambas moléculas
:
vQ
( ) [ ] ( )1 ··· * vD AE Q E Q≡
( ) ( )
( ) ( )1 1 1 2 3 6 1 2 3 6
* *
, ,... , , ,...A
A A A D D DN
A D A A D Delec elec v v v v
E Q E q q q q q q
E E E Q E Q
−=
= + + +
DN −
)
(6.47)
donde el significado de los términos es análogo a la ecuación (6.46).
Nos interesa conocer la dependencia de la función diferencia de energía entre los dos
estados electrónicos respecto de las coordenadas internas de ambas moléculas, es
decir, nos interesa conocer la función:
( E∆
( ) ( )
( ) ( ) ( ) (1 0
*A D A A A A D D D DT T v v v v v v v v
E E Q E Q
E E E Q E Q E Q E Q
∆ ≡ −
= − + − + − )* (6.48)
donde y ( )*A A AT elec elecE E E≡ − ( )*D D D
T elec elecE E E≡ − .
De ahora en adelante, consideraremos que la diferencia: ( ) (* )D D D Dv v v vE Q E Q− se puede
tomar aproximadamente igual a cero, o análogamente que se cumple:
( ) ( ) ( ) ( )* *A A A A D D D Dv v v v v v v vE Q E Q E Q E Q− − , es decir, la molécula dadora solamente es
capaz de ceder la energía DTE . Esta aproximación simplifica notablemente el problema,
y como ya se indicó en la sección 6.3, implica que la contribución del dador al descenso
de energía del estado final del complejo respecto del inicial es despreciable frente a la
del aceptor. De esta manera, si realizamos una serie de experimentos en los que se mide
la constante de velocidad de transferencia de energía de distintos dadores a un mismo
aceptor, en principio es posible encontrar moléculas dadoras con un esqueleto rígido, de
tal manera que la presente aproximación sea correcta. En cualquier caso, la validez de
esta aproximación se discutirá más adelante (ver punto 6.4.4)
Como ya se ha indicado anteriormente, para explicar la dependencia del factor
exponencial de la constante de velocidad con la energía triplete del dador ( DTE ), se debe

6.4 TET con Superficies de Energía Potencial Completas 273
conocer la ecuación (6.48), ya que una vez conocida la dependencia de E∆ con las
coordenadas internas de la molécula A, y conocida también la dependencia de la energía
potencial de la molécula en el estado S0 con las coordenadas, es decir, conocida la
función ( )A Av vE Q , se puede obtener el cruce de mínima energía entre las superficies
adiabáticas, y una vez realizada la corrección debida al acoplamiento electrónico,
obtener la energía de activación del proceso, como ya se indicó en la sección 6.3.
La expansión de la ecuación (6.48) hasta primer orden en coordenadas internas ha sido
tratada en la sección 6.3, se ha visto cómo describe correctamente el proceso de TET en
el COT, y se ha concluido que dicha teoría debe ser capaz de explicar también el
comportamiento de muchos quenchers flexibles. Sin embargo, en el presente estudio se
desea hacer uso de la SEP completas, sin restricción alguna, y de esta manera poder
estudiar todos los posibles comportamientos de los quenchers en la TET. La pregunta a
responder por lo tanto es la siguiente: Para un dador de energía DTE conocida, ¿Cuál es
la geometría del complejo activado?, es decir, ¿Cuál es la distorsión geométrica de
mínima energía en el estado fundamental que debe adoptar la molécula aceptora para
ser capaz de recibir dicha energía? Es más, podemos preguntarnos si es posible construir
una curva continua en el espacio de configuraciones nucleares de tal manera que cada
uno de los puntos de la misma corresponda a una geometría del complejo activado para
una determinada energía triplete del dador. La teoría desarrollada anteriormente en la
sección 6.3 nos ofrece un importante parámetro que mide el grado de no-verticalidad en
la TET, el parámetro ( 1/ 2212gγ κ= ) donde g es la norma del gradiente en el estado T1
calculada para la geometría de equilibrio de la molécula aceptora en S0, y es una
combinación de constantes de fuerza en la dirección del gradiente, también para la
misma geometría en S
1κ
0. Sin embargo, esta aproximación de primer orden en la
diferencia de energías entre las SEPs de los dos estados, es tanto más válida cuanto
mayor sea la norma del gradiente g, y por lo tanto más acusado el comportamiento no-
vertical del proceso. Es decir, cuanto mayor sea el término lineal en la expansión de la
función diferencia de energía potencial entre los dos estados electrónicos implicados
[*D···A] y [D···A*], esto es:
( ) ( ) t t1
2A D
T TE Q E E∆ = − + + +x g x Hx … (6.49)

274 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
Donde x es el vector posición del complejo que contiene las coordenadas internas del
mismo, g es el vector gradiente de la energía potencial y H es la matriz hessiana de la
energía. Sin embargo, puede ocurrir que el término de segundo orden ( )t12 x Hx o
términos de orden superior, tengan una contribución no despreciable frente al término
lineal en (6.49). En este caso, es posible desarrollar nuevas aproximaciones que
incluyan distintos términos de orden superior. Sin embargo, es más conveniente usar la
superficie de energía potencial completa y no las propiedades locales de la misma en un
pequeño entorno cuando se desean introducir correcciones de segundo orden o
superiores, debido no sólo a la mayor simplicidad de los cálculos, sino también a la
mayor precisión de los resultados obtenidos.
Se ha desarrollado un algoritmo (basado en el cálculo de gradientes) para la
determinación de la energía de activación de un proceso de transferencia de energía
triplete, que introduce restricciones con el método de los multiplicadores de Lagrange,
usando las SEP completas de los estados electrónicos involucrados en el proceso.
Este algoritmo se ha denominado “Coordenada del Complejo Activado de Transferencia
de Energía Triplete” (TET-ACC), ya que nos ofrece las coordenadas internas del
complejo activado para la transferencia de energía triplete en función de la fuerza
dirigente: , suple una de las carencias de la aproximación de primer orden para el
cálculo de la dependencia de la energía de activación del proceso TET con la energía
triplete de la molécula dadora (Sección 6.3). Esto es debido a que se hace uso de toda la
SEP para uno y otro estado electrónico del complejo, y no únicamente de las
propiedades de las mismas en un entorno pequeño. Por lo tanto el algoritmo permite
explicar casos de TET en los que sólo la aproximación de primer orden no sea
suficiente, lo cual ocurre en el caso de SEP complejas.
TE∆
De manera sumaria, se puede decir que el algoritmo está conformado por una serie de
pasos de tal forma que se van calculando diversos puntos de una curva en el espacio de
configuraciones nucleares para la molécula aceptora, siendo cada punto de la curva (en
este espacio), un punto de mínima energía en S0 sujeto a la condición de que la
diferencia de energía entre S0 y T1 es una constante. Dicho de otro modo, la geometría
molecular hallada cumple que es de mínima energía (mínimo relativo) en S0 para un
salto energético S0 T1 fijado. Para distintas diferencias de energía (S0/T1), se van

6.4 TET con Superficies de Energía Potencial Completas 275
obteniendo diversos puntos, los cuales conforman una curva en el espacio de
configuraciones nucleares de la molécula aceptora.
A la curva que comienza en la configuración de mínima energía en S0 para la molécula
aceptora y cumple los requisitos descritos anteriormente, la denominamos curva TET-
ACC.
En el punto de cruce entre las dos superficies se debe cumplir:
( ) (*0 )A A AT v v v v
AE E E Q E Q∆ = = ∆ + − (6.50)
donde . A DT TE E E∆ = − T
Por lo tanto, la curva que queremos construir debe cumplir para todo punto de la misma
que la función lagrangiana:
( ) ( ) ( ) ( )( *0
A A A A A Av v v v v vL Q E Q E Q E Q Eλ= + − −∆ )T (6.51)
sea un extremo relativo, esto es, se deben satisfacer las siguientes igualdades:
0 1,2,...,3 6Ai
L i Nq∂
= = −∂
(6.52)
(6.53) ( ) ( )*A Av vE Q E Q E− = T∆
donde se ha usado por simplicidad la notación: AvQ Q≡ , ya que las únicas coordenadas
importantes son las del aceptor.
Existen varias posibilidades para construir una curva que cumpla estas características,
(Fig. 6.5) por ejemplo, se puede ir variando TE∆ paulatinamente, y calcular las 3NA-6
igualdades de la primera de las condiciones de la optimización (ecuación 6.52). Sin
embargo, esta posibilidad presenta algunos problemas computacionales, el principal es
que existen dos condiciones independientes: la primera es TE cte∆ = , y la segunda, que
los vectores gradiente de ambas SEP (E0 y E1) sean paralelos, esto es:
(6.54) 0 1E E∇ ×∇ = 0
y ambas condiciones deben converger (dentro de los criterios de convergencia)
simultáneamente al ir variando la diferencia de energías triplete ( )TE∆ . Una posible
solución a este problema consiste en dejar la variable TE∆ sin fijar, y únicamente usar
la condición dada por la ecuación (6.52), de tal manera que se construya la curva
variando las coordenadas del sistema sometidas a una cierta restricción, que como se
pasará a explicar con detalle más adelante, consiste en fijar la norma del desplazamiento
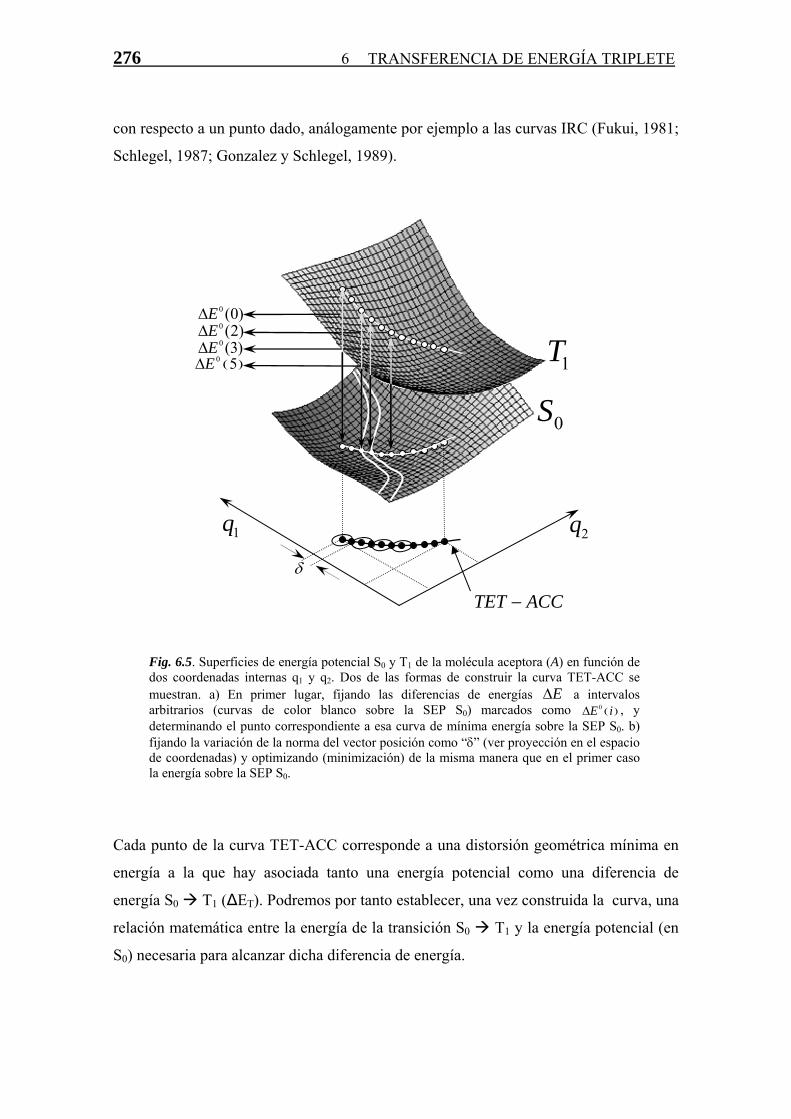
276 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
con respecto a un punto dado, análogamente por ejemplo a las curvas IRC (Fukui, 1981;
Schlegel, 1987; Gonzalez y Schlegel, 1989).
1q 2q
δ
0S1T
TET RC−TET ACC−
0 (0)E∆0 (2)E∆0 (3)E∆( )0 5E∆
Fig. 6.5. Superficies de energía potencial S0 y T1 de la molécula aceptora (A) en función de dos coordenadas internas q1 y q2. Dos de las formas de construir la curva TET-ACC se muestran. a) En primer lugar, fijando las diferencias de energías E∆ a intervalos arbitrarios (curvas de color blanco sobre la SEP S0) marcados como ( )0E i∆ , y determinando el punto correspondiente a esa curva de mínima energía sobre la SEP S0. b) fijando la variación de la norma del vector posición como “δ” (ver proyección en el espacio de coordenadas) y optimizando (minimización) de la misma manera que en el primer caso la energía sobre la SEP S0.
Cada punto de la curva TET-ACC corresponde a una distorsión geométrica mínima en
energía a la que hay asociada tanto una energía potencial como una diferencia de
energía S0 T1 (∆ET). Podremos por tanto establecer, una vez construida la curva, una
relación matemática entre la energía de la transición S0 T1 y la energía potencial (en
S0) necesaria para alcanzar dicha diferencia de energía.

6.4 TET con Superficies de Energía Potencial Completas 277
6.4.1.2 Descripción del algoritmo§
Inicialmente se parte de la geometría de equilibrio de la molécula aceptora, que en el
espacio de configuraciones denotaremos como ( )00R , donde el superíndice (0) indica el
número de paso en el cálculo de la curva (Fig. 6.6).
A este vector ( )00R le sumamos el vector pivotante: , de norma dada (la cual se
mantendrá constante durante toda la primera etapa), que en este caso es un vector con la
dirección y sentido inicial del vector diferencia de fuerzas entre T
1R
1 ( )1TF y S0 . De
esta manera, el vector corresponde a la geometría molecular inicial,
desplazada respecto de la posición de equilibrio en S
( 0SF )
1
)
( )00 +R R
0 en la dirección de máximo
decrecimiento de energía , entre dichos estados. (∆F
(0)0R
(1)0R
( 1)0R −
1R
1q
2q
3q
1 2
1−
2−
Curva TET-ACC en laregión exotérmica
Curva TET-ACC en laregión endotérmica
Puntos optimizadosde la curva TET-ACCCentro de la Hiperesfera
Fig. 6.6. Representación esquemática de una curva TET-ACC en la región “endotérmica” (curva en negro) así como la región “exotérmica” (curva gris). Se muestra el origen de la primera de las hyperesferas ( )0
0R correspondiente a la geometría de equilibrio de la molécula aceptora en S0. El origen de las dos hyperesferas correspondientes al primer paso en la dirección “endotérmica” y “exotérmica” corresponden a y respectivamente. Los puntos optimizados de la curva se representan por los números 2,1,-1 y -2, los positivos pertenecen a la región “endotérmica” y los negativos a la “exotérmica”.
(1)0R (-1)
0R
§ Ver Apéndice G con el código Fortran de la programación del algoritmo.

278 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
Este vector inicial es el mejor que podemos elegir, sin embargo la nueva geometría no
tiene por qué cumplir la condición que estamos buscando, esto es, la ecuación (6.54).
Para que se cumpla esta condición, el vector al que se le ha eliminado la proyección
sobre , y que denominaremos
0SF
∆F0S⊥F , debe ser nulo, ya que de otra manera los vectores
y no serían paralelos. Éste es el paso fundamental del proceso de optimización
del algoritmo TET-ACC. Puesto que
0SF1TF
0S⊥F puede no ser nulo, para que lo sea se debe
hacer un desplazamiento de coordenadas internas de la molécula mediante la rotación
del vector dentro de la hiperesfera de centro 1R ( )00R y radio 1R en la dirección del
vector hasta que dicha proyección se anule, con lo que se satisface la condición
buscada. Sin embargo, es inmediato demostrar que si es paralelo a , también
debe serlo , por lo que igualmente podríamos tomar como vector desplazamiento el
vector eliminando la proyección sobre
0S⊥F
0SF ∆F
1TF
1TF ∆F . Por lo tanto la suma de ambos vectores
también se puede tomar como vector de desplazamiento (una vez eliminada la
correspondiente proyección). Así pues, se toma el vector 1T S0+F F , y se elimina la
proyección sobre (vector en la Figura 6.7) , realizando un desplazamiento
en esa dirección.
∆F1T⊥ +F F
0S⊥
Este desplazamiento, lógicamente, se escala para conseguir que éste sea coherente con
la aproximación de primer orden de la energía potencial, es decir, para conseguir que el
desplazamiento no sea mayor de lo que la expansión de primer orden de la SEP predice
dentro de un error razonable. Además si el desplazamiento supera un máximo
(generalmente se ha usado la mitad del radio de la “hiperesfera”) se escala para alcanzar
dicho valor (por simplicidad en la Figura 6.7, se ha utilizado como factor de
escalamiento del vector, la unidad).
Por otro lado para que la nueva geometría posea la distancia deseada al origen (que
recordemos, viene dada por la norma de ) tenemos que escalar nuevamente el vector,
proyectándolo sobre la “hiperesfera” de radio
1R
1R , con lo que obtenemos una nueva
geometría (denotada como “2” en la Figura 6.7).
Este proceso se repite iterativamente hasta que se alcanza un punto que cumple la
condición dada por la ecuación (6.54). En este caso los vectores y son paralelos 1TF
0SF

6.4 TET con Superficies de Energía Potencial Completas 279
entre sí, y por lo tanto, la componente de los mismos fuera del vector ∆ ( y F1T⊥F
0S⊥F
respectivamente) es nula, por lo que el desplazamiento también lo es (Fig. 6.8).
1q
2q
3q
0SF1TF
∆F
12
0 1S T⊥ ⊥+F F
0R
1RCurva TET-ACC en laregión exotérmica
Curva TET-ACC en laregión endotérmica
Centro de la hiperesfera
Puntos del procesode optimización
Fig. 6.7. Representación esquemática del primer paso del algoritmo TET-ACC, donde se observa el desplazamiento de la geometría desde el punto “1” al punto “2” mediante la proyección del vector sobre la “hiperesfera” de centro y de radio
0S⊥ +F F
1T⊥
0R 1R . Asimismo, se puede observar cómo este último vector se obtiene como suma de las proyecciones sobre el plano perpendicular al vector ∆F de los vectores fuerza en S0 y T1:
y . 0SF
1TF
El siguiente paso es análogo al que se realizó para el primer punto, de tal manera que se
desplazan las coordenadas internas en la dirección del vector (Fig. 6.8) con una
norma de desplazamiento igual a
1R
1R . Esta nueva geometría pasa a ser el vector ( )10R ,
donde recordemos que el superíndice (1) indica que es el paso de la curva TET-ACC en
el que estamos. Este vector ( )10R , a su vez es el centro de la nueva hiperesfera de radio
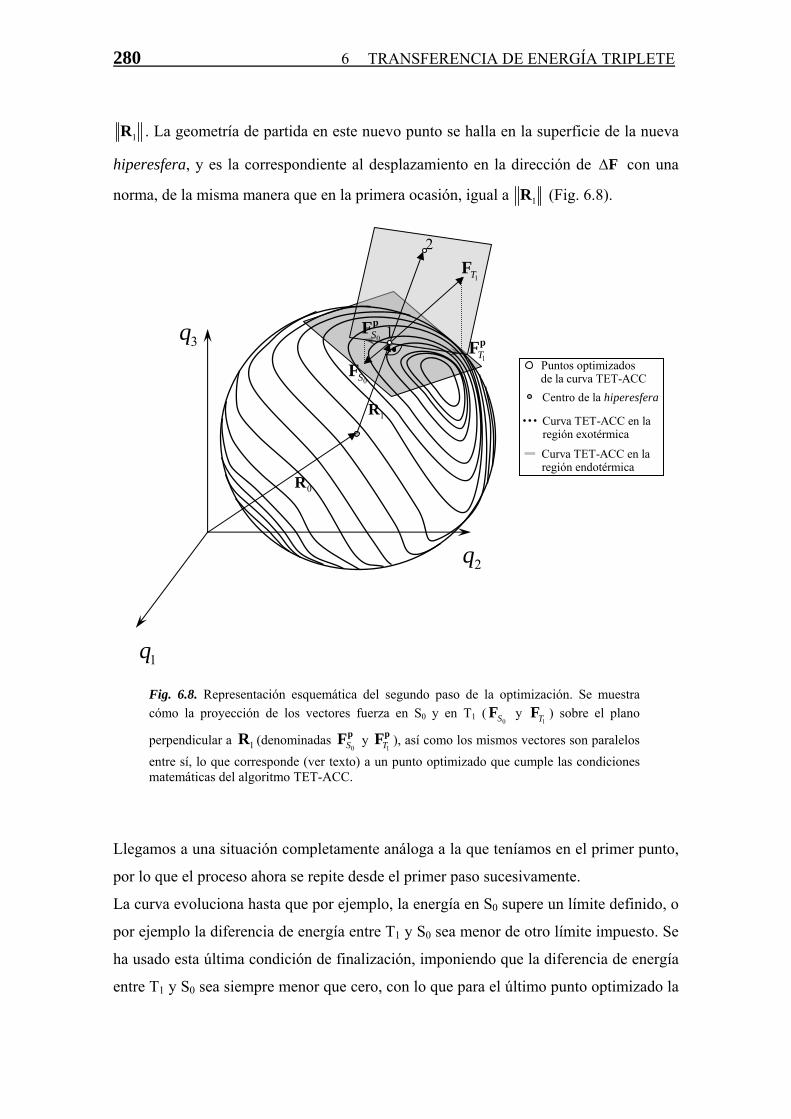
280 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
1R . La geometría de partida en este nuevo punto se halla en la superficie de la nueva
hiperesfera, y es la correspondiente al desplazamiento en la dirección de con una
norma, de la misma manera que en la primera ocasión, igual a
∆F
1R (Fig. 6.8).
1TpF
1q
2q
3q
1R
0R
0SpF
0SF
1TF2
1
Curva TET-ACC en laregión exotérmica
Curva TET-ACC en laregión endotérmica
Puntos optimizadosde la curva TET-ACCCentro de la hiperesfera
Fig. 6.8. Representación esquemática del segundo paso de la optimización. Se muestra cómo la proyección de los vectores fuerza en S0 y en T1 ( y ) sobre el plano
perpendicular a (denominadas y ), así como los mismos vectores son paralelos entre sí, lo que corresponde (ver texto) a un punto optimizado que cumple las condiciones matemáticas del algoritmo TET-ACC.
0SF1TF
1R0S
pF1TpF
Llegamos a una situación completamente análoga a la que teníamos en el primer punto,
por lo que el proceso ahora se repite desde el primer paso sucesivamente.
La curva evoluciona hasta que por ejemplo, la energía en S0 supere un límite definido, o
por ejemplo la diferencia de energía entre T1 y S0 sea menor de otro límite impuesto. Se
ha usado esta última condición de finalización, imponiendo que la diferencia de energía
entre T1 y S0 sea siempre menor que cero, con lo que para el último punto optimizado la

6.4 TET con Superficies de Energía Potencial Completas 281
diferencia de energía es cercana a cero y nos encontramos por lo tanto en un punto de
cruce de sistemas (ISC) mínimo en energía.
6.4.1.3 Análisis y Topología de las curvas TET-ACC
En este punto discutiremos la topología de las curvas TET-ACC así como el análisis de
las mismas con el fin de clasificar los resultados obtenidos. En una primera parte se
analiza la topología de la curva, su comienzo y finalización, mientras que después se
estudia la información que se puede obtener del análisis de estas curvas respecto de la
estructura y energía de los complejos activados en el proceso de TET.
1. Topología
Toda curva TET-ACC comienza por definición en la geometría de equilibrio para la
molécula aceptora en su estado fundamental (S0). Para esta geometría de equilibrio, la
energía de transición corresponde exactamente a la transición espectroscópica FC
S0→T1. En este punto inicial, la curva bifurca, ya que existe una misma dirección con
dos sentidos opuestos en los cuales se cumplen las condiciones impuestas por el
algoritmo. Por lo tanto el recorrer la curva en un sentido u otro depende únicamente del
sentido inicial que tome el vector (véase arriba). De este modo, si dicho vector lleva
la dirección del vector fuerza en T
1R
1, la energía en T1 disminuirá y con ella la diferencia
de energía entre el estado S0 y el T1, esto es, disminuirá la energía necesaria ( )DTE que
la molécula dadora (D) debe ceder a la aceptora (A) para que se de dicha transición,
respecto de la energía de la molécula aceptora correspondiente a la transición S0 → T1
( )ATE . Por lo tanto la curva en esta dirección explora la región “endotérmica” o normal
de transferencia de energía, para la que se cumple ( ) 0D AT TE E− ≤ . Sin embargo, si se
toma como vector inicial ( )1R el gradiente en T1 (es decir, el opuesto al vector fuerza
en T1), el gap energético entre los estados S0 y T1 del aceptor aumenta, de tal manera
que la energía necesaria de una molécula dadora ( )DTE para ceder energía a la aceptora

282 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
aumenta, lo que conlleva una situación opuesta, esto es: ( ) 0D AT TE E− ≥ , y que
corresponde a la región “exotérmica”.
Las curvas TET-ACC finalizan cuando el último de los pasos del algoritmo (ver punto
6.4.1.2) no se puede realizar, lo cual ocurre cuando el vector diferencia de gradientes es
igual a cero. Esta circunstancia se puede dar principalmente en dos casos. El primero de
ellos, cuando ambos vectores gradiente de la energía son nulos (y por lo tanto su
diferencia también). Esta posibilidad debe ser excepcional, ya que no es común que la
geometría de equilibrio de la molécula en el estado fundamental (S0) sea exactamente la
misma que la de equilibrio en T1. En cualquier caso podría presentarse
excepcionalmente en moléculas aceptoras para las que existan varias conformaciones
accesible en S0, y una misma curva TET-ACC pase por varias de estas conformaciones.
La otra de las posibilidades para que la curva finalice, es que ambos vectores sean
iguales, es decir, que se cumpla: 0E 1E∇ =∇ , con lo que la diferencia es nula. Esta
circunstancia se presenta con bastante frecuencia en zonas de degeneración energética
entre los estados S0 y T1 (cruce de sistemas), por lo que es normal que una curva finalice
en este tipo de puntos, y es de esperar que sea la situación más frecuente. De hecho,
aunque los gradientes no sean iguales, la curva debe terminar en el punto de cruce de
energías S0/T1, ya que la energía de la transición S0 → T1 en este punto es nula y por lo
tanto la energía triplete del dador presenta un mínimo: 0DTE = . Esta circunstancia se
eligió como criterio de finalización de la curva TET-ACC (ver punto 6.4.1.2). Por lo
tanto, la mayoría de las curvas TET-ACC terminan en un punto de cruce de sistemas, en
el que la diferencia de energías entre un estado y otro se hace nula. Es interesante hacer
notar que este punto de cruce de sistemas, debido a las condiciones impuestas en el
algoritmo, es mínimo en energía en la región de cruce de ambos estados, por lo que
eventualmente puede suponer a la vez un canal de desactivación energética no-radiante
de la molécula aceptora para regresar al estado fundamental y recuperar así su
capacidad aceptora inicial.
2. Análisis
La principal información que se puede extraer de la curva TET-ACC viene referida a
dos aspectos, el energético y el estructural.

6.4 TET con Superficies de Energía Potencial Completas 283
Respecto del primero, se puede obtener la energía de activación de la molécula aceptora
necesaria para poder tomar energía de una molécula dadora. Es decir, podemos
establecer un relación entre la energía del dador ( )DTE y la mínima energía potencial del
aceptor ( )E≠ , necesaria para que pueda tomar esa energía electrónica, que se
corresponde con la mínima energía de cruce entre las PES, la cual, una vez aplicada la
corrección del acoplamiento electrónico y la de la energía vibracional residual, se
corresponde con la energía de activación. Conociendo pues esta relación entre energía
de activación ( )E≠ y energía del dador ( )DTE , es fácil obtener una dependencia
matemática entre ambas magnitudes a partir de los resultados obtenidos mediante el
cálculo ab-initio de esta curva.
Para la aproximación lineal o de primer orden, la energía de activación , presenta
una dependencia con
E≠
DTE de la siguiente manera (ver sección 6.3):
( )2
2
A DT TE E
Eγ
≠−
= , donde 2
2
1
2gγκ
= . Si dicha aproximación es correcta para un rango
grande de diferencia de energías triplete: ( )A DT TE E− , de la representación de versus
obtendríamos una recta de pendiente
E≠
( 2A DT TE E− ) 2γ − (Fig. 6.9).
Usando el algoritmo TET-ACC, la energía de activación puede dejar de ser una función
lineal del cuadrado de la diferencia de energías triplete: ( )2A DT TE E− , para ser en general
una función más compleja de dicha magnitud. Por ejemplo, a partir de la representación
de vs. ( procedente de los datos obtenidos en la curva TET-ACC,
podemos ajustar los datos a polinomios en la variable
E≠ )2A DT TE E−
( )2A DT TE E− de distinto grado.
Según los casos que se han estudiado (ver más adelante a lo largo de este capítulo)
generalmente una expansión hasta segundo orden de la energía de activación en función
de ( suele ser suficiente, por lo tanto, tenemos: )2A DT TE E−
( ) ( )
2221
2 2T
T
E CE Eγ
≠ ∆ ⎡ ⎤= + ∆⎣ ⎦ (6.55)
donde γ el es parámetro de distorsión geométrica, y es una constante procedente del
ajuste.
1C
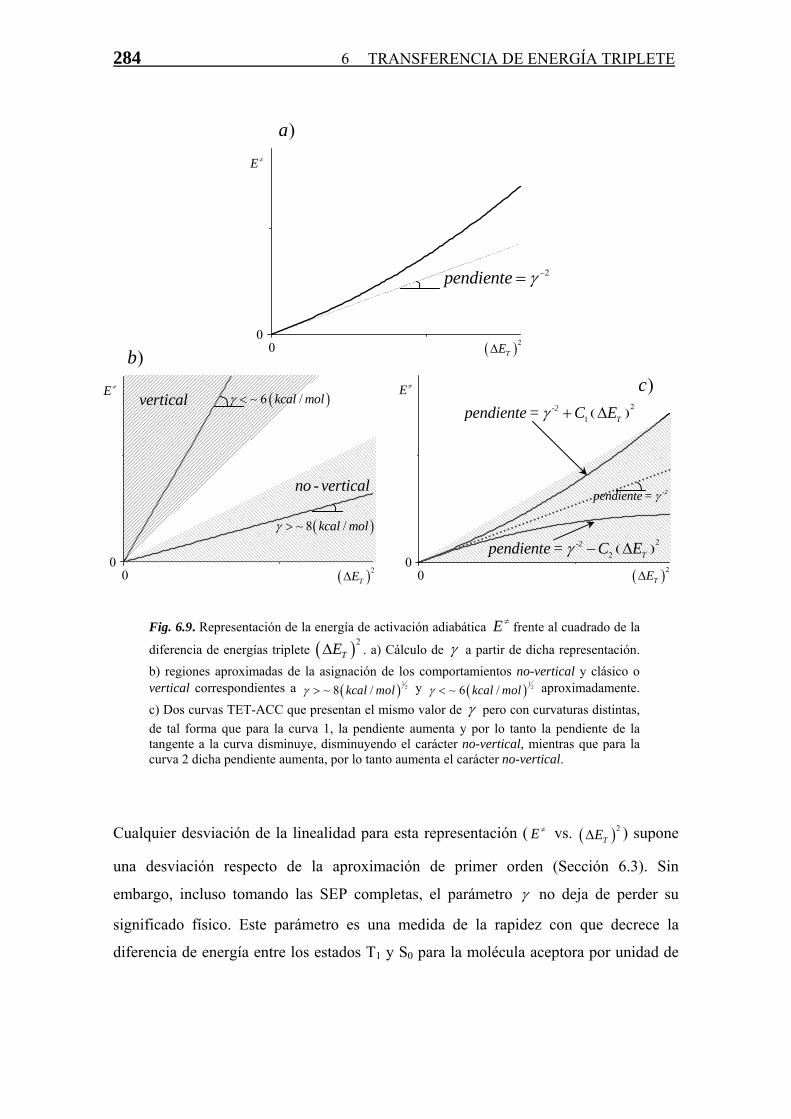
284 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
)c
E≠
00 ( )2
TE∆
)a
2pendiente γ −=
E≠
00
( )6 /kcal molγ < ∼
( )8 /kcal molγ > ∼
( )2TE∆
)b
vertical
no - vertical
E≠
00 ( )2
TE∆
( )2
1-2
Tpendiente = C Eγ + ∆
( )2
2-2
Tpendiente= C Eγ − ∆
-2pendiente = γ
Fig. 6.9. Representación de la energía de activación adiabática E≠ frente al cuadrado de la
diferencia de energías triplete ( )2TE∆ . a) Cálculo de γ a partir de dicha representación.
b) regiones aproximadas de la asignación de los comportamientos no-vertical y clásico o vertical correspondientes a ( )
128 /kcal molγ > ∼ y ( )
126 /kcal molγ < ∼ aproximadamente.
c) Dos curvas TET-ACC que presentan el mismo valor de γ pero con curvaturas distintas, de tal forma que para la curva 1, la pendiente aumenta y por lo tanto la pendiente de la tangente a la curva disminuye, disminuyendo el carácter no-vertical, mientras que para la curva 2 dicha pendiente aumenta, por lo tanto aumenta el carácter no-vertical.
Cualquier desviación de la linealidad para esta representación ( E ≠ vs. ( ) supone
una desviación respecto de la aproximación de primer orden (Sección 6.3). Sin
embargo, incluso tomando las SEP completas, el parámetro
)2TE∆
γ no deja de perder su
significado físico. Este parámetro es una medida de la rapidez con que decrece la
diferencia de energía entre los estados T1 y S0 para la molécula aceptora por unidad de

6.4 TET con Superficies de Energía Potencial Completas 285
energía de activación, se puede calcular através de la pendiente a la curva vs.
en un punto cualquiera. (Fig. 6.9).
E≠
( 2A DT TE E− )
Distintos comportamientos se pueden observar en función de los datos que se obtienen
de la representación. En primer lugar, de la pendiente a la curva en ( )
podemos obtener el parámetro
20A D
T TE E− =
0γ (de ahora en adelante usaremos la nomenclatura TEγ ∆
donde el subíndice TE∆ indica el valor de ( )A DT T TE E E∆ = − para el cual se calcula γ ,
de esta forma, para la aproximación de primer orden, se tiene que 0γ γ= ) , y por lo
tanto podemos medir el grado de no-verticalidad que va a presentar la molécula
aceptora (Fig. 6.9b). En los casos de SEP complejas, es posible que incluso para
diferencias de energía triplete bajas, se obtenga un comportamiento que
rápidamente se aleja de la linealidad, en tal caso la aproximación lineal deja de ser
válida y el uso de las SEP completas se hace imprescindible. Por otro lado, las curvas
que presentan un comportamiento lineal pueden curvarse al aumentar la diferencia de
energías triplete. En tal caso, si la curvatura ocurre hacia arriba, indica que el
comportamiento no-vertical disminuye a medida que la diferencia de energías triplete
aumenta (Fig. 6.9c ). Si por el contrario, se curva hacia abajo, la pendiente va
disminuyendo, aumentando por lo tanto
( 2A DT TE E− )
TEγ ∆ y concurrentemente el comportamiento
no-vertical (Fig. 6.9c ) con lo que el carácter de no-verticalidad aumenta a medida que
aumenta. ( 2A DT TE E− )
En conclusión, el comportamiento no-vertical se puede evaluar a veces recurriendo
únicamente al parámetro 0γ (γ calculado en ( )20A D
T TE E− = ). Sin embargo una
descripción detallada de dicho comportamiento requiere un análisis de toda la curva,
esto es, tanto la pendiente inicial (aproximación de primer orden) como la curvatura a lo
largo de la curva E ≠ vs. (aproximaciones de orden superior) (Fig. 6.9). ( 2TE∆ )
)
Por último, en lo que respecta a la información sobre la energía, es importante destacar
que el algoritmo introduce una ventaja muy importante con respecto a la aproximación
de primer orden, ya que al tener ahora una curva paramétricamente dependiente de
( 2TE∆ que nos da la estructura molecular óptima para que ocurra la transferencia de

286 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
energía, podemos calcular en todos los puntos de la misma las funciones de partición de
la molécula aceptora, y de esta manera obtener la contribución de esta molécula a las
funciones termodinámicas de activación. Debido a la especial importancia de este punto
se discutirá más adelante (punto 6.4.2) desde una perspectiva teórica, así como en la
aplicación al estudio de la TET en los estilbenos (punto 6.4.3).
Hemos visto qué información se puede extraer acerca de la energética del proceso a
través de los resultados obtenidos de las curvas TET-ACC. Ahora abordaremos la
información estructural.
Las curvas TET-ACC son curvas en el espacio de configuraciones nucleares calculadas
computacionalmente con un determinado método ab initio. Por lo tanto es importante
remarcar que cada punto de dicha curva se corresponde con una estructura de la
molécula aceptora que cumple la condición de que la energía en el estado fundamental
(respecto de la de equilibrio) es la menor de todas las que poseen una transición vertical
S0→T1 igual a la que tiene dicha geometría. De esta manera, esta geometría molecular
es la óptima (la de menor energía en S0) que puede dar lugar a la transición y por lo
tanto al proceso de TET con un dador que posea una energía triplete igual a aquella.
Cada geometría o configuración nuclear dada por cada punto optimizado de la curva
TET-ACC corresponde por lo tanto a un estado de transición para el proceso de TET. Se
puede entonces, estudiando las distorsiones a lo largo de la curva TET-ACC, obtener las
deformaciones óptimas que la molécula aceptora debe sufrir para verificar el proceso de
TET en función de la energía del dador ( DTE ).
6.4.1.4 Transferencia de Energía Triplete Exotérmica. La Región
Invertida
Cuando la diferencia D AT TE E− es mayor que cero, la energía del dador disponible para
ceder del dador ( DTE ) es mayor que la energía triplete del aceptor ( )A
TE , por lo tanto se
habla de un proceso “exotérmico” aunque se debe hacer notar que el término no tiene el
significado correcto, ya que la diferencia de energías triplete ( )D AT TE E− no se
corresponde en general con la energía libre del proceso.

6.4 TET con Superficies de Energía Potencial Completas 287
Como ya se apuntó antes, las curvas TET-ACC toman dos sentidos opuestos desde el
mínimo de energía en S0. Por un lado la curva crece en la dirección en que
, lo que corresponde a la región “endotérmica” y por otro lado en la
dirección en que , que corresponde a la “exotérmica” (Fig. 6.10a). Para esta
última situación la curva sigue la dirección del gradiente, es decir, de crecimiento de la
energía en T
0D AT TE E− <
0D AT TE E− >
1. Esta situación generalmente corresponde a un aumento progresivo del
gradiente, ya que la curvatura de la SEP en la coordenada de transferencia de energía
suele ser positiva§, por lo que el valor de TEγ ∆ en general aumenta, y la no-verticalidad
en la región “exotérmica” aumenta también. Esto conlleva que el decrecimiento de la
constante de velocidad de transferencia de energía en la región “exotérmica” sea más
lento que en la región normal (Fig. 6.10b). Por el contrario, la aproximación lineal
predice un decrecimiento igual tanto para una región como para la otra (Fig. 6.10a). De
manera general, se predice una región invertida análoga a la predicha para transferencia
electrónica en la región “exotérmica”. Se han encontrado sistemas en los que esta región
invertida se observa en procesos de transferencia de energía triplete (Sigman y Closs,
1991; Parola et al.,1996; Serpa et al., 2003). Sin embargo, en mucho otros casos no se
llega a observar, siendo la constante de velocidad de TET igual a la de difusión. Esto es
debido principalmente a tres circunstancias, la primera de ellas es la que se acaba de
apuntar, esto es, el decrecimiento de la constante de velocidad es en general más lento
en la región invertida. Sólo algunos casos, en los que un aumento de la energía en T1
lleva consigo una disminución del gradiente pueden favorecer el que sea observable
experimentalmente la región invertida. Por otro lado, la presencia de estados excitados
triplete de mayor energía que T1, así como de otras vías de desactivación pueden servir
como estados finales de la molécula aceptora en el proceso (Formosinho et al., 1998;
Bodesheim et al., 1994) (Fig. 6.10c).
Además, debido a que la anchura de potencial en la región invertida es mucho menor
que en la región normal, el efecto túnel va a ser muy importante en el aumento de la
eficiencia de la transferencia en esta región, como ya se demostró para los procesos de
transferencia electrónica (Ulstrup y Jortner, 1975).
§ La curvatura de la PES en T1 tras la transición FC suele ser positiva si la geometría de equilibrio en T1 no está muy
desplazada respecto de la S0, sin embargo en algunos casos de moléculas aceptoras con alto grado de no-verticalidad es posible encontrar curvaturas negativas, por lo que esta premisa dejaría de ser válida.
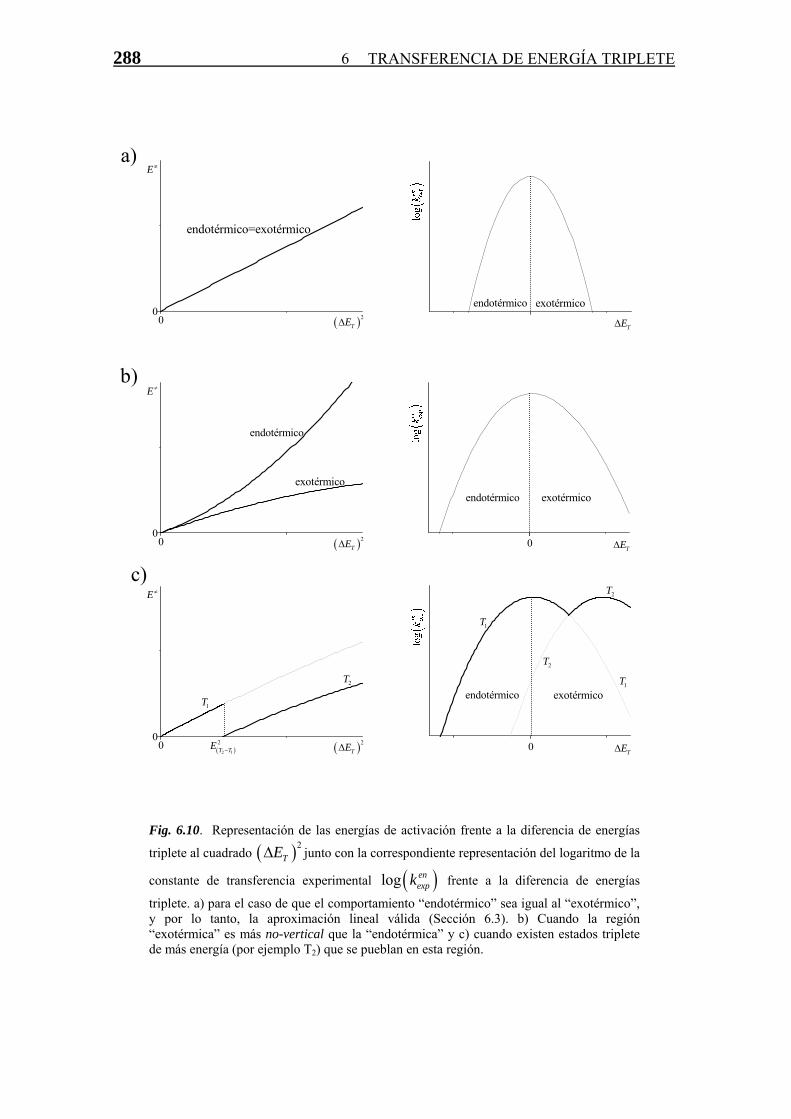
288 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
Fig. 6.10. Representación de las energías de activación frente a la diferencia de energías
triplete al cuadrado ( 2T )E∆ junto con la correspondiente representación del logaritmo de la
constante de transferencia experimental ( )log enexpk frente a la diferencia de energías
triplete. a) para el caso de que el comportamiento “endotérmico” sea igual al “exotérmico”, y por lo tanto, la aproximación lineal válida (Sección 6.3). b) Cuando la región “exotérmica” es más no-vertical que la “endotérmica” y c) cuando existen estados triplete de más energía (por ejemplo T2) que se pueblan en esta región.
E≠
( )2TE∆0
0 8.0
8.2
8.4
8.6
8.8
9.0
9.2
9.4
9.6
9.8
10.0
TE∆
exotérmico
endotérmico=exotérmico
endotérmico
a)
E≠
( )2TE∆0
0 8.0
8.2
8.4
8.6
8.8
9.0
9.2
9.4
9.6
9.8
10.0
TE∆0
exotérmico
endotérmico
exotérmicoendotérmico
b)
E≠
( )2TE∆
1T
2T
( )2 1
2T TE −0
0 8.0
8.2
8.4
8.6
8.8
9.0
9.2
9.4
9.6
9.8
10.0
TE∆0
1T
1T2T
2T
exotérmicoendotérmico
c)

6.4 TET con Superficies de Energía Potencial Completas 289
Por lo tanto, en las moléculas que presentan una gran diferencia de energía entre los
estados triplete, especialmente entre T1 y T2, puede ser observable la región invertida.
Estas dos circunstancias junto con el efecto túnel hacen que en general sea complicado
observar experimentalmente la región invertida, sin embargo si se dan las tres, esto es:
que el gradiente en T1 disminuya al aumentar la energía en S0 en la región “exotérmica”,
que la diferencia energética entre T2 y T1 sea grande, y que el efecto túnel no sea
decisivo (quizá esta última circunstancia sea la más inusual) debe ser posible observar
dicha región invertida en la zona “exotérmica”.
6.4.2 Funciones Termodinámicas de Activación a partir de las curvas
TET-ACC calculadas
Como se ha visto a lo largo de esta sección 6.4, el algoritmo TET-ACC permite la
determinación de una curva en el espacio de coordenadas internas de la molécula que
nos ofrece una dependencia funcional entre la energía de activación (la componente
electrónica de la misma, es decir, la calculada en base a las SEP) y la diferencia para
dicha geometría entre las energías de los estados T1 y S0. Cada punto de esta curva así
obtenida representa un estado de transición para una determinada transferencia de
energía, o dicho de otra manera, cada geometría representa la estructura molecular de
mínima activación de la molécula aceptora para estar en resonancia energética con la
molécula dadora y así poderse verificar la transferencia de energía.
La teoría variacional del estado de transición (Truhlar y Garrett, 1979) hace uso de las
curvas IRC para el cálculo del estado de transición generalizado. Según esta teoría cada
punto de la curva IRC es un posible TS. Para determinar cuál de los puntos del IRC es
el TS se calculan las funciones de partición de la molécula (en general del complejo
activado) usando para ello como coordenadas internas aquéllas ortogonales a la curva,
es decir, las traslaciones, rotaciones, y las vibraciones resultantes de los modos
normales del hessiano proyectado (con respecto a la trayectoria de la curva). Una vez
determinadas las funciones de partición, se calculan las magnitudes termodinámicas de
activación, esto es, H ≠∆ , y S ≠∆ G≠∆ . Optimizando esta última (la energía libre de
activación) se obtiene el estado de transición generalizado (GTS).

290 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
Análogamente a esta teoría, la curva TET-ACC representa una colección de TS cada
uno de ellos correspondiente a una reacción distinta de transferencia de energía (con
distinta molécula dadora). De la misma manera, al disponer de cada una de las
geometrías de la curva TET-ACC es posible calcular las funciones de partición en cada
punto, y a partir de ellas obtener la contribución de la molécula aceptora a las funciones
termodinámicas de activación del proceso.
A continuación se describe brevemente el cálculo de las funciones de partición y
funciones termodinámicas de activación tal y como han sido calculadas con el paquete
de programas Gaussian 98 (Gaussian 98, 1998).
La función de partición para el movimiento de traslación viene dada por la expresión:
( )3 2
2
2 Bt
mk Tqh
π= V . Calculando el volumen a partir de la ecuación de los gases ideales,
tenemos: BV k T P= , por lo que la función de partición es: ( )3 2
2
2 Btr
mk T k Tqh P
π= B . Por
lo tanto, la contribución entrópica del movimiento de traslación es:
( ) ( ), ln 1 3t tS T P E q= + + 2 (6.56)
Por otro lado, la contribución a la energía térmica interna es:
( )2 ln 3( )2
tt A B
V
qE T N k T RTT
∂= =
∂ (6.57)
El movimiento rotacional, tiene la siguiente función de partición (expresada en función
de los momentos de inercia de la molécula):
( )
1 2 3 2
1 2rA B C
Tq πσ
⎛ ⎞= ⎜ ⎟⎝ ⎠Θ Θ Θ
, donde 2
28AA B
hI kπ
Θ = siendo , ,A B CI los momentos principales
de inercia, y σ el número de simetría (número de rotaciones que dan lugar a la misma
geometría molecular, que para los moléculas como los estilbenos con simetría C2 es
igual a 2) . Por lo que la contribución a la entropía de este movimiento es:
( ) ( 32lnr rS T R q= + ) (6.58)
Y la contribución a la energía térmica interna:
( ) ( )2 ln 32
rr
V
qE T RT RTT
∂= =
∂ (6.59)

6.4 TET con Superficies de Energía Potencial Completas 291
La función de partición de la energía vibracional dentro de la aproximación armónica
puede ser escrita como: 11 i
vib Ti
qe−Θ
=−∏ , donde i ih kBνΘ = . La contribución a la
entropía debida al movimiento vibracional viene dada por la expresión:
( ) ( )( )ln 11
i
i
Tiv T
i
TS T R ee
−Θ
Θ
Θ= − −
−∑ (6.60)
donde la suma está extendida a todos los modos normales. La contribución a la energía
térmica interna es:
( )1 12 1i
vib i Ti
E ReΘ
= Θ +−∑ (6.61)
Por último, la energía electrónica no contribuye a ninguna de las funciones
termodinámicas.
De esta manera, es posible calcular a partir de los momentos de inercia de la molécula y
de su hessiano (segundas derivadas de la energía potencial) las funciones de partición y
por lo tanto las funciones termodinámicas.
Como ya se ha dicho antes, puesto que cada punto de la curva TET-ACC representa un
posible TS correspondiente a una reacción de transferencia de energía, para calcular la
función de partición vibracional debemos proyectar sobre el vector gradiente, el
hessiano de la energía. De esta manera obtenemos un hessiano reducido con un total de
3N-7 modos normales de vibración, que son los que contribuyen a las funciones de
partición en el estado de transición y por lo tanto al cálculo de las magnitudes
termodinámicas. Esto permite por fin obtener una dependencia entre las magnitudes
termodinámicas de activación (contribución a las mismas de la molécula aceptora) y la
energía triplete (energía de resonancia entre aceptor y dador) de la molécula dadora.
Por lo tanto la curva TET-ACC no solamente permite identificar la geometría óptima (y
por lo tanto la energía electrónica de activación) que debe adoptar la molécula aceptora
para que se de la transferencia de energía entre ella y un dador con una energía triplete
determinada, sino que además, nos permite calcular las funciones de partición para
dicha geometría y por lo tanto las funciones termodinámicas de activación. Esto es de
gran utilidad, ya que nos permite discutir qué factores geométricos y energéticos
contribuyen decisivamente a la transferencia de energía no-vertical. En concreto, con
respecto a los factores energéticos, se puede discernir entre factores entrópicos y

292 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
entálpicos en la contribución a la constante cinética de transferencia de energía, cuestión
que como se verá, es de gran interés.
Las moléculas paradigmáticas en la discusión de la no-verticalidad en los procesos de
TET son sin duda el cis- y trans-estilbeno, han sido con diferencia las más estudiadas
por lo que en el siguiente apartado se analizarán con los métodos expuestos hasta ahora
y se intentará dar una explicación a su comportamiento en base a las teorías presentadas.
6.4.3 Transferencia de Energía Triplete No-Vertical en los Estilbenos
El cis- y trans-estilbeno (c-Stb y t-Stb en adelante) fueron las primeras moléculas en las
que se estudió la transferencia de energía triplete no-verticalidad (Hammond y Saltiel,
1963; Saltiel y Hammond, 1963). De hecho, el c-Stb presenta un claro comportamiento
no-vertical, ya que las velocidades de transferencia de energía frente a diversos dadores
son excepcionalmente altas en comparación con las predichas por la cinética de Sandros
(Sandros, 1964). La naturaleza, así como la cuantificación del comportamiento no-
vertical en la transferencia de energía triplete, sigue siendo aún hoy tema de estudio,
incluso, en los muy estudiados estilbenos, tanto experimental (Schanze et al., 1998)
como teóricamente (Catalán y Saltiel, 2001).
Como ya se apuntó en la introducción de este capítulo, existen varias explicaciones al
comportamiento anómalo TET no-vertical. La primera de ellas, debida a Hammond y
Saltiel (Hammond y Saltiel, 1963; Saltiel y Hammond, 1963), utilizaba el concepto de
“phamtom triplet”, una especie química no muy bien definida que servía tal vez como
intermedio inestable en la reacción de transferencia de energía. Se abandonó dicha
interpretación para dar explicaciones más elaboradas, recurriendo a la regla de oro de
Fermi y atribuyendo a los factores de Franck-Condon o a la densidad de estados
modulada por la población de las bandas calientes vibracionales, la responsabilidad de
dicho comportamiento. Por otro lado, y en base a resultados experimentales de variación
de la constante de transferencia de energía en función de la temperatura, Saltiel y
colaboradores (Saltiel et al., 1984) atribuyeron el comportamiento no-vertical del c-Stb
a la entropía de activación y no a la entalpía de activación, contrariamente a lo que
muchos autores habían mantenido hasta entonces.
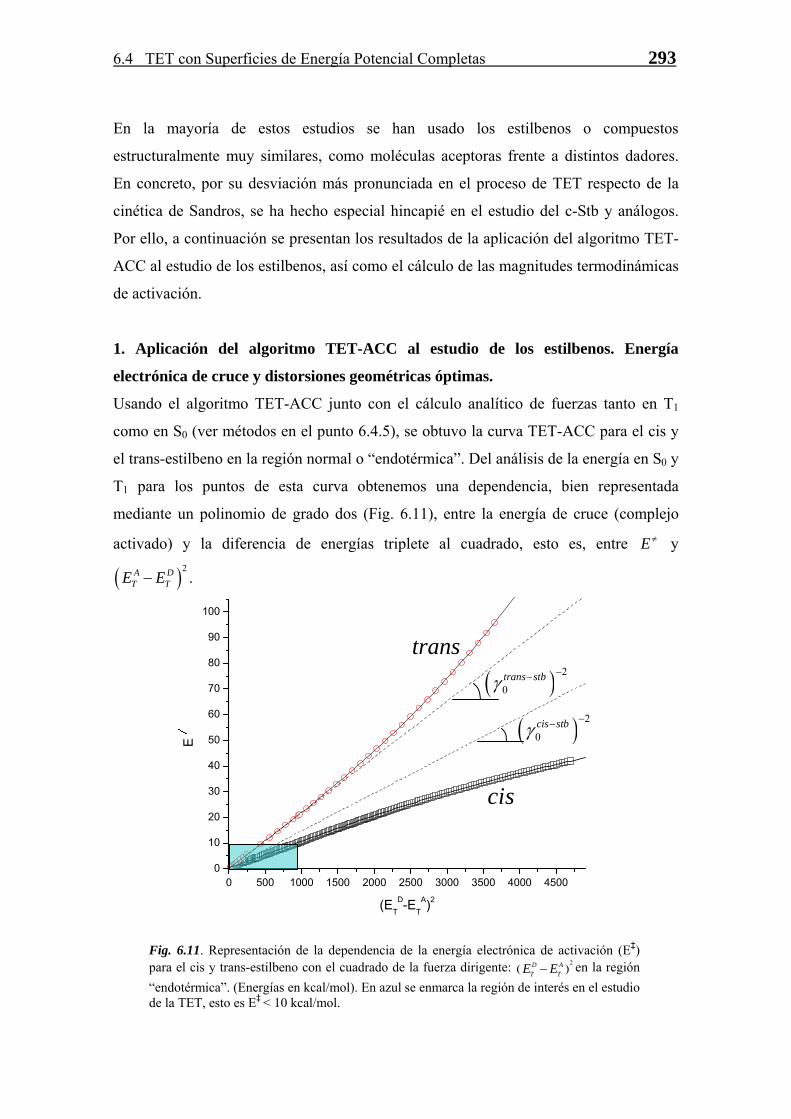
6.4 TET con Superficies de Energía Potencial Completas 293
En la mayoría de estos estudios se han usado los estilbenos o compuestos
estructuralmente muy similares, como moléculas aceptoras frente a distintos dadores.
En concreto, por su desviación más pronunciada en el proceso de TET respecto de la
cinética de Sandros, se ha hecho especial hincapié en el estudio del c-Stb y análogos.
Por ello, a continuación se presentan los resultados de la aplicación del algoritmo TET-
ACC al estudio de los estilbenos, así como el cálculo de las magnitudes termodinámicas
de activación.
1. Aplicación del algoritmo TET-ACC al estudio de los estilbenos. Energía
electrónica de cruce y distorsiones geométricas óptimas.
Usando el algoritmo TET-ACC junto con el cálculo analítico de fuerzas tanto en T1
como en S0 (ver métodos en el punto 6.4.5), se obtuvo la curva TET-ACC para el cis y
el trans-estilbeno en la región normal o “endotérmica”. Del análisis de la energía en S0 y
T1 para los puntos de esta curva obtenemos una dependencia, bien representada
mediante un polinomio de grado dos (Fig. 6.11), entre la energía de cruce (complejo
activado) y la diferencia de energías triplete al cuadrado, esto es, entre y
.
E≠
( )2A DT TE E−
0 500 1000 1500 2000 2500 3000 3500 4000 45000
10
20
30
40
50
60
70
80
90
100
E
(ETD-ET
A)2
( ) 2
0cis stbγ
−−
( ) 2
0trans stbγ
−−
trans
cis
Fig. 6.11. Representación de la dependencia de la energía electrónica de activación (E‡) para el cis y trans-estilbeno con el cuadrado de la fuerza dirigente: ( en la región “endotérmica”. (Energías en kcal/mol). En azul se enmarca la región de interés en el estudio de la TET, esto es E
)2D A
T TE E−
‡ < 10 kcal/mol.

294 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
Como podemos observar (Fig. 6.11), la pendiente a la curva en el origen para ambos
compuestos es distinta, el cis-estilbeno presenta una pendiente menor que el trans-
Estilebeno, lo que se traduce en un valor mayor de 0γ para el cis- que para el trans-
estilbeno. Por lo tanto la aproximación lineal ya predice, a este nivel de cálculo, un
comportamiento no-vertical mayor en el isómero cis que en el trans, en concordancia
con estudios experimentales (por ejemplo: Saltiel et al., 1984; Schanze et al., 1998).
Además, a medida que aumenta la diferencia de energías triplete (∆ET), la curva
correspondiente al t-Stb se curva hacia arriba, mientras que la del c-Stb lo hace hacia
abajo. Esto quiere decir que el c-Stb aumenta su carácter no-vertical a medida que la
transferencia de energía se hace más “endotérmica”, mientras que el t-Stb hace justo lo
contrario. En definitiva, tomando el parámetro de distorsión geométrica (γ) como
medida del grado de no-verticalidad, el c-Stb presenta inicialmente (es decir, γ0) un
comportamiento no-vertical un 13% mayor que el t-Stb ( ( )1
20 6.78 /trans kcal molγ = ,
( )1
20 8.24 /cis kcal molγ = , donde como ya se ha dicho el subíndice cero indica
0 /A DT TE E kcal m− = ol ).
La figura 6.11 representa una región muy amplia de diferencias de energía triplete,
mucho mayor de la que es necesaria para discutir la transferencia de energía TET, ya
que la menor constante de velocidad observada experimentalmente es del orden de siete
órdenes de magnitud menor que la máxima (la de difusión, kd), lo que supone, a
temperatura ambiente, una energía de activación próxima a las 10 kcal/mol (la región de
interés para la TET se ha marcado con un rectángulo azul en la figura 6.11). Se
muestran el resto de la curvas TET-ACC para ambos estilbenos (figura 6.11) para las
cuales los últimos puntos de ambas curvas corresponden aproximadamente a los puntos
de cruce de sistemas (degeneración energética entre los estados S0 y T1 de la molécula)
que como ya se discutió (ver arriba) el único significado físico que pueden tener es el de
servir eventualmente como vías de decaimiento al estado fundamental. Pasaremos ahora
a analizar en detalle y por separado las curvas TET-ACC del c-Stb y t-Stb.
·Curva TET-ACC para el cis-estilbeno
Analizando la curva TET-ACC en la región importante para el proceso de transferencia
de energía triplete, es decir E‡ < 10 kcal/mol, se observa cómo la molécula es capaz de
hacer descender la diferencia de energía entre los estados T1 y S0 en unas 30 kcal/mol a
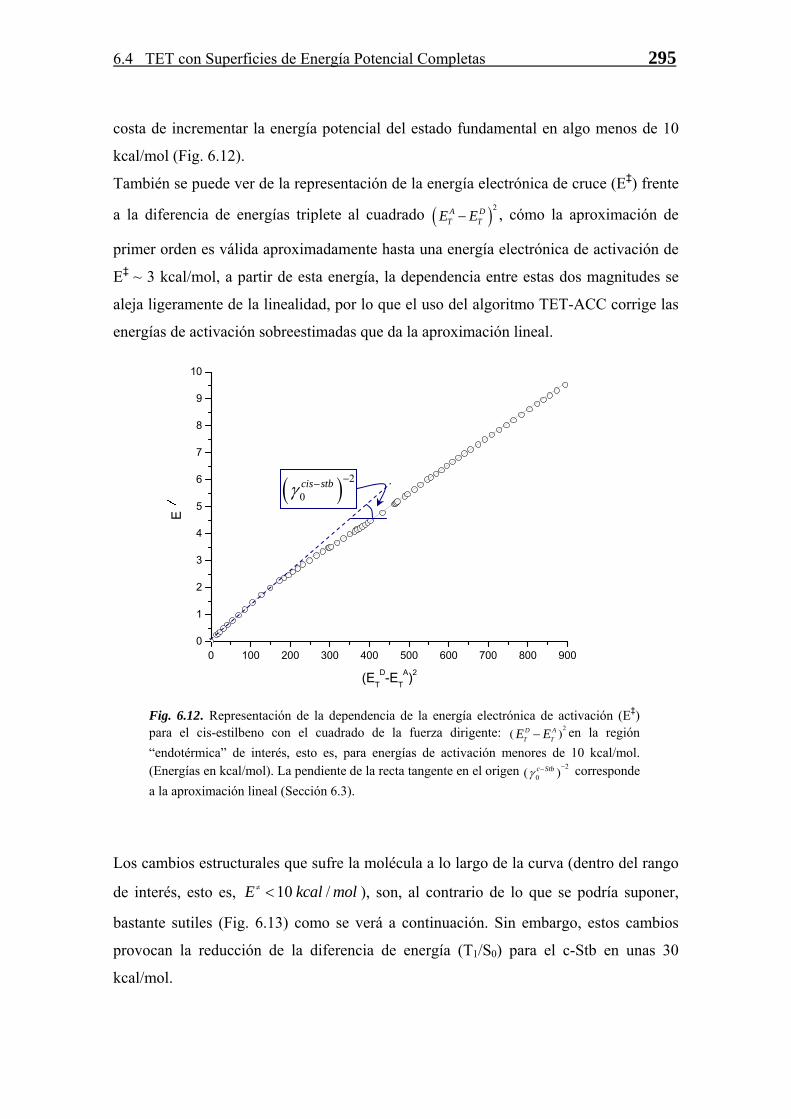
6.4 TET con Superficies de Energía Potencial Completas 295
costa de incrementar la energía potencial del estado fundamental en algo menos de 10
kcal/mol (Fig. 6.12).
También se puede ver de la representación de la energía electrónica de cruce (E‡) frente
a la diferencia de energías triplete al cuadrado ( )2A DT TE E− , cómo la aproximación de
primer orden es válida aproximadamente hasta una energía electrónica de activación de
E‡ ~ 3 kcal/mol, a partir de esta energía, la dependencia entre estas dos magnitudes se
aleja ligeramente de la linealidad, por lo que el uso del algoritmo TET-ACC corrige las
energías de activación sobreestimadas que da la aproximación lineal.
0 100 200 300 400 500 600 700 800 9000
1
2
3
4
5
6
7
8
9
10
E
(ETD-ET
A)2
( ) 2
0cis stbγ
−−
Fig. 6.12. Representación de la dependencia de la energía electrónica de activación (E‡) para el cis-estilbeno con el cuadrado de la fuerza dirigente: ( en la región “endotérmica” de interés, esto es, para energías de activación menores de 10 kcal/mol. (Energías en kcal/mol). La pendiente de la recta tangente en el origen ( corresponde a la aproximación lineal (Sección 6.3).
)
ol , so
2D AT TE E−
)2
0c Stbγ −−
Los cambios estructurales que sufre la molécula a lo largo de la curva (dentro del rango
de interés, esto es, n, al contrario de lo que se podría suponer,
bastante sutiles (Fig. 6.13) como se verá a continuación. Sin embargo, estos cambios
provocan la reducción de la diferencia de energía (T
10 /E kcal m≠ < )
1/S0) para el c-Stb en unas 30
kcal/mol.

296 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
Profundizando un poco más en los cambios geométricos óptimos en la molécula de c-
Stb (que por otra parte es la que presenta el mayor carácter no-vertical de los dos
estilbenos) podemos estudiar la variación de dos diedros importantes en la discusión de
los factores estructurales que modulan el proceso, el de torsión del grupo bencilo (D2) y
el de torsión entorno al doble enlace (D1) (Fig. 6.13).
1D
2D
0 /kcal mol5 /kcal mol
10 /kcal mol42 /kcal mol
Fig. 6.13. Estructura de equilibrio en S0 del cis-estilbeno (izquierda). Se muestran los ángulos de torsión D1 y D2, importantes en la discusión de la no-verticalidad en este compuesto. Se muestran también (derecha) cuatro geometrías correspondientes a una energía de activación electrónica (E‡) de 0 (geometría de equilibrio en S0 marcada en verde), 5, 10 y 42 (geometría de cruce de sistemas T1/S0) kcal/mol aproximadamente.
En lo que respecta a las distorsiones geométricas asociadas al comportamiento no-
vertical en compuestos orgánicos insaturados con dobles enlaces alternos (entre los que
el c-Stb es el más destacado), se ha asociado la fácil activación térmica de un modo
normal de vibración en el estado fundamental como la responsable de la disminución de
energía entre los estados T1 y S0 de la molécula aceptora. En concreto, algunos autores
han atribuido la variación de D1 al comportamiento no-vertical (por ejemplo: Hammond
y Saltiel, 1963; Catalán y Saltiel, 2001), mientras que otros lo han hecho a la variación
de D2 (por ejemplo: Davies et al., 1995; Gorman et al., 1991; Gorman et al., 1993).
Gracias a los resultados obtenidos a partir de la curva TET-ACC podemos detallar los
cambios geométricos que sufre el c-Stb y compararlos con los que existen en la
bibliografía. Para entender mejor la variación de estos dos ángulos diedro (D1 y D2) en
el proceso de transferencia de energía en el c-Stb, se han representado los valores de
éstos para las geometrías optimizadas de la curva TET-ACC en función del cuadrado de
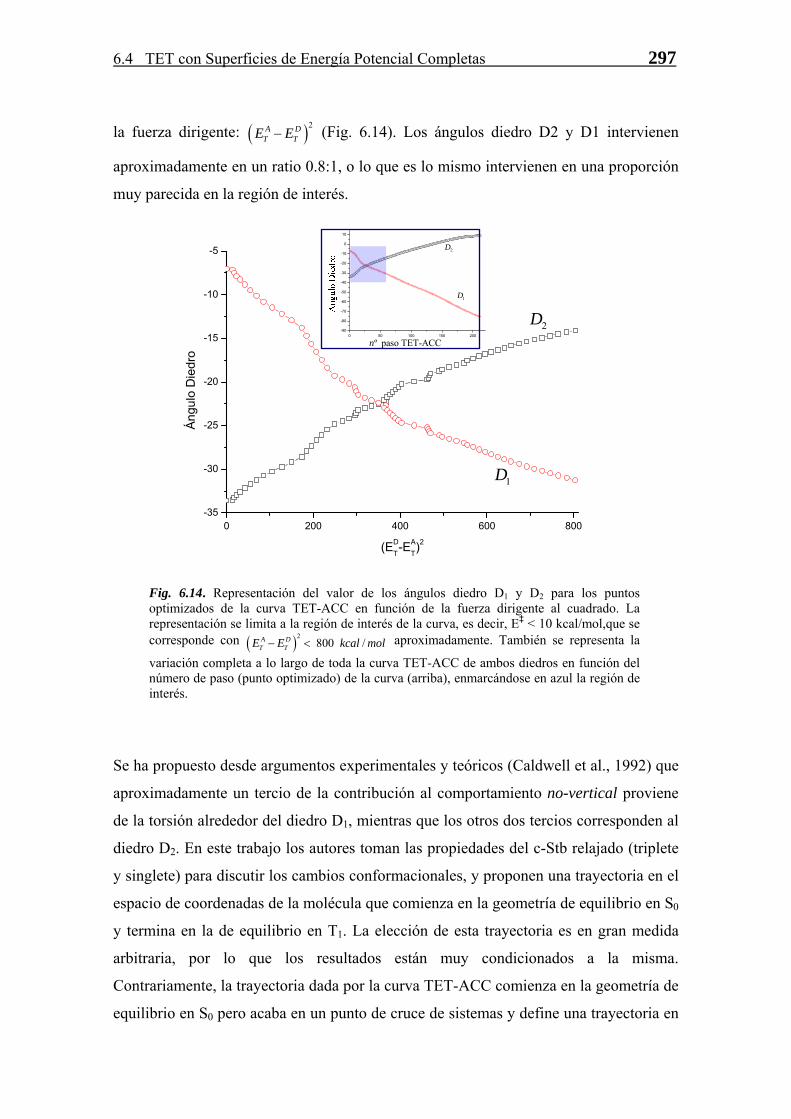
6.4 TET con Superficies de Energía Potencial Completas 297
la fuerza dirigente: ( (Fig. 6.14). Los ángulos diedro D2 y D1 intervienen
aproximadamente en un ratio 0.8:1, o lo que es lo mismo intervienen en una proporción
muy parecida en la región de interés.
)
l a
2A DT TE E−
0 200 400 600 800-35
-30
-25
-20
-15
-10
-5
Áng
ulo
Die
dro
(EDT-EA
T)2
2D
1D
0 50 100 150 200-90
-80
-70
-60
-50
-40
-30
-20
-10
0
10
2D
1D
º paso TET-ACCn
Fig. 6.14. Representación del valor de los ángulos diedro D1 y D2 para los puntos optimizados de la curva TET-ACC en función de la fuerza dirigente al cuadrado. La representación se limita a la región de interés de la curva, es decir, E‡ < 10 kcal/mol,que se corresponde con ( ) proximadamente. También se representa la
variación completa a lo largo de toda la curva TET-ACC de ambos diedros en función del número de paso (punto optimizado) de la curva (arriba), enmarcándose en azul la región de interés.
2800 /A D
T TE E kcal mo− <
Se ha propuesto desde argumentos experimentales y teóricos (Caldwell et al., 1992) que
aproximadamente un tercio de la contribución al comportamiento no-vertical proviene
de la torsión alrededor del diedro D1, mientras que los otros dos tercios corresponden al
diedro D2. En este trabajo los autores toman las propiedades del c-Stb relajado (triplete
y singlete) para discutir los cambios conformacionales, y proponen una trayectoria en el
espacio de coordenadas de la molécula que comienza en la geometría de equilibrio en S0
y termina en la de equilibrio en T1. La elección de esta trayectoria es en gran medida
arbitraria, por lo que los resultados están muy condicionados a la misma.
Contrariamente, la trayectoria dada por la curva TET-ACC comienza en la geometría de
equilibrio en S0 pero acaba en un punto de cruce de sistemas y define una trayectoria en

298 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
la que en todo momento se cumple la condición de ser la de menor energía de activación
con un mayor descenso de energía.
Como ya se ha expuesto a lo largo de todo este capítulo, la zona de la SEP triplete que
importa en el proceso de TET corresponde solamente a una pequeña región entorno a la
configuración de equilibrio en S0, que en el caso del c-Stb supone no más de un giro de
20 grados tanto para D1 como D2 (Fig. 6.14). Esta pequeña variación contrasta con las
variaciones del orden de 90 y 45 grados para D1 y D2 respectivamente que suelen
discutirse en la bibliografía. Para concluir, hay que señalar que el tratamiento
experimental (Caldwell et al., 1992) parece tener también una serie de obstáculos que
hacen llegar a conclusiones probablemente erróneas. Básicamente esto es debido a que
los experimentos se llevaron a cabo sobre una molécula aceptora análoga al c-Stb en la
que se bloqueó la rotación entorno al doble enlace del cis-estilbeno (D1) con un puente
carbonado (norborneno) que presenta cierta tensión, por lo que al observar un
comportamiento no-vertical los autores atribuyen dicho comportamiento a la rotación
entorno a D2. Algunos autores han señalado en base a estudios teóricos que esto es
erróneo, ya que la inclusión de este puente no bloquea notablemente dicha rotación
(Catalán y Saltiel, 2001). Sin embargo, a pesar de que realmente se consiguiese
bloquear esta rotación, en general puede ser posible observar comportamiento no-
verticalidad bloqueando tanto D1 como D2. Mientras haya al menos una distorsión que
pueda satisfacer las condiciones ya mencionadas (alto valor del gradiente en T1 y bajas
constantes de fuerza en S0 en la dirección del gradiente en T1) podrá observarse
comportamiento no-vertical. Por lo tanto, como se remarcará más adelante, los criterios
de no-verticalidad no pueden ser geométricos sino energéticos, de tal manera que la
presencia de dobles enlaces o enlaces simples en una molécula ni asegura ni descarta la
posibilidad de que presente un comportamiento no-vertical en el proceso de TET.
Por otro lado, recientemente se ha estudiado con métodos ab initio similares§ a los
usados en este punto la PES del c-Stb (Catalán y Saltiel, 2001). En este trabajo, se llega
a la conclusión de que prácticamente en su totalidad el diedro D1 es el responsable de la
no-verticalidad (aproximadamente en un 75% frente a un 25% de D2), aunque no
descartan la participación (en menor medida) de otras coordenadas o distorsiones
moleculares. Existe por lo tanto una gran discrepancia entre los valores de variación de
§ Los autores usaron el métod B3Lyp con funciones de base 6-31+G(d,p)

6.4 TET con Superficies de Energía Potencial Completas 299
los ángulos diedro D1 y D2 obtenidos usando el algoritmo TET-ACC (56% D1 y 44%
D2) frente a los obtenidos por los autores (75% D1 y 25% D2) (Catalán y Saltiel, 2001),
a pesar de usar prácticamente el mismo método ab initio en la determinación de las
superficies de energía potencial.
Catalán y Saltiel llegan a estas conclusiones se llega a través del siguiente
procedimiento: Se calcula en primer lugar la energía de relajación vibracional en el c-
Stb en T1 correspondiente a la relajación desde la geometría de la transición FC hasta la
geometría de equilibrio en este estado, que es según los autores de unas 15 kcal/mol.
Partiendo de la geometría FC y congelando el ángulo diedro D1, optimizan en T1 el
resto de variables estructurales, obteniendo un descenso de energía de 2.7 kcal/mol. De
este estudio deducen que aproximadamente ¾ del descenso de energía se deben al
ángulo diedro bloqueado, y ¼ aproximadamente al resto de coordenadas.
Las conclusiones a las que llegan los autores (Catalán y Saltiel, 2001) están en clara
contradicción con los resultados a los que se han llegado aplicando el algoritmo TET-
ACC por los siguientes motivos:
-En primer lugar, solamente una región de las SEP (T1 y S0) entorno a la geometría de
equilibrio en S0 es necesaria para estudiar el proceso de TET no-vertical, por lo que
dicho proceso es insensible a las propiedades del la SEP entorno a la región de
equilibrio en T1 en el caso del c-Stb, ya que esta región de la SEP solamente se puede
alcanzar con energías de activación cercanas a las 40 kcal/mol (ver figura 6.11) que son
completamente inaccesibles a temperatura ambiente. Por lo tanto no se pueden discutir
el comportamiento no-vertical en base a las propiedades del c-Stb relajado en T1.
-En segundo lugar, el bloqueo de una coordenada lleva implícita la elección de una
trayectoria en T1 a partir de la cual se analiza el proceso de TET. La elección de la
trayectoria es fundamental, y de su elección depende la bondad de los resultados. Esta
trayectoria no coincide con la que se presenta en esta Tesis como óptima (TET-ACC)
por lo que ésta es una segunda causa de discrepancia.
-Por último, se ha mostrado a lo largo de este capítulo que una combinación de
propiedades de las dos SEP involucradas en el proceso (S0 y T1) es la que determina el
comportamiento no-vertical, lo que se pone de manifiesto de manera cuantitativa, en el
parámetro de distorsión geométrica, que depende tanto de propiedades en T1 (gradiente
de la energía) como de S0 (combinación lineal de constantes de fuerza recuperadora).
Por este motivo, el análisis de la energía de relajación en T1 por sí sola es insuficiente en

300 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
la determinación de las distorsiones moleculares óptimas que permiten el proceso de
transferencia de energía triplete no-vertical, y consecuentemente, ceñirse al estudio de
T1 debe ser una fuente más de error en la interpretación de los resultados.
Similares procedimientos a los levados a cabo por Catalán y Saltiel (Catalán y Saltiel,
2001) han sido realizados recientemente por otros autores, desde un punto de vista
teórico en otro tipos de compuestos (Lalevée et al., 2002; Lalevée et al., 2002b)
presentando las mismas objeciones en el análisis e interpretación de las coordenadas que
influyen en el proceso de TET.
Concluyendo, la desviación respecto de la cinética de Sandros en el proceso de TET no
puede atribuirse únicamente a la posibilidad dé que se de una de las distorsiones antes
mencionadas (rotación de los diedros D1 y D2) o incluso las dos, al contrario, lo que
determina la no-verticalidad es el binomio: coordenada con un bajo valor de la
constante de fuerza recuperadora en S0 en la dirección del vector gradiente en T1, y un
alto valor de dicho gradiente. No se puede simplificar el problema en términos
estructurales, recurriendo únicamente al estudio de las coordenadas de “torsión entorno
al doble enlace” o “torsión de los grupos fenilos”. Cada molécula presenta unas
características propias que hacen que presente un comportamiento distinto, a pesar de
poder presentar semejanza estructural con otras en las que el comportamiento no-
vertical está claramente establecido. Es importante remarcar que: “El criterio para la
identificación de moléculas aceptoras que presentan comportamiento no-vertical debe
ser siempre energético y nunca geométrico, y además debe tener en cuenta las
propiedades de las SEP T1 y S0 en la región de interés, esto es, la que puede ser
alcanzada por activación térmica en el estado fundamental”.
· Curva TET-ACC para el trans-estilbeno
Los resultados del estudio de la curva TET-ACC para el trans-estilbeno (t-Stb) a nivel
B3Lyp/6-31G(d) confirman cualitativamente los resultados experimentales más
recientes que existen sobre el mismo en lo que respecta a su comportamiento no-
vertical, que señalan el bajo carácter no-vertical del mismo en el proceso de
transferencia de energía triplete (Schanze et al., 1998).
Sin embargo, en este compuesto hay que tener especial cuidado en el análisis de la
curva TET-ACC debido a la gran contribución de la correlación dinámica a la energía
electrónica que es de esperar en un compuesto cercano a la planaridad y con una

6.4 TET con Superficies de Energía Potencial Completas 301
distribución de carga π extendida por toda la molécula como es el c-Stb (Molina et al.,
1997).
De hecho, tanto la determinación experimental como teórica de la estructura en el
estado fundamental del trans-estilbeno son complicadas. Mientras que algunos autores
afirman que la estructura en el estado fundamental es plana C2h (Champagne et al.,
1990; Waldeck, 1991; Chiang y Laane, 1994) otros proponen una estructura ligeramente
distorsionada (C2) en la que los grupos fenilos se hayan ligeramente fuera del plano
(Traetteberg et al., 1975; Kobayashi et al., 1982). Usando el método MP2 con unas
funciones de base gaussianas 6-31G(d) y derivadas segundas de la SEP analíticas para
caracterizar los puntos estacionarios, se encuentra que la estructura plana es un estado
de transición con una energía de tan solo 0.67 kcal/mol por encima del mínimo C2, con
un valor del modo normal imaginario de ~40 cm-1. De la misma manera, usando el
método B3Lyp/6-31G(d) se encontró una situación análoga (cualitativamente) a la
encontrada a nivel MP2/6-31G(d).
Puesto que el cálculo de la curva TET-ACC se efectuó con el método B3Lyp/6-31G(d),
para prevenir posibles errores del método en la determinación de la SEP del estado
fundamental, se realizaron dichas curvas comenzando tanto desde la estructura C2h
plana como desde la C2 (ligeramente menor en energía). Ambas curva prácticamente se
superponen, lo que pone de manifiesto que la elección de la estructura plana C2h para la
determinación de la curva TET-ACC es correcta como ya se puso de manifiesto en la
determinación teórica del espectro electrónico (Molina et al., 1997).
Además los parámetros estructurales determinados a nivel B3Lyp/6-31G(d) están más
cercanos a los encontrados a nivel CASSCF(12 OM, 10e) que a nivel HF. En concreto
uno de los parámetros estructurales más sensibles a la inclusión de correlación
electrónica en la energía es la distancia C=C central, que a nivel HF es de 1.332 Å, a
nivel CASSCF(12,10) es de 1.351Å (Molina et al., 1997) y a nivel B3Lyp es de 1.349
Å.
Teniendo en cuenta que el método usado (B3Lyp/6-31G(d)) coincide cualitativamente
en la descripción de la topología de la SEP con métodos superiores (ver arriba), así
como con algunas evidencias experimentales (ver arriba), los resultados obtenidos de la
aplicación del algoritmo TET-ACC deben tener al menos un carácter cualitativo.
La dependencia de la energía electrónica de cruce con la diferencia de energías triplete
viene marcada por una gran dependencia lineal en la zona de interés (E‡ < 10 kcal/mol)
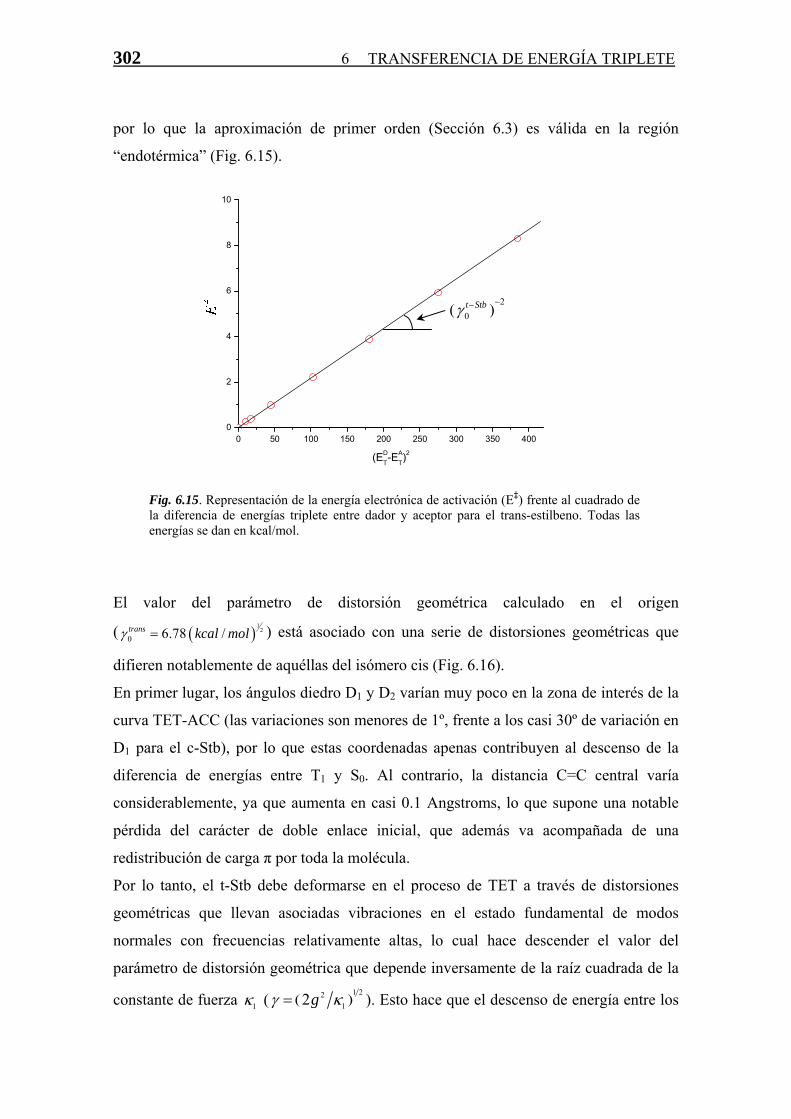
302 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
por lo que la aproximación de primer orden (Sección 6.3) es válida en la región
“endotérmica” (Fig. 6.15).
0 50 100 150 200 250 300 350 4000
2
4
6
8
10
(EDT-EA
T)2
( )2
0t Stbγ −−
Fig. 6.15. Representación de la energía electrónica de activación (E‡) frente al cuadrado de la diferencia de energías triplete entre dador y aceptor para el trans-estilbeno. Todas las energías se dan en kcal/mol.
El valor del parámetro de distorsión geométrica calculado en el origen
( ( )1
20 6.78 /trans kcal molγ = ) está asociado con una serie de distorsiones geométricas que
difieren notablemente de aquéllas del isómero cis (Fig. 6.16).
En primer lugar, los ángulos diedro D1 y D2 varían muy poco en la zona de interés de la
curva TET-ACC (las variaciones son menores de 1º, frente a los casi 30º de variación en
D1 para el c-Stb), por lo que estas coordenadas apenas contribuyen al descenso de la
diferencia de energías entre T1 y S0. Al contrario, la distancia C=C central varía
considerablemente, ya que aumenta en casi 0.1 Angstroms, lo que supone una notable
pérdida del carácter de doble enlace inicial, que además va acompañada de una
redistribución de carga π por toda la molécula.
Por lo tanto, el t-Stb debe deformarse en el proceso de TET a través de distorsiones
geométricas que llevan asociadas vibraciones en el estado fundamental de modos
normales con frecuencias relativamente altas, lo cual hace descender el valor del
parámetro de distorsión geométrica que depende inversamente de la raíz cuadrada de la
constante de fuerza 1κ ( ( )1 22
12gγ κ= ). Esto hace que el descenso de energía entre los
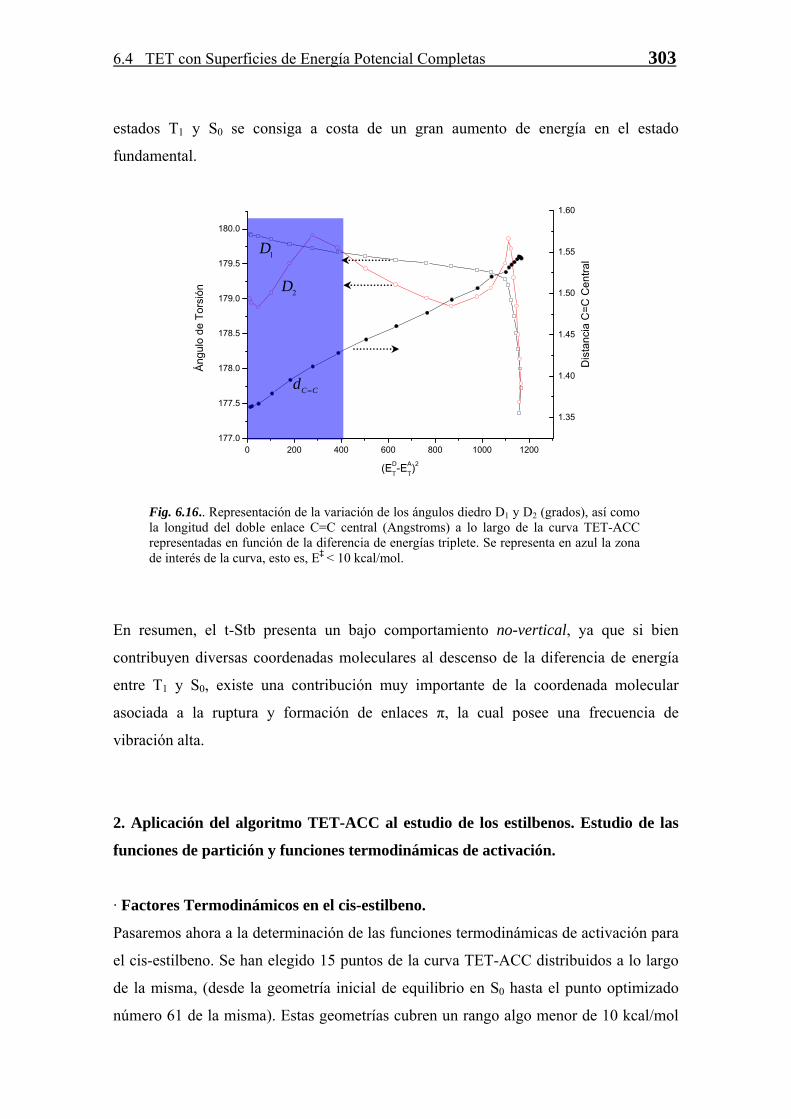
6.4 TET con Superficies de Energía Potencial Completas 303
estados T1 y S0 se consiga a costa de un gran aumento de energía en el estado
fundamental.
0 200 400 600 800 1000 1200177.0
177.5
178.0
178.5
179.0
179.5
180.0
Áng
ulo
de T
orsi
ón
(EDT-EA
T)2
1.35
1.40
1.45
1.50
1.55
1.60
Dis
tanc
ia C
=C C
entra
l1D
2D
C Cd =
Fig. 6.16.. Representación de la variación de los ángulos diedro D1 y D2 (grados), así como la longitud del doble enlace C=C central (Angstroms) a lo largo de la curva TET-ACC representadas en función de la diferencia de energías triplete. Se representa en azul la zona de interés de la curva, esto es, E‡ < 10 kcal/mol.
En resumen, el t-Stb presenta un bajo comportamiento no-vertical, ya que si bien
contribuyen diversas coordenadas moleculares al descenso de la diferencia de energía
entre T1 y S0, existe una contribución muy importante de la coordenada molecular
asociada a la ruptura y formación de enlaces π, la cual posee una frecuencia de
vibración alta.
2. Aplicación del algoritmo TET-ACC al estudio de los estilbenos. Estudio de las
funciones de partición y funciones termodinámicas de activación.
· Factores Termodinámicos en el cis-estilbeno.
Pasaremos ahora a la determinación de las funciones termodinámicas de activación para
el cis-estilbeno. Se han elegido 15 puntos de la curva TET-ACC distribuidos a lo largo
de la misma, (desde la geometría inicial de equilibrio en S0 hasta el punto optimizado
número 61 de la misma). Estas geometrías cubren un rango algo menor de 10 kcal/mol
304 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
en energía de activación (componente electrónica), y a una diferencia de energías
triplete de aproximadamente 30 kcal/mol. Como ya se dijo antes, la función de
partición vibracional se ha calculado con los autovalores del hessiano proyectado a lo
largo del vector gradiente, por lo tanto uno de los modos normales de vibración se
elimina debido a la proyección (ver Punto 6.4.2).
TE∆
Para el c-Stb, los resultados obtenidos se recogen en la tabla 6.1 en la cual se presentan
los valores absolutos de las funciones termodinámicas entalpía y entropía (H y S), los
valores de las funciones termodinámicas de activación (H‡, S‡), así como los
incrementos de activación (∆H‡, ∆S‡, ∆G‡). Se da también la diferencia de energías
triplete: . TE∆
Tabla 6.1.Valores, para distintas geometrías de la curva TET-ACC calculada (ver texto), de las funciones termodinámicas H y S, de las funciones termodinámicas de activación (H‡, S‡, ∆H‡, ∆S‡ y ∆G‡), así como la diferencia de energías electrónicas entre T1 y S0 (∆ET) para el cis-estilbeno en función del punto (N) a lo largo de la curva TET-ACC a.
a El Hessiano total en S0 presenta todos los autovalores positivos.
N Hb,c
Sd
H‡ c
S‡ d
∆H‡ e
∆S‡,d,f ∆G‡ e
∆ET
e
0 -0.474298 105.857 - - - - - 68.667
2 -0.474056 105.089 -0.477676 105.056 -2.273 -0.434 (0.130)
-2.143 64.975
3 -0.474006 104.885 -0.477512 104.829 -2.171 -0.661 (0.197)
-1.974 64.432
5 -0.473781 104.534 -0.477104 104.438 -1.914 -1.052 (0.313)
-1.601 63.086
10 -0.472377 103.983 -0.475600 103.840 -0.971 -1.65 (0.492)
-0.479 58.401
15 -0.470790 103.846 -0.473945 103.649 0.221 -2.208 (0.658)
0.879 54.692
20 -0.469653 103.758 -0.472764 103.505 0.963 -2.352 (0.701)
1.664 52.332
25 -0.468933 103.609 -0.471994 103.324 1.446 -2.533 (0.755)
2.201 50.820
30 -0.468221 103.438 -0.471253 103.137 1.911 -2.72 (0.811)
2.722 49.437
35 -0.467794 103.135 -0.470761 102.803 2.219 -3.054 (0.911)
3.13 48.589
40 -0.466608 103.445 -0.469557 103.116 2.975 -2.741 (0.817)
3.792 46.982
45 -0.465385 103.205 -0.468360 102.877 3.726 -2.98 (0.888)
4.614 45.282
50 0.464210 103.617 -0.467145 103.284 4.488 -2.573 (0.767)
5.255 43.975
56 -0.463238 103.375 -0.466149 103.043 4.960 -2.447 (0.729)
5.689 42.702
61 -0.461297 103.330 -0.464133 103.007 6.224 -2.483 (0.740)
6.964 40.313
bH=-540 + (valor en la tabla) cEn unidades atómicas dEn unidades de cal·K-1mol-1. eEn unidades de kcal· mol-1. fEntre paréntesis el valor de (-T∆S‡) en kcal/mol para T=298K
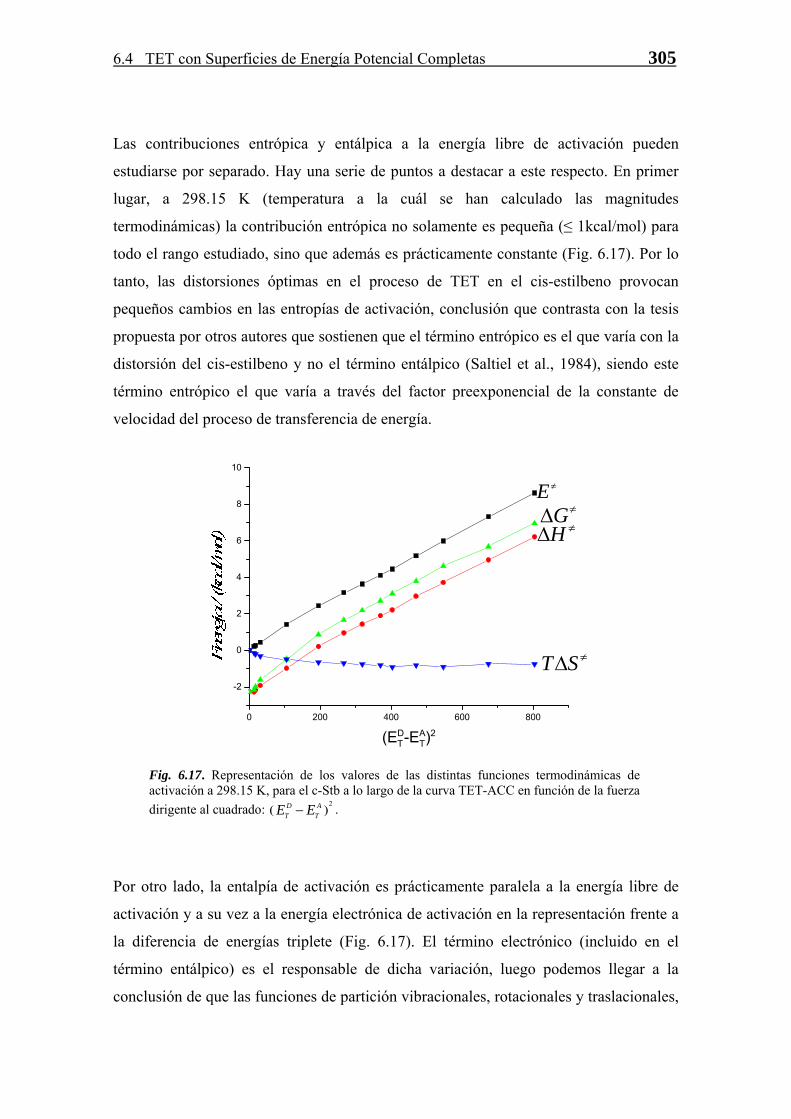
6.4 TET con Superficies de Energía Potencial Completas 305
Las contribuciones entrópica y entálpica a la energía libre de activación pueden
estudiarse por separado. Hay una serie de puntos a destacar a este respecto. En primer
lugar, a 298.15 K (temperatura a la cuál se han calculado las magnitudes
termodinámicas) la contribución entrópica no solamente es pequeña (≤ 1kcal/mol) para
todo el rango estudiado, sino que además es prácticamente constante (Fig. 6.17). Por lo
tanto, las distorsiones óptimas en el proceso de TET en el cis-estilbeno provocan
pequeños cambios en las entropías de activación, conclusión que contrasta con la tesis
propuesta por otros autores que sostienen que el término entrópico es el que varía con la
distorsión del cis-estilbeno y no el término entálpico (Saltiel et al., 1984), siendo este
término entrópico el que varía a través del factor preexponencial de la constante de
velocidad del proceso de transferencia de energía.
0 200 400 600 800
-2
0
2
4
6
8
10
Ener
gy/(k
cal/m
ol)
(EDT-EA
T)2
H ≠∆G≠∆
T S ≠∆
E≠
Fig. 6.17. Representación de los valores de las distintas funciones termodinámicas de activación a 298.15 K, para el c-Stb a lo largo de la curva TET-ACC en función de la fuerza dirigente al cuadrado: ( )
2D AT TE E− .
Por otro lado, la entalpía de activación es prácticamente paralela a la energía libre de
activación y a su vez a la energía electrónica de activación en la representación frente a
la diferencia de energías triplete (Fig. 6.17). El término electrónico (incluido en el
término entálpico) es el responsable de dicha variación, luego podemos llegar a la
conclusión de que las funciones de partición vibracionales, rotacionales y traslacionales,

306 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
apenas contribuyen al comportamiento no-vertical en el cis-estilbeno y por extensión,
en compuestos similares. Estas funciones de partición que en el formalismo de la teoría
del estado de transición (en su formulación microcanónica) están contenidos en el
término pre-exponencial no varían apenas en el proceso de excitación de la molécula
aceptora, siendo el término exponencial (entálpico) el que a través de la energía
potencial varía con dicho proceso de activación y por lo tanto con . ( )2
TE∆
Todos los resultados presentados a lo largo de este capítulo indican que la activación
térmica, a través del término entálpico (en el marco de la TST en su formulación
termodinámica) o equivalentemente del término de energía potencial de cruce entre
estados (en el marco de la TST en su formulación microcanónica), es la responsable del
comportamiento no-vertical.
Aplicando el algoritmo TET-ACC se ha demostrado cómo para el c-Stb las
deformaciones óptimas en el estado fundamental que dan lugar a un descenso máximo
de la diferencia de energías (T1/S0) demandan un aumento de la energía térmica del
sistema a través de la componente entálpica. La variación en esta componente se debe
prácticamente a la variación de la energía electrónica (potencial) del sistema, luego en
este caso al menos, y muy probablemente en aceptores flexibles similares al c-Stb, no es
necesario recurrir al cálculo de las funciones de partición, ya que las SEP del mismo son
suficientes para estudiar las propiedades del aceptor en el proceso de transferencia de
energía triplete-triplete.
· Factores Termodinámicos en el trans-estilbeno.
Como ya se vio antes, el t-Stb no presenta un alto valor del parámetro de distorsión
geométrica ( 0γ ) por lo que tampoco debe presentar un gran carácter no-vertical en su
comportamiento como aceptor. Los cambios geométricos que favorecen el descenso de
energía entre los estados T1 y S0 son bastante sutiles, por lo que es de esperar que no
haya cambios notorios en las funciones de partición y por lo tanto en las funciones
termodinámicas de activación. De hecho, esto es lo que reflejan los cálculos realizados
de las magnitudes termodinámicas de activación a lo largo de la curva TET-ACC (Tabla
6.2).
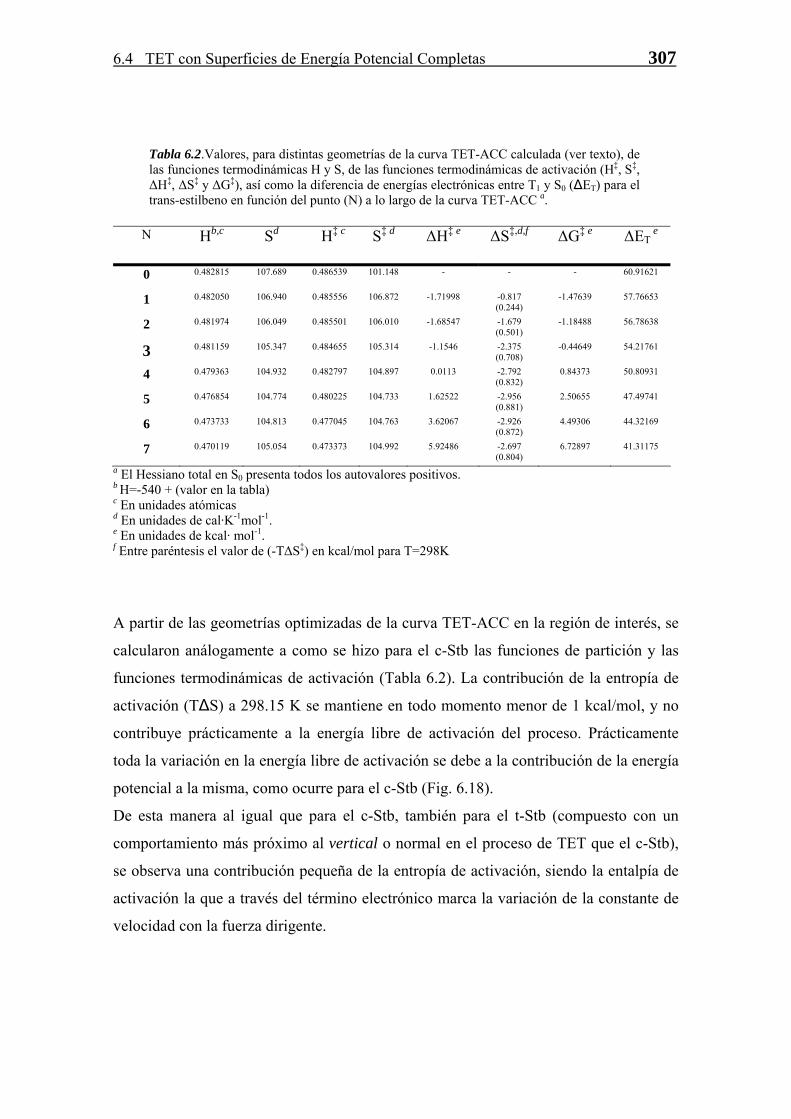
6.4 TET con Superficies de Energía Potencial Completas 307
Tabla 6.2.Valores, para distintas geometrías de la curva TET-ACC calculada (ver texto), de las funciones termodinámicas H y S, de las funciones termodinámicas de activación (H‡, S‡, ∆H‡, ∆S‡ y ∆G‡), así como la diferencia de energías electrónicas entre T1 y S0 (∆ET) para el trans-estilbeno en función del punto (N) a lo largo de la curva TET-ACC a.
a El Hessiano total en S0 presenta todos los autovalores positivos.
N Hb,c Sd
H‡ c
S‡ d ∆H‡ e
∆S‡,d,f ∆G‡ e
∆ET
e
0 0.482815 107.689 0.486539 101.148 - - - 60.91621
1 0.482050 106.940 0.485556 106.872 -1.71998 -0.817 (0.244)
-1.47639 57.76653
2 0.481974 106.049 0.485501 106.010 -1.68547 -1.679 (0.501)
-1.18488 56.78638
3 0.481159 105.347 0.484655 105.314 -1.1546 -2.375 (0.708)
-0.44649 54.21761
4 0.479363 104.932 0.482797 104.897 0.0113 -2.792 (0.832)
0.84373 50.80931
5 0.476854 104.774 0.480225 104.733 1.62522 -2.956 (0.881)
2.50655 47.49741
6 0.473733 104.813 0.477045 104.763 3.62067 -2.926 (0.872)
4.49306 44.32169
7 0.470119 105.054 0.473373 104.992 5.92486 -2.697 (0.804)
6.72897 41.31175
b H=-540 + (valor en la tabla) c En unidades atómicas d En unidades de cal·K-1mol-1. e En unidades de kcal· mol-1. f Entre paréntesis el valor de (-T∆S‡) en kcal/mol para T=298K
A partir de las geometrías optimizadas de la curva TET-ACC en la región de interés, se
calcularon análogamente a como se hizo para el c-Stb las funciones de partición y las
funciones termodinámicas de activación (Tabla 6.2). La contribución de la entropía de
activación (T∆S) a 298.15 K se mantiene en todo momento menor de 1 kcal/mol, y no
contribuye prácticamente a la energía libre de activación del proceso. Prácticamente
toda la variación en la energía libre de activación se debe a la contribución de la energía
potencial a la misma, como ocurre para el c-Stb (Fig. 6.18).
De esta manera al igual que para el c-Stb, también para el t-Stb (compuesto con un
comportamiento más próximo al vertical o normal en el proceso de TET que el c-Stb),
se observa una contribución pequeña de la entropía de activación, siendo la entalpía de
activación la que a través del término electrónico marca la variación de la constante de
velocidad con la fuerza dirigente.
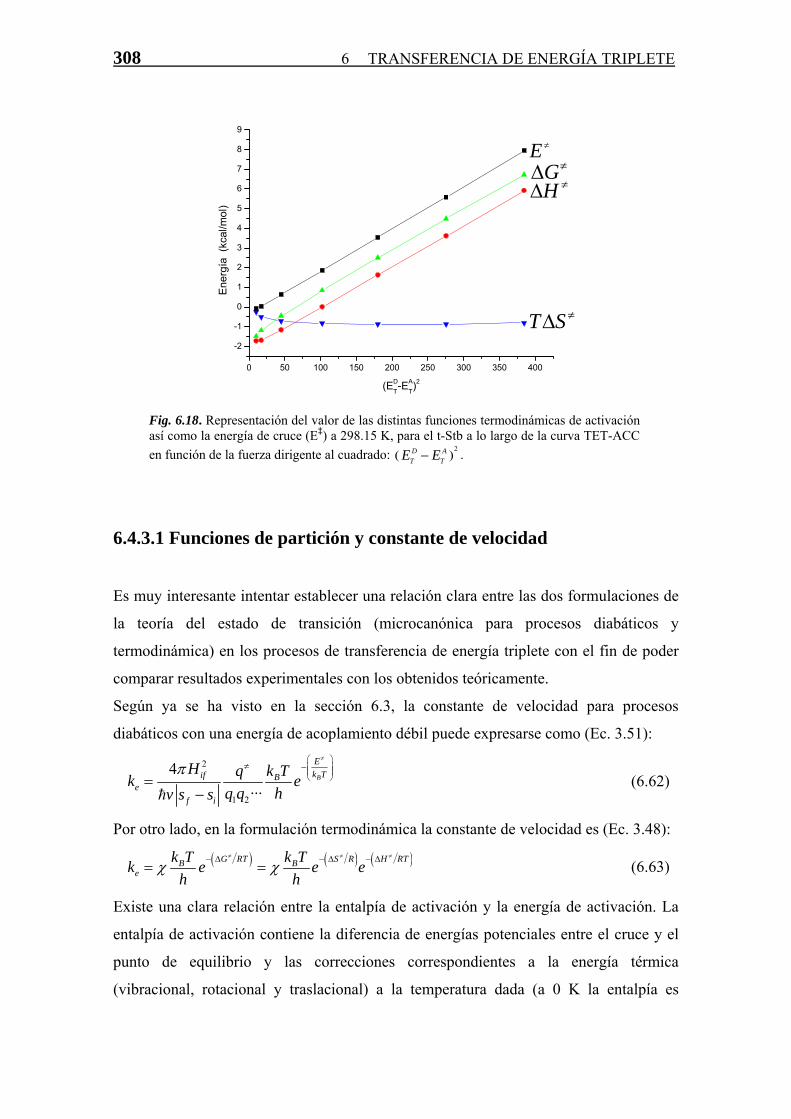
308 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
G≠∆H ≠∆
T S ≠∆
0 5 1 15 2 25 3 30 00 0 00 0 00 50 400
-2
-1
0
1
2
3
4
5
6
7
8
9
Ener
gía
(kca
l/mol
)
DT-EA
T)2
E
(E
≠
Fig. 6.18. Representación del valor de las distintas funciones termodinámicas de activación así como la energía de cruce (E‡) a 298.15 K, para el t-Stb a lo largo de la curva TET-ACC en función de la fuerza dirigente al cuadrado: ( )
2D AT TE E− .
6.4.3.1 Funciones de partición y constante de velocidad
Es muy interesante intentar establecer una relación clara entre las dos formulaciones de
la teoría del estado de transición (microcanónica para procesos diabáticos y
termodinámica) en los procesos de transferencia de energía triplete con el fin de poder
comparar resultados experimentales con los obtenidos teóricamente.
Según ya se ha visto en la sección 6.3, la constante de velocidad para procesos
diabáticos con una energía de acoplamiento débil puede expresarse como (Ec. 3.51):
2
1 2
4···
B
Ek Tif B
ef i
H k Tqkq q hv s s
π≠⎛ ⎞
≠ −⎜ ⎟⎜⎝=
−e
⎟⎠ (6.62)
Por otro lado, en la formulación termodinámica la constante de velocidad es (Ec. 3.48):
( ) ( ) ( )G RT S R H RTB Be
k T k Tk e e eh h
χ χ≠ ≠− ∆ − ∆ − ∆
= =≠
(6.63)
Existe una clara relación entre la entalpía de activación y la energía de activación. La
entalpía de activación contiene la diferencia de energías potenciales entre el cruce y el
punto de equilibrio y las correcciones correspondientes a la energía térmica
(vibracional, rotacional y traslacional) a la temperatura dada (a 0 K la entalpía es

6.4 TET con Superficies de Energía Potencial Completas 309
simplemente la energía electrónica más la corrección de la energía vibracional residual).
La primera de estas contribuciones de la entalpía de activación (ecuación 6.63) se
corresponde con E‡ (ecuación 6.64). En este sentido, es fácil correlacionar el término
exponencial de la ecuación (6.62) con el mismo de la ecuación (6.63).
Sin embargo, establecer una relación entre las constantes preexponenciales de ambas
ecuaciones, así como con la entropía de activación calculada con el algoritmo TET-
ACC no es inmediato.
La contribución a la entropía de activación tiene varias componentes, la de la molécula
aceptora, la de la dadora, la del disolvente y las posibles derivadas de las interacciones
entre molécula aceptora y dadora principalmente:
(6.64) A D DteS S S S S≠ ≠ ≠ ≠∆ = ∆ + ∆ + ∆ + ∆ AD≠
donde A denota aceptor, D dador, Dte disolvente y AD la pareja Aceptor-Dador.
Como se ha comprobado para los estilbenos, el primer término no solamente es pequeño
sino que además varía muy poco. El término debido a la molécula aceptora es de esperar
que tome un valor bajo, ya que para simplificar el problema hemos supuesto que la
molécula dadora presenta una estructura rígida y sus funciones de partición apenas
cambian tras la excitación. Por otro lado, el disolvente puede tener un papel importante
en la entropía de activación, sin embargo, no hay motivo para que dicha contribución
dependa de la diferencia de energías triplete, o lo que es lo mismo, de la coordenada de
reacción ya que los cambios estructurales a lo largo de dicha coordenada son pequeños
(como se ha visto en los estilbenos) y no deben afectar sensiblemente a la energía de
solvatación de la molécula aceptora.
Por último está la contribución de la interacción entre las moléculas dadora y aceptora a
la entropía de activación. El término principal del factor preexponencial en la ecuación
(6.62) es el de acoplamiento electrónico (Hif), ya que es este acoplamiento el que
permite que la reacción ocurra. Como se vio en el punto 6.2.1, este acoplamiento
depende de las coordenadas de posición relativa de las dos moléculas, y en modo alguno
depende de la energía de excitación del aceptor y/o del dador. La cuestión es por lo
tanto si este término debe incluirse en el coeficiente de transmisión (χ) o en la entropía
de activación a través de la interacción entre dador y aceptor ADS ≠∆ . Probablemente este
término tenga una contribución tanto en el coeficiente de transmisión como en la
entropía de activación, ya que por un lado se ha demostrado una clara dependencia entre

310 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
el valor del acoplamiento y la orientación y posición relativa de los grupos cromóforos
en procesos de transferencia de energía triplete intramolecular (Speiser, 1996) por lo
que existe una relación entre la organización de las moléculas en el complejo
(componente entrópica) y el valor del acoplamiento y por lo tanto de la constante
cinética. Por otro lado, debe existir un valor del acoplamiento (Hif) máximo,
independiente de la orientación y posición relativa de los grupos cromóforos, y que
solamente depende de la naturaleza de la función de onda (a través de la extensión
espacial de los orbitales que intervienen principalmente en el proceso) de dador y
aceptor como ya se ha visto en la determinación de Hif (punto 6.2.1), por lo que no
depende de la organización de las moléculas en el complejo de encuentro.
Es interesante también analizar los cambios que debe haber en el término
preexponencial de la constante cinética a través de la variación de Hif y su relación con
la diferencia de energías triplete (∆ET), es decir, la variación de Hif a lo largo de la
correspondiente coordenada TET-ACC. Para analizar esta variación en la entropía de
activación vamos a usar los resultados obtenidos para el c-Stb, molécula que posee un
gran comportamiento no-vertical. Hemos visto cómo en el c-Stb ocurren una serie de
cambios estructurales, y cómo el término entrópico de activación debido a la
contribución de la molécula aceptora por sí sola apenas contribuye a la energía libre de
activación, pero cabría preguntarse en qué sentido varía la entropía de activación debida
al acoplamiento entre estados. A lo largo de la coordenada de transferencia de energía la
forma y extensión de los orbitales HOMO y LUMO del c-Stb no cambian de manera
importante (hasta tal punto que su forma y extensión es prácticamente idéntica en toda
la región de interés para la TET), por lo que estas distorsiones geométricas no deben
introducir cambios importantes en el término de acoplamiento electrónico.
Por otro lado tampoco debe haber cambios importantes en el coeficiente de transmisión
a lo largo de la coordenada de reacción, ya que las deformaciones geométricas no
incrementan ni disminuyen la eficiencia del acoplamiento. Concluimos por lo tanto que
la entropía de activación no debe cambiar notablemente en la reacción cuando se
realizan una serie de experimentos de transferencia de energía triplete desde distintos
dadores similares a un mismo aceptor y en un mismo disolvente.
Por lo tanto, si bien el término Hif varía inevitablemente con el cambio de dador
(manteniendo siempre el mismo aceptor y el mismo disolvente) ya que la distribución
de la carga varía de un dador a otro, eligiendo dadores que presenten una distribución de

6.4 TET con Superficies de Energía Potencial Completas 311
carga similar evitamos que el valor Hif se aleje excesivamente de un valor medio
correspondiente a todos los dadores usados en la serie de experimentos. Esta variación
en Hif va asociada a un cambio en el factor preexponencial de la constante cinética a
través probablemente de cambios tanto en S ≠∆ como en el coeficiente de transmisión
χ . Por otro lado, a la par que varía el dador también varía la energía triplete del mismo.
Sería erróneo atribuir la variación en la constante preexponencial de la constante
cinética al cambio en la energía triplete del aceptror ( DTE ), en vez de al cambio en Hif.
Este podría ser una posible objeción al trabajo de Saltiel y colaboradores (Saltiel et al.,
1984) en el que atribuyen el comportamiento no-vertical a la variación en la entropía de
activación con el cambio en la energía triplete del dador.
6.4.4 Limitaciones del algoritmo e interpretación de resultados
En este punto se analizarán las limitaciones del algoritmo, las precauciones que han de
tomarse a la hora de interpretar los resultados computacionales y la relación con los
datos experimentales. Primeramente se expondrán las limitaciones debidas al método de
cálculo y al algoritmo, y en segundo lugar se hará lo mismo con respecto a la
comparación con los resultados experimentales.
-En primer lugar, el algoritmo hace uso de gradientes calculados para los estados
electrónicos S0 y T1 de la molécula aceptora para geometrías que se pueden hallar
relativamente lejanas a las de equilibrio, por lo tanto, se necesita usar un método que
describa bien toda la superficie de energía potencial. En general el método de Hartree-
Fock no resulta ser el adecuado debido a que no describe correctamente los sistemas de
capa abierta, y puede fallar también en la descripción de la SEP del estado fundamental
en regiones relativamente lejanas a la de equilibrio. En algunas ocasiones es posible
usar métodos como los basados en las teorías del funcional de la densidad (DFT), como
se ha hecho en el estudio de los estilbenos, métodos que se han demostrado útiles en la
descripción de estados triplete (T1) en hidrocarburos insaturados (Brink et al., 1998;
Brink et al., 2001). Teorías más elaboradas y de mayor coste computacional como son
las perturbacionales (por ejemplo MP2) o las multiconfiguracionales (por ejemplo
CASSCF) son a priori métodos que van a dar buenos resultados, pero que en algunos

312 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
casos son difíciles de utilizar debido al gran coste computacional. Por otro lado el
cálculo de la energía correspondiente a la transición S0 → T1 es posible realizarlo de
manera precisa con el método CASPT2 (Molcas v.5, 1999) que se ha aplicado al estudio
del comportamiento no-vertical en el 1,3,5,7-Ciclooctatetraeno (ver punto 6.3.2),
aunque en algunos casos, como los estilbenos, no ha sido posible aplicarlo. En cualquier
caso, si se pretende únicamente comparar la no-verticalidad de distintos dadores, se
puede recurrir al estudio de la dependencia de la energía de activación con la diferencia
( )A DT TE E− , con lo que esta posible causa de error (la incorrecta determinación de la
diferencia de energía S0→T1 para la transición vertical FC) se elimina, al no depender
este estudio de los valores absolutos de energía triplete del aceptor ( ATE ), sino relativos
( A DT TE E− ).
-Por otro lado el algoritmo hace uso de SEP que no incluyen la corrección de la energía
de activación debida al acoplamiento electrónico entre estados. Por lo tanto, la inclusión
de este efecto debe disminuir la energía de activación, aunque muy ligeramente, del
orden de las centenas de J/mol (~0.1 kcal/mol) (Levy y Speiser, 1992; Speiser, 1996),
siempre dependiendo de la naturaleza del aceptor, ya que en función de la capacidad de
solape electrónico con la molécula dadora, el acoplamiento y por lo tanto la energía del
mismo variarán.
-Muy importante también es la puntualización que se debe hacer sobre el cálculo de las
magnitudes termodinámicas de activación. Estos cálculos, tal y como se han realizado
para los estilbenos antes, son la contribución a las funciones termodinámicas
únicamente de la molécula dadora, es decir, cuando calculamos la energía libre de
activación (ver por ejemplo el punto anterior) estamos calculando únicamente la
contribución de la molécula aceptora (el correspondiente estilbeno en el punto anterior)
a dicha función termodinámica. A cada magnitud habría que añadir la contribución de la
molécula dadora, del disolvente además de tener en cuenta en su cálculo la interacción
entre todos ellos. Por lo tanto estas magnitudes calculadas no pueden en principio
correlacionarse con los valores experimentales.
Como ya se dijo antes, la influencia del disolvente en la constante de velocidad del
proceso es sin duda grande, pero afecta principalmente a la constante preexponencial de
la misma. Por otro lado, si elegimos una molécula dadora que no de lugar a importantes
reorganizaciones energéticas en el proceso de transferencia de energía triplete, sus

6.4 TET con Superficies de Energía Potencial Completas 313
funciones de partición en el estado fundamental y excitado serán parecidas, por lo que la
contribución de la misma a las funciones termodinámicas de activación va a ser
despreciables frente a la de la molécula aceptora. De esta manera, si bien el término
preexponencial no puede ser predicho en base al estudio de las SEP con el algoritmo
TET-ACC, el factor exponencial sí que puede ser descrito cuantitativamente.
Por último, se considera que la molécula dadora sólo puede ceder la energía DTE , que es
equivalente a suponer que el espectro de emisión S0←T1 está centrado en esta energía y
presenta una banda estrecha. Esta aproximación no debe presentar grandes dificultades,
incluso es beneficiosa desde el punto de vista conceptual, ya que nos permite estudiar
las propiedades de la molécula acpetora prescindiendo al máximo de la naturaleza de la
molécula dadora. En general se puede encontrar un dador que presente una emisión T1
→ S0 muy poco dispersa, de esta manera, la energía de la transición no depende
prácticamente de las coordenadas de la molécula a la temperatura de estudio, y por lo
tanto, se puede considerar constante. No obstante, si se quisiera estudiar la transferencia
entre un par de moléculas determinadas, simplemente ha de realizarse el cálculo del
gradiente para el complejo [*D···A] en vez de para [A] únicamente, y para el complejo
[D···A*] en vez de para [A*].
En lo que respecta a la relación con los datos experimentales, consideremos un
experimento en el que se miden las constantes de transferencia de energía desde
distintos dadores con energías triplete diversas al aceptor estudiado. Los resultados de
aplicar el algoritmo predicen la energía de activación del proceso en función de la
energía triplete del dador, sin embargo, no predicen los valores de la constante de
transferencia de energía.
La constante de velocidad experimental del proceso de transferencia de energía se puede
expresar según la ecuación (6.42) como:
exp
1
en d
d e
e e
kk k kk k− −
=+ +
(6.69)
Considerando que el proceso de transferencia reversible no compite con el de
transferencia directa, llegamos a la expresión:
( )exp 0log log log 1 exp Ten d
de
E Ekk kk RT
≠−
⎡ ⎤⎡ ⎤∆= − +⎢ ⎥⎢ ⎥
⎢ ⎥⎣ ⎦⎣ ⎦ (6.70)

314 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
donde se conoce la dependencia de ( )TE E≠ ∆ pero no el cociente 0d
e
kk− , mientras que
es conocido debido a que es aproximadamente el límite de velocidad del proceso de
transferencia.
dk
Por otro lado, si consideramos que los dadores poseen una estructura electrónica
similar, se puede demostrar (ver sección 6.3) que la contribución al término pre-
exponencial es muy similar para todos ellos, por lo que la ecuación (6.40) adopta
la forma:
( 0ek )
( ) ( )0 exp Te
eB
E Ek k T
k T
≠⎛ ⎞∆−⎜ ⎟⎝ ⎠
(6.71)
por lo tanto, el cociente 0d
e
kk− , puede tomarse como constante para dicha serie de
dadores. Conocida la dependencia funcional de la energía de activación con la energía
triplete del dador a través de los resultados obtenidos con la aplicación del algoritmo
TET-ACC, se pueden relacionar por último las medidas de las constantes de velocidad
con los resultados obtenidos ab initio de manera similar a como se hizo en el punto 6.3
para el COT.
6.4.5 Métodos
Se ha usado el funcional híbrido de tres parámetro de Becke (Becke, 1988; Becke,
1993) en combinación con el funcional de correlación de Lee, Yang y Parr (Lee et al.,
1988) tal y como está implementado en Gaussian98 (Gaussian98, 1998), junto con las
funciones de base 6-31G(d), para todos los cálculos de los estilbenos. En concreto,
energías y gradientes analíticos son los que se han calculado tanto en S0 como en T1
para construir la curva TET-ACC. La geometría de equilibrio en S0 de ambos estilbenos
se ha determinado al mismo nivel de cálculo usando gradientes analíticos y segundas
derivadas analíticas con el fin de caracterizar la naturaleza de los puntos estacionarios.
Asimismo las funciones de partición vibracionales a lo largo de la curva TET-ACC se
calcularon con hessianos de la energía analíticos al mismo nivel de cálculo, mientras
que el resto de funciones de partición no dependen del método usado en el cálculo de la
función de onda electrónica.

6.5 Conclusiones 315
66..55 CCoonncclluussiioonneess
El modelo propuesto en la primera parte de este capítulo que se aplicó al estudio del
comportamiento no-vertical del 1,3,5,7-Ciclooctatetraeno, así como el desarrollo del
algoritmo TET-ACC que generaliza el modelo inicial y que se ha aplicado al estudio del
cis- y trans-estilbeno, permiten establecer en base a los resultados obtenidos una serie de
conclusiones:
-En primer lugar hay que destacar que en el cálculo de la curva TET-ACC así como las
funciones de partición a lo largo de la misma para los estilbenos, es el término entálpico
el que marca el comportamiento no-vertical, y no el entrópico a través del factor
preexponencial de la constante cinética. Esto está en acuerdo con las conclusiones a las
que numerosos autores (ver texto) han llegado en el estudio de la no-verticalidad en el
cis-estilbeno y otros compuestos.
Además, puesto que la entropía de activación debe jugar un papel minoritario, en
general no será necesario (a no ser que se quiera una mayor precisión en los cálculos
teóricos) calcular las funciones termodinámicas de activación, y el análisis de la energía
potencial (electrónica) debe ser suficiente.
-El parámetro de distorsión geométrica (γ0) es en general un buen índice del
comportamiento no-vertical de una molécula aceptora, ya que para este tipo de TET se
espera un alto valor del gradiente en T1, siendo por lo tanto válida al aproximación de
primer orden. Cuando la molécula presenta unas SEP más complejas en las que la
aproximación de primer orden ya no sea válida es recomendable el uso de la curva TET-
ACC para determinar la dependencia del término exponencial de la constante de
transferencia de energía con la fuerza dirigente. Ambos métodos nos ofrecen las
distorsiones geométricas óptimas que debe sufrir la molécula aceptora para que se
verifique el proceso de TET. De hecho, puesto que el cálculo de γ0 suele ser bastante
sencillo, es posible establecer computacionalmente y al menos de forma cualitativa el
carácter no-vertical de una molécula aceptora.
-Los resultados de la aplicación de la aproximación lineal al cálculo de las propiedades
de COT están en acuerdo con los datos experimentales y muestran el muy marcado

316 6 TRANSFERENCIA DE ENERGÍA TRIPLETE
carácter no-vertical del COT como molécula aceptora en reacciones de TET. En esta
molécula en la que la rotación entorno a los enlaces dobles está prohibida el
comportamiento no-vertical está asociado a dos coordenadas principalmente: la
redistribución de la carga π alargando enlaces dobles C=C y acortando los simples C-C,
y la rotación entorno a los enlaces sencillos C-C.
-El cis-Estilbeno presenta un carácter no-vertical menor que el COT, aunque muy
marcado también. Al igual que en el COT, la redistribución de carga π es una de las
coordenadas importantes en el comportamiento no-vertical, al igual que la torsión
entorno al doble enlace central así como la correspondiente a la rotación de los fenilos.
Además la variación de estas dos coordenadas en el proceso de TET es muy parecida,
por lo que ninguna de las dos puede ser considerada como principal.
El trans-Estilbeno, que presenta un carácter no-vertical menor que el cis-Estilbeno,
también presenta una gran redistribución de carga π que hace disminuir notablemente la
diferencia de energías entre los estados S0 y T1.
-Por último remarcar que los criterios que determinan el carácter no-vertical de un
aceptor deben ser energéticos y no estructurales, y un buen criterio energético puede ser
el parámetro γ0, o la curva TET-ACC.


















