
DILANTIN* Dilantin DESCRIPCIÓN - Sitio Web del … · SJS/ NET, no se debe reanudar el uso de este...
28
1 DILANTIN* (Fenitoína sódica) Dilantin ® (cápsulas de fenitoína sódica de liberación prolongada) DESCRIPCIÓN Cada cápsula de Dilantin 100 mg (cápsula de fenitoína sódica de liberación prolongada) contiene 100 mg de fenitoína sódica. El comportamiento in vivo del producto se caracteriza por una tasa de absorción lenta y prolongada con concentraciones de pico plasmático previstas entre las 4 y 12 horas en contraste con las Cápsulas de fenitoína sódica rápidas, que tienen una rápida tasa de absorción con una concentración de pico plasmático prevista entre las 1½ y 3 horas. INDICACIONES Y USO Fenitoína está indicada para el control de las convulsiones tónico clónicas (gran mal) y de las convulsiones psicomotoras (lóbulo temporal) y en la prevención y el tratamiento de convulsiones ocurridas durante o después de una neurocirugía. Sería necesario realizar determinaciones del nivel de fenitoína sérica para el ajuste de la dosis óptima (consulte DOSIFICACIÓN Y ADMINISTRACIÓN). CONTRAINDICACIONES La fenitoína está contraindicada en pacientes con antecedentes de hipersensibilidad a la fenitoína, a sus componentes inactivos, o a otras hidantoínas. La administración concomitante de Dilantin está contraindicada con delavirdina debido a la posible pérdida de respuesta virológica y posible resistencia a la delavirdina, o a la clase de inhibidores de la transcriptasa inversa no nucleósidos. ADVERTENCIAS Efectos de la interrupción abrupta La fenitoína no se puede interrumpir abruptamente en pacientes epilépticos ya que puede precipitar el estado epiléptico. Cuando a juicio del clínico surja la necesidad de reducción de la dosificación, interrupción o sustitución por otra medicación anticonvulsivante alternativa, la
Transcript of DILANTIN* Dilantin DESCRIPCIÓN - Sitio Web del … · SJS/ NET, no se debe reanudar el uso de este...

1
DILANTIN*
(Fenitoína sódica)
Dilantin®
(cápsulas de fenitoína sódica de liberación prolongada)
DESCRIPCIÓN
Cada cápsula de Dilantin 100 mg (cápsula de fenitoína sódica de liberación prolongada)
contiene 100 mg de fenitoína sódica. El comportamiento in vivo del producto se caracteriza por
una tasa de absorción lenta y prolongada con concentraciones de pico plasmático previstas entre
las 4 y 12 horas en contraste con las Cápsulas de fenitoína sódica rápidas, que tienen una rápida
tasa de absorción con una concentración de pico plasmático prevista entre las 1½ y 3 horas.
INDICACIONES Y USO
Fenitoína está indicada para el control de las convulsiones tónico clónicas (gran mal) y de las
convulsiones psicomotoras (lóbulo temporal) y en la prevención y el tratamiento de convulsiones
ocurridas durante o después de una neurocirugía.
Sería necesario realizar determinaciones del nivel de fenitoína sérica para el ajuste de la dosis
óptima (consulte DOSIFICACIÓN Y ADMINISTRACIÓN).
CONTRAINDICACIONES
La fenitoína está contraindicada en pacientes con antecedentes de hipersensibilidad a la
fenitoína, a sus componentes inactivos, o a otras hidantoínas.
La administración concomitante de Dilantin está contraindicada con delavirdina debido a la
posible pérdida de respuesta virológica y posible resistencia a la delavirdina, o a la clase de
inhibidores de la transcriptasa inversa no nucleósidos.
ADVERTENCIAS
Efectos de la interrupción abrupta
La fenitoína no se puede interrumpir abruptamente en pacientes epilépticos ya que puede
precipitar el estado epiléptico. Cuando a juicio del clínico surja la necesidad de reducción de la
dosificación, interrupción o sustitución por otra medicación anticonvulsivante alternativa, la

2
implementación deberá ser gradual. En el caso de una reacción alérgica o de hipersensibilidad se
podría requerir la sustitución rápida por una terapia alternativa. En este caso, el tratamiento
alternativo debe consistir en una droga anticonvulsivante que no pertenezca a la clase química de
las hidantoínas.
Comportamiento suicida e ideación
Los fármacos antiepilépticos (FAEs), incluido Dilantin, aumentan el riesgo de ideas o
comportamientos suicidas en pacientes tratados con estos fármacos para cualquier indicación.
Los pacientes tratados con cualquier FAE para cualquier indicación deben ser monitoreados ante
la aparición o el empeoramiento de depresión, pensamientos o comportamientos suicidas, o
cualquier otro cambio inusual en el estado de ánimo o comportamiento.
Los análisis conjuntos de 199 ensayos clínicos controlados con placebo (monoterapia y terapia
adyuvante) de 11 FAEs diferentes mostraron que los pacientes asignados al azar a alguno de los
FAEs tuvieron aproximadamente el doble del riesgo (Riesgo Relativo ajustado 1.8, 95% IC: 1.2,
2.7) de tener ideación o comportamiento suicida comparados con los pacientes asignados al azar
con el grupo placebo. En estos ensayos, que tenían una duración de tratamiento media de 12
semanas, la tasa de incidencia estimada de comportamiento o ideación suicida era del 0.43%
entre 27863 pacientes tratados con FAEs comparada con una tasa del 0.24% entre 16029
pacientes tratados con placebo; se muestra el aumento de aproximadamente un caso de
pensamientos o comportamiento suicidas por cada 530 pacientes tratados. Ocurrieron cuatro
suicidios entre los pacientes tratados con la droga durante los ensayos y ninguno entre los
pacientes que recibían tratamiento con placebo, pero el número es demasiado pequeño como para
permitir cualquier conclusión sobre el efecto de la droga en el suicidio.
El mayor riesgo de ideación o comportamiento suicida con FAEs se observó tan pronto como en
la primera semana de comenzado el tratamiento con FAEs y continuó durante el tratamiento bajo
evaluación. No se pudo evaluar el riesgo de ideación o comportamiento suicida después de las 24
semanas porque la mayoría de los ensayos incluidos en los análisis no se extendían más allá de
las 24 semanas.
El riesgo de ideación o comportamiento suicida tenía, por lo general, consonancia entre las
drogas de la información analizada. El hallazgo de un mayor riesgo con FAEs de diferentes
mecanismos de acción y a través de un rango de indicaciones sugiere que este riesgo implica a
todos los FAEs utilizados para cualquier indicación. El riesgo no varió en forma sustancial en
relación a la edad (5 a 100 años) en los ensayos clínicos analizados.
La Tabla 1 muestra el riesgo absoluto y relativo por indicación para todos los FAEs evaluados.
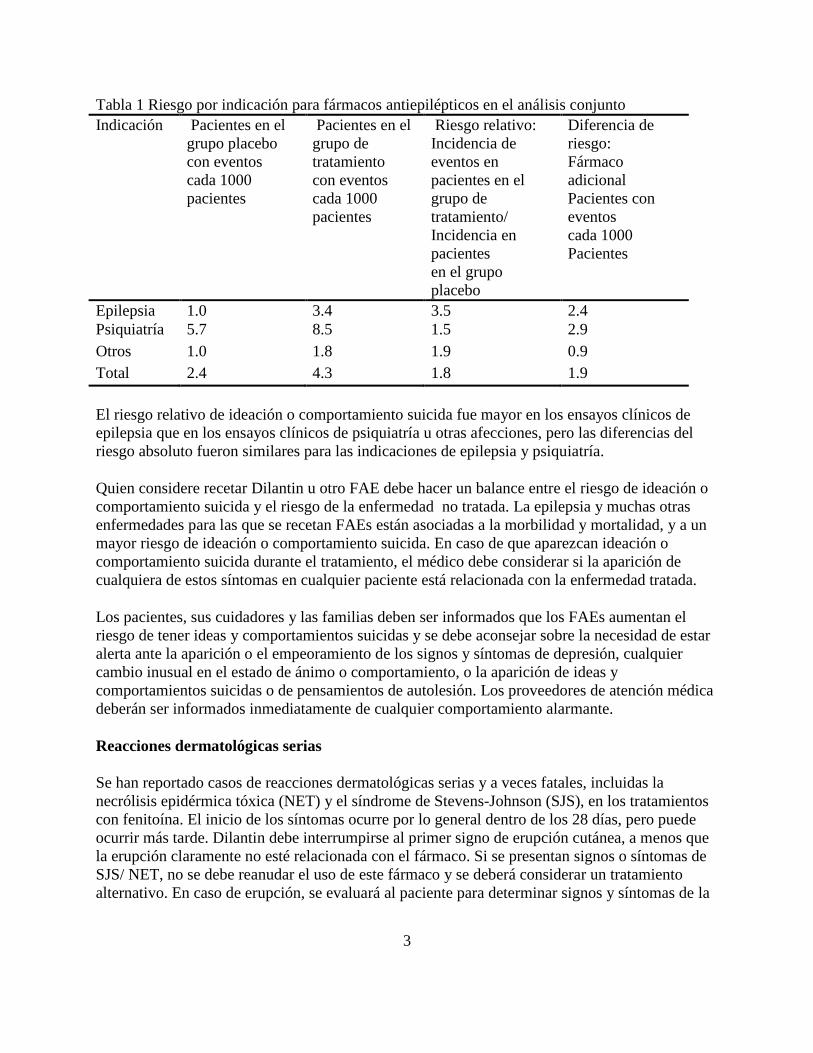
3
Tabla 1 Riesgo por indicación para fármacos antiepilépticos en el análisis conjunto
Indicación Pacientes en el
grupo placebo
con eventos
cada 1000
pacientes
Pacientes en el
grupo de
tratamiento
con eventos
cada 1000
pacientes
Riesgo relativo:
Incidencia de
eventos en
pacientes en el
grupo de
tratamiento/
Incidencia en
pacientes
en el grupo
placebo
Diferencia de
riesgo:
Fármaco
adicional
Pacientes con
eventos
cada 1000
Pacientes
Epilepsia 1.0 3.4 3.5 2.4
Psiquiatría 5.7 8.5 1.5 2.9
Otros 1.0 1.8 1.9 0.9
Total 2.4 4.3 1.8 1.9
El riesgo relativo de ideación o comportamiento suicida fue mayor en los ensayos clínicos de
epilepsia que en los ensayos clínicos de psiquiatría u otras afecciones, pero las diferencias del
riesgo absoluto fueron similares para las indicaciones de epilepsia y psiquiatría.
Quien considere recetar Dilantin u otro FAE debe hacer un balance entre el riesgo de ideación o
comportamiento suicida y el riesgo de la enfermedad no tratada. La epilepsia y muchas otras
enfermedades para las que se recetan FAEs están asociadas a la morbilidad y mortalidad, y a un
mayor riesgo de ideación o comportamiento suicida. En caso de que aparezcan ideación o
comportamiento suicida durante el tratamiento, el médico debe considerar si la aparición de
cualquiera de estos síntomas en cualquier paciente está relacionada con la enfermedad tratada.
Los pacientes, sus cuidadores y las familias deben ser informados que los FAEs aumentan el
riesgo de tener ideas y comportamientos suicidas y se debe aconsejar sobre la necesidad de estar
alerta ante la aparición o el empeoramiento de los signos y síntomas de depresión, cualquier
cambio inusual en el estado de ánimo o comportamiento, o la aparición de ideas y
comportamientos suicidas o de pensamientos de autolesión. Los proveedores de atención médica
deberán ser informados inmediatamente de cualquier comportamiento alarmante.
Reacciones dermatológicas serias
Se han reportado casos de reacciones dermatológicas serias y a veces fatales, incluidas la
necrólisis epidérmica tóxica (NET) y el síndrome de Stevens-Johnson (SJS), en los tratamientos
con fenitoína. El inicio de los síntomas ocurre por lo general dentro de los 28 días, pero puede
ocurrir más tarde. Dilantin debe interrumpirse al primer signo de erupción cutánea, a menos que
la erupción claramente no esté relacionada con el fármaco. Si se presentan signos o síntomas de
SJS/ NET, no se debe reanudar el uso de este fármaco y se deberá considerar un tratamiento
alternativo. En caso de erupción, se evaluará al paciente para determinar signos y síntomas de la

4
reacción por drogas con eosinofilia y síntomas sistémicos (consulte abajo DRESS/
hipersensibilidad multiorgánica).
Estudios en pacientes de origen chino han detectado una fuerte asociación entre el riesgo de
desarrollar SJS/NET y la presencia del HLA-B*1502, una variante alélica hereditaria del gen
HLA B, en pacientes que usan carbamazepina. Es escasa la evidencia que sugiere que el HLA-
B*1502 puede ser un factor de riesgo para el desarrollo de SJS/NET en pacientes de origen
asiático que consumen otros fármacos antiepilépticos asociados al SJS/NET, incluida la
fenitoína. Se debe considerar evitar suministrar fenitoína como tratamiento alternativo a la
carbamazepina en pacientes que son positivos para el HLA-B*1502.
El uso del genotipado del alelo HLA-B*1502 tiene limitaciones significativas y nunca deberá
sustituir el control clínico adecuado y el tratamiento del paciente. No se ha estudiado el rol de
otros posibles factores en el desarrollo y morbilidad del SJS/NET, como por ejemplo, dosis de
fármacos antiepilépticos (FAEs), cumplimiento, medicamentos concomitantes, comorbilidades y
el nivel de monitoreo dermatológico.
Reacción por drogas con eosinofilia y síntomas sistémicos (DRESS/hipersensibilidad
multiorgánica)
Se han reportado casos de reacción por drogas con eosinofilia y síntomas sistémicos (DRESS),
también conocida como hipersensibilidad multiorgánica, en pacientes que consumen fármacos
antiepilépticos, incluido Dilantin. Algunos de estos eventos han resultado fatales o
potencialmente mortales. Por lo general el síndrome DRESS se presenta, aunque no
exclusivamente, con fiebre, erupción o linfadenopatía, en asociación con otro sistema de
órganos, como hepatitis, nefritis, anormalidades hematológicas, miocarditis o miositis a veces
semejantes a una infección viral aguda. A menudo se desarrolla la eosinofilia. Dado que este
trastorno varía en su expresión, puede que haya otros sistemas de órganos involucrados que aquí
no se mencionaron. Es importante destacar que se pueden presentar manifestaciones tempranas
de hipersensibilidad, como fiebre y linfadenopatía, sin que se evidencie una erupción. Si se
presentan estos signos o síntomas, se deberá evaluar al paciente en forma inmediata. Se debe
interrumpir Dilantin si no se puede determinar una etiología alternativa de los signos o síntomas.
Hipersensibilidad
Dilantin y otras hidantoínas están contraindicados en pacientes que han experimentado
hipersensibilidad a la fenitoína (consulte CONTRAINDICACIONES). Además, considere
alternativas a fármacos estructuralmente similares como las carboxamidas (por ejemplo,
carbamazepina), barbitúricos, succinimidas y oxazolidinadionas (por ejemplo, la trimetadiona)
para estos mismos pacientes. De manera similar, considere alternativas al tratamiento con
Dilantin en caso de registrarse antecedentes de reacciones de hipersensibilidad a estos fármacos
de similar estructura en el paciente o familiares directos.

5
Lesión hepática
Se han reportado casos de hepatotoxicidad con Dilantin, incluidos casos poco frecuentes de
disfunción hepática aguda. Estos eventos pueden ser parte del espectro del DRESS o pueden
ocurrir en forma aislada. Otras manifestaciones comunes son ictericia, hepatomegalia, niveles
elevados de transaminasas en suero, leucocitosis y eosinofilia. El curso clínico de la
hepatotoxicidad aguda por fenitoína varía desde una recuperación rápida a resultados fatales. En
estos pacientes con hepatotoxicidad aguda, se deberá interrumpir Dilantin inmediatamente y no
se suministrará otra vez.
Sistema hematopoyético
Ocasionalmente, se han reportado complicaciones hematopoyéticas relacionadas con la
administración de Dilantin. Algunas de estas complicaciones fueron trombocitopenia,
leucopenia, granulocitopenia, agranulocitosis y pancitopenia con o sin supresión de la médula
ósea.
Varios informes sugieren una relación entre la fenitoína y el desarrollo de linfadenopatía (local o
generalizada) incluyendo hiperplasia nodular linfoide benigna, pseudolinfoma, linfoma y
enfermedad de Hodgkin. Si bien no se ha establecido una relación causa y efecto, la ocurrencia
de linfadenopatías señala la necesidad de diferenciar esta afección de otros tipos de patologías de
los ganglios linfáticos. La afectación de los ganglios linfáticos puede ocurrir con o sin síntomas o
signos de DRESS.
Para todos los casos de linfadenopatía, se indica un período extenso de seguimiento y se deberá
realizar el mejor esfuerzo para lograr controlar las convulsiones a través de fármacos
antiepilépticos alternativos.
Efectos sobre la vitamina D y huesos
El uso crónico de fenitoína en pacientes con epilepsia ha sido asociado con una disminución de
la densidad mineral ósea (osteopenia, osteoporosis y osteomalacia) y fractura de huesos. La
fenitoína induce enzimas metabolizadoras hepáticas. De esta manera puede reforzar el
metabolismo de la vitamina D y disminuir los niveles de vitamina D; y podría resultar en
deficiencia de vitamina D, hipocalcemia e hipofosfatemia. Se deberá considerar evaluar con
pruebas de hueso de laboratorio y exámenes radiológicos, según corresponda, e iniciar los planes
de tratamiento conforme a los lineamientos establecidos.
Efectos del consumo de alcohol en los niveles séricos de fenitoína
La ingestión aguda de alcohol puede aumentar los niveles séricos de fenitoína, mientras que el
uso crónico del alcohol puede disminuirlos.

6
Exacerbación de la porfiria
Dado que existen informes aislados que asocian a la fenitoína con la exacerbación de la porfiria,
se deberá tener precaución al utilizar este medicamento en pacientes con esa enfermedad.
Uso durante el embarazo: Clínica:
Riesgos para la madre. Puede ocurrir un aumento en la frecuencia de las convulsiones durante el
embarazo debido a la alteración de la farmacocinética de la fenitoína. La medición periódica de
las concentraciones de fenitoína plasmática puede ser de importancia en el tratamiento de
mujeres embarazadas como guía para un apropiado ajuste de la dosificación (consulte
PRECAUCIONES, Pruebas de Laboratorio). Sin embargo, es probable que se indique la
restitución de la dosificación original después del parto.
Riesgos para el feto. La paciente que consuma este fármaco durante el embarazo o la paciente
que quede embarazada durante el tratamiento de fenitoína deberá ser notificada de los posibles
daños al feto.
La exposición prenatal a la fenitoína puede aumentar los riesgos de malformaciones congénitas y
otros resultados adversos del desarrollo. Se ha informado sobre una mayor incidencia de
malformaciones congénitas importantes (tales como hendiduras orofaciales y defectos
cardíacos), anormalidades menores (rasgos faciales dismórficos, hipoplasia de uñas y dedos),
anormalidades del crecimiento (incluyendo la microcefalia) y deficiencias mentales en hijos de
mujeres epilépticas que consumían fenitoína solamente o de manera concomitante con otras
drogas anticonvulsivantes durante el embarazo. También se han reportado varios casos de
cáncer, incluyendo neuroblastoma, en niños cuyas madres consumían fenitoína durante el
embarazo. La incidencia total de malformaciones en niños de madres epilépticas tratadas con
fármacos antiepilépticos (fenitoína u otros) durante el embarazo es de aproximadamente el 10%
o del doble al triple que en la población general. Sin embargo, se desconocen los aportes
relativos de los fármacos antiepilépticos y otros factores asociados con la epilepsia a este mayor
riesgo, y en la mayoría de los casos no ha sido posible atribuir anormalidades del desarrollo
específicas a un fármaco antiepiléptico en particular.
Los pacientes deberán consultar con su médico para evaluar los riesgos y beneficios de la
fenitoína durante el embarazo.
Periodo postparto. Puede ocurrir un trastorno de sangrado potencialmente mortal relacionado
con niveles disminuidos de vitamina K dependientes de factores de coagulación en neonatos
expuestos a la fenitoína en el útero. Se puede prevenir esta afección inducida por el fármaco con
la administración de vitamina K a la madre antes del parto y al neonato después del parto.
No clínico.
La administración de fenitoína en animales preñadas provocó teratogenicidad (aumento de las
incidencias de malformaciones fetales) y otras toxicidades del desarrollo (incluida la mortalidad
embriofetal, el deterioro del crecimiento y las alteraciones del comportamiento) en varias

7
especies animales en dosis de importancia clínica.
PRECAUCIONES
General: El hígado es el sitio principal de biotransformación de la fenitoína; los pacientes con
función hepática disminuida, los de edad avanzada o los que se encuentran muy enfermos
podrían presentar signos tempranos de toxicidad.
Un porcentaje menor de los pacientes que recibieron tratamiento con fenitoína han metabolizado
el fármaco lentamente. El metabolismo lento puede deberse a una disponibilidad enzimática
limitada y falta de inducción; parece estar determinado genéticamente. Si aparecen señales
tempranas de toxicidad en el SNC relacionadas con el fármaco, se deberán revisar los niveles de
plasma inmediatamente.
Se ha documentado hiperglucemia como resultado del efecto inhibidor del fármaco en la
secreción de insulina. La fenitoína también puede elevar el nivel de glucosa plasmática en
pacientes diabéticos.
La fenitoína no está indicada en convulsiones debido a causas hipoglucémicas u otras
metabólicas. Los procedimientos de diagnóstico apropiados deberán realizarse según las
indicaciones.
La fenitoína no es efectiva en convulsiones de ausencias (pequeño mal). Si se presentan
convulsiones tónico clónicas (gran mal) y de ausencia (pequeño mal), se necesita un tratamiento
concomitante de fármacos.
Los niveles séricos de la fenitoína sostenidos sobre la escala óptima pueden producir estados de
confusión conocidos como “delirio”, “psicosis” o “encefalopatía” o raramente disfunción
cerebelosa irreversible y/o atrofia cerebelosa. En consecuencia, al primer signo de toxicidad
aguda, se recomienda la determinación de los niveles plasmáticos. Se recomienda una reducción
en la dosis del tratamiento de fenitoína, si los niveles plasmáticos son excesivos; y si los
síntomas persisten, se recomienda la finalización de la terapia con fenitoína (consulte
ADVERTENCIAS).
Información para los pacientes:
Indicar a los pacientes que deben tomar Dilantin solamente como fue prescripto.
A los pacientes que consumen fenitoína se les debe advertir sobre la importancia de seguir
estrictamente el régimen de dosificación indicado y de informar a su médico sobre cualquier
afección clínica que imposibilite tomar el fármaco oralmente, según se indicó, por ejemplo,
cirugía, etc.

8
Los pacientes deben conocer los signos de toxicidad temprana y los síntomas de posibles
reacciones hematológicas, dermatológicas, hipersensibles o hepáticas. Estos síntomas pueden
incluir, pero no limitarse a, fiebre, dolor de garganta, erupción, úlceras en la boca, fácil
formación de hematomas, linfadenopatía y hemorragia petequial o púrpura, y en el caso de
reacciones hepáticas, anorexia, náusea/vómito o ictericia. El paciente deberá entender que, como
estos signos y síntomas pueden indicar una reacción grave, tiene que informar cualquier
incidente al médico. Aún más, el paciente deberá saber que estos signos y síntomas deberán
reportarse aún si son leves o si ocurren después del uso prolongado.
Los pacientes deberán ser advertidos sobre la utilización de otros fármacos o bebidas alcohólicas
sin consultar primero a su médico.
Es importante enfatizar la importancia de la higiene dental para minimizar el desarrollo de
hiperplasia gingival y sus complicaciones.
Los pacientes, sus cuidadores y las familias deben ser informados que los FAEs aumentan el
riesgo de tener ideas y comportamientos suicidas, y se debe aconsejar sobre la necesidad de estar
alerta ante la aparición o el empeoramiento de los signos y síntomas de depresión, cualquier
cambio inusual en el estado de ánimo o comportamiento, o la aparición de ideas y
comportamientos suicidas o de pensamientos de autolesión. Las conductas preocupantes deben
ser reportadas de inmediato a los encargados del cuidado de la salud.
No consumir comprimidos que estén descoloridos.
Pruebas de laboratorio: Determinar el nivel sérico de fenitoína podría ser necesario para
realizar ajustes óptimos en la dosis. Las dosis de fenitoína se suelen seleccionar para obtener las
concentraciones de fenitoína sérica terapéutica totales entre 10 y 20 µg/ml (concentraciones de
fenitoína no ligada entre 1 y 2 µg/ml).
Interacciones medicamentosas: La fenitoína se liga ampliamente a las proteínas plasmáticas y
tiende al desplazamiento competitivo. Las enzimas CYP2C9 y CYP2C19 del citocromo hepático
P450 metabolizan la fenitoína, la cual es muy susceptible a la interacción con fármacos
inhibidores porque está sujeta al metabolismo saturable. La inhibición del metabolismo podría
producir aumentos significativos en las concentraciones de fenitoína circulante y aumentar el
riesgo de toxicidad del fármaco. La fenitoína es un inductor potente de las enzimas hepáticas que
metabolizan los fármacos. Las determinaciones de los niveles séricos de fenitoína son de gran
ayuda cuando se sospecha que existen interacciones farmacológicas.
Las interacciones farmacológicas más comunes se mencionan más abajo:
Nota: No se espera que la lista sea inclusiva o completa. Se deberá consultar el prospecto
individual de cada fármaco.

9
Fármacos que afectan las concentraciones de fenitoína:
Los fármacos que podrían aumentar los niveles séricos de fenitoína incluyen: consumo
agudo de alcohol, amiodarona, agentes antiepilépticos (etosuximida, felbamato,
oxcarbazepina, metsuximida, topiramato), azoles (fluconazol, ketoconazol, itraconazol,
miconazol, voriconazol), capecitabina, cloranfenicol, clordianzepóxido, disulfiram,
estrógenos, fluorouracilo, fluoxetina, fluvastatina, fluvoxamina, antagonistas H2 (por
ejemplo, cimetidina), halotano, isoniacida, metilfenidato, omeprazol, fenotiazinas,
salicilatos, sertralina, succinimida, sulfonamidas (por ejemplo, sulfametizol, sulfafenazol,
sulfadiacina, sulfametoxazol-trimetoprim), ticlopidina, tolbutamida, trazodona y
warfarina.
Los fármacos que podrían disminuir los niveles de fenitoína incluyen: medicamentos para
el cáncer usualmente en combinación (por ejemplo, bleomicina, carboplatino, cisplatino,
doxorrubicina, metotrexato), carbamazepina, el abuso crónico de alcohol, diazepam,
diazóxido, ácido fólico, fosamprenavir, nelfinavir, reserpina, rifampina, ritonavir, hierba
de San Juan, sucralfato, teofilina y vigabatrin.
La administración de fenitoína con preparados que aumentan el pH gástrico (por ejemplo,
suplementos o antiácidos que contienen carbonato de calcio, hidróxido de aluminio e
hidróxido de magnesio) pueden afectar la absorción de la fenitoína. En la mayoría de los
casos en que se han visto las interacciones, el efecto es una disminución en los niveles de
fenitoína cuando los medicamentos se toman al mismo tiempo. Cuando sea posible, no
deben tomarse fenitoína y estos productos a la misma hora del día.
Los fármacos que podrían disminuir o aumentar los niveles séricos de fenitoína incluyen:
fenobarbital, valproato de sodio y ácido valproico. De manera similar, los efectos de la
fenitoína sobre el nivel sérico de fenobarbital, ácido valpróico y valproato ácido son
impredecibles.
La incorporación o interrupción de estos agentes en pacientes en tratamiento con
fenitoína puede necesitar un ajuste en la dosis de fenitoína para lograr un resultado
clínico óptimo.
Fármacos influenciados por la fenitoína:
Fármacos que no deberán administrarse junto con la fenitoína: Delavirdina (ver
CONTRAINDICACIONES).
Fármacos que disminuyen su eficacia con la fenitoína incluyen: azoles (fluconazol,
ketoconazol, itraconazol, voriconazol), posaconazol, corticosteroides, doxiciclina,
estrógenos, furosemida, irinotecán, anticonceptivos orales, paclitaxel, paroxetina,
quinidina, rifampicina, sertralina, tenipósido, teofilina y vitamina D.
Se han documentado respuestas PT/INR aumentadas o disminuidas cuando se
administran fenitoína y warfarina juntas.
La fenitoína disminuye las concentraciones plasmáticas de metabolitos activos de
albendazol, algunos antivirales para el tratamiento del VIH (efavirenz,
lopinavira/ritonavir, indinavir, nelfinavir, ritonavir, saquinavir), agentes antiepilépticos
(carbamazepina, felbamato, lamotrigina, topiramato, oxcarbazepina, quetiapina),

10
atorvastatina, cloropropamida, clozapina, ciclosporina, digoxina, fluvastatina, ácido
fólico, metadona, mexiletina, nifedipina, nimodipina, nisoldipina, praziquantel,
simvastatina y verapamilo.
Cuando se administra fenitoína con sólo fosamprenavir puede disminuir la concentración
de amprenavir, el metabolito activo. Cuando se administra fenitoína con la combinación
de fosamprenavir y ritonavir, puede aumentar la concentración de amprenavir.
Ha ocurrido resistencia a la acción de bloqueo neuromuscular de los agentes
bloqueadores neuromusculares no despolarizantes pancuronio, vecuronio, rocuronio y
cisatracurio en pacientes a los que se administra fenitoína a largo plazo. Se desconoce si
la fenitoína tiene o no el mismo efecto sobre otros agentes no despolarizantes. Debe
monitorearse estrechamente a los pacientes para detectar una recuperación del bloqueo
neuromuscular más rápida de lo esperada y los requisitos de tasa de infusión pueden ser
más altos.
La incorporación o interrupción de la fenitoína durante el tratamiento concomitante con
estos agentes puede requerir ajustes en la dosis de estos agentes para obtener un resultado
clínico óptimo.
Interacción del medicamento con preparaciones nutritivas/ alimentación enteral: La
literatura sugiere que los pacientes que han recibido preparaciones alimenticias enterales y/o
suplementos nutricionales relacionados presentan un menor nivel de fenitoína plasmática. Por lo
tanto, se sugiere que no se administre concomitantemente fenitoína con preparaciones
alimenticias enterales. Podrían necesitarse controles más frecuentes de los niveles plasmáticos de
fenitoína en estos pacientes.
Interacciones con pruebas de laboratorio/medicamentosas: La fenitoína puede disminuir las
concentraciones séricas de T4. Puede producir valores por debajo de lo normal en las pruebas de
dexametasona y metirapona. La fenitoína puede producir niveles séricos elevados de glucosa,
fosfatasa alcalina y gama glutamil transpeptidasa (GGT).
Se deberá tener cuidado al utilizar métodos inmunoanalíticos para medir las concentraciones
plasmáticas de fenitoína.
Carcinogénesis, mutagénesis, deterioro de la fertilidad
Carcinogénesis: Consulte la sección de ADVERTENCIAS (sistema hematopoyético)
En los estudios de carcinogenicidad, se administró fenitoína con la dieta a ratones (10, 25 o
45 mg/kg/día) y a ratas (25, 50 o 100 mg/kg/día) durante 2 años. La incidencia de tumores
hepatocelulares aumentó en ratones machos y hembras con la dosis más alta. No se observaron
incidencias de tumores en ratas. Las dosis más altas analizadas en estos estudios se asociaron con
picos en los niveles plasmáticos de fenitoína por debajo de las concentraciones terapéuticas en
seres humanos.
En los estudios sobre carcinogenicidad informados en la bibliografía, se administró fenitoína con
la dieta durante 2 años en dosis de hasta 600 ppm (aproximadamente 90 mg/kg/día) a ratones y
de hasta 2400 ppm (aproximadamente 120 mg/kg/día) a ratas. Las incidencias de tumores

11
hepatocelulares aumentaron en ratones hembra en todas las dosis analizadas, excepto en la dosis
más baja. En ratas no se observaron aumentos en la incidencia de tumores.
Mutagénesis
La fenitoína fue negativa en la prueba de Ames y en el ensayo in vitro de clastogenicidad en las
células ováricas del hámster chino (CHO).
En los estudios informados en la bibliografía, la fenitoína fue negativa en el ensayo in vitro de
linfoma en ratones y en el ensayo in vivo de micronúcleos en ratones. La fenitoína fue
clastogénica en el ensayo in vitro de intercambio de cromátidas hermanas en las células CHO.
Fertilidad
No se realizó una evaluación adecuada de los efectos de la fenitoína en la fertilidad en machos ni
en hembras.
Embarazo:
Consulte la sección ADVERTENCIAS.
Madres lactantes: No se recomienda que las mujeres que utilizan este fármaco amamanten
niños porque la fenitoína parece secretarse en bajas concentraciones en la leche humana.
Uso pediátrico: Consulte la sección de DOSIS Y ADMINISTRACIÓN.
Uso geriátrico: La depuración de la fenitoína tiende a disminuir con el aumento de la edad.
REACCIONES ADVERSAS
Cuerpo en general: Se han observado reacciones alérgicas en forma de erupción y raramente en
formas más graves (consulte el párrafo Piel y apéndices abajo) y el DRESS (consulte
ADVERTENCIAS). También se ha documentado anafilaxia.
También se ha informado tosquedad en los rasgos faciales, lupus eritematoso sistémico,
periarteritis nodosa y alteraciones en la inmunoglobulina.
Sistema nervioso: Las reacciones adversas más comunes presentadas durante el tratamiento con
fenitoína son reacciones en el sistema nervioso y suelen estar relacionadas con la dosis. Las
reacciones incluyen nistagmo, ataxia, dificultad al hablar, disminución en la coordinación y
confusión mental. También se han documentado mareos, vértigo, insomnio, nerviosismo
transitorio, espasmos motores, parestesias y dolores de cabeza. También se ha informado
raramente que la fenitoína inducía discinesias, que incluyen corea, distonia, temblor y asterixis
similares a las inducidas por la fenotiazina y otros fármacos neurolépticos. Se han informado
casos de atrofia cerebelosa, que parece producirse más probablemente en entornos con niveles
elevados de fenitoína y/o uso de fenitoína a largo plazo.

12
Se ha observado una polineuropatía periférica predominantemente sensorial en pacientes con
tratamiento de fenitoína a largo plazo.
Sistema digestivo: Disfunción hepática aguda, hepatitis tóxica, lesión hepática, náuseas, vómito,
estreñimiento, aumento de tamaño de los labios e hiperplasia gingival.
Piel y apéndices: Las manifestaciones dermatológicas, a veces acompañadas por fiebre,
incluyen salpullidos escarlatiniformes o morbiliformes. Un salpullido morbiliforme (parecido a
la rubeola) es el más común, los otros tipos de dermatitis son más raros. Otras formas más graves
que podrían resultar mortales incluyen dermatitis bullosa, exfoliativa o purpúrica, síndrome de
Stevens-Johnson y necrólisis epidérmica tóxica (consulte ADVERTENCIAS). También se han
documentados casos de hipertricosis.
Sistema hematológico y linfático: Se han documentado ocasionalmente complicaciones
hematopoyéticas, algunas mortales, asociadas con la administración de fenitoína. Estas
incluyeron trombocitopenia, leucopenia, granulocitopenia, agrunolocitosis y pancitopenia con o
sin supresión de la médula ósea. Aunque ha habido casos de macrocitosis o anemia
megaloblástica, estas patologías responden al tratamiento con ácido fólico. Se ha documentado
linfadenopatía que incluye hiperplasia de los ganglios linfáticos, pseudolinfoma, linfoma y
enfermedad de Hodgkin (consulte ADVERTENCIAS).
Sentidos especiales: El sentido alterado del gusto incluye gusto metálico.
Urogenital: Enfermedad de Peyronie
SOBREDOSIFICACIÓN
No se conoce la dosis mortal en pacientes pediátricos. La dosis mortal en adultos se estima entre
2 y 5 gramos. Los síntomas iniciales son nistagmo, ataxia y disartria. Otros signos son temblor,
hiperreflexia, letargo, habla dificultosa, visión borrosa, náusea y vómito. El paciente puede entrar
en estado comatoso y sufrir hipotensión. La muerte se debe a la disminución de la respiración y
circulación.
Hay variaciones marcadas entre los individuos con respecto a los niveles plasmáticos de
fenitoína en los que ocurre la toxicidad. El nistagmo o mirada lateral usualmente aparece con 20
mcg/ml, la ataxia con 30 mcg/ml, la disartria y letargia aparecen cuando la concentración
plasmática es > 40 mcg/ml; sin embargo, se han documentado concentraciones tan altas como 50
mcg/ml sin evidencia de toxicidad . Se han tomado cantidades de hasta 25 veces la dosis
terapéutica, dando como resultado concentraciones plasmáticas de más de 100 mcg/ml con una
recuperación completa. Se han informado casos de disfunción y atrofia cerebelosa irreversible.
Tratamiento: El tratamiento es no específico dado que no hay antídoto conocido.
Se debe observar cuidadosamente la capacidad de los sistemas respiratorios y circulatorios y
emplear medidas de apoyo apropiadas. Se puede considerar la hemodiálisis dado que la fenitoína

13
no se liga completamente a las proteínas plasmáticas. Se ha utilizado la transfusión total de
intercambio como tratamiento para la intoxicación grave en pacientes pediátricos.
En caso de sobredosificación aguda se deberá considerar la posible presencia de otros supresores
del SNC como el alcohol.
DOSIS Y ADMINISTRACIÓN
Deberán monitorearse las concentraciones séricas para cambiar de Dilantin (cápsula de fenitoína
sódica de liberación prolongada) a las Cápsulas de fenitoína sódica de acción rápida, y de las
sales sódicas a la forma libre del ácido de fenitoína.
Dilantin (cápsula de fenitoína sódica de liberación prolongada) está formulado con la sal sódica
de la fenitoína. La forma libre del ácido de fenitoína se utiliza en la suspensión de Dilantin-125
y en las cápsulas infantiles, Dilantin Infatabs. Como existe aproximadamente un aumento del 8%
en el contenido del fármaco con la forma libre de ácido sobre la de sal sódica, ajustes en la
dosificación y un monitoreo del nivel sérico pueden ser necesarios cuando se hace el cambio de
un producto formulado con ácido libre a un producto formulado con sal sódica y viceversa.
General: La dosificación deberá ser individual para garantizar el máximo beneficio. En algunos
casos, las determinaciones del nivel sérico podrán ser necesarias para los ajustes de dosis
óptimos; el nivel plasmático clínico efectivo suele ser de 10 a 20 mcg/ml. Con la dosis
recomendada, puede ser necesario un periodo de siete a diez días para alcanzar niveles
plasmáticos de fenitoína en estado estacionario y los cambios en la dosis (aumento o
disminución) no deberán realizarse con intervalos menores de siete a diez días.
Dosificación en adultos:
Dosis diaria dividida: Los pacientes que no han recibido tratamiento previo pueden comenzar
con un Dilantin 100 mg (cápsula de fenitoína sódica de liberación prolongada) tres veces por día
y la dosis puede ajustarse para cumplir con requerimientos individuales. Para la mayoría de los
adultos, la dosis de mantenimiento satisfactoria será una cápsula entre tres y cuatro veces diarias.
En caso de ser necesario, se puede aumentar a dos cápsulas tres veces por día.
Dosificación una vez por día: En los adultos, si el control convulsivo se establece con dosis
divididas de tres Dilantin 100 mg (cápsula de fenitoína sódica de liberación prolongada) diaria,
podría considerarse una dosis diaria de Dilantin 300 mg (cápsula de fenitoína sódica de
liberación prolongada). Estudios comparativos entre las dosis divididas de 300 mg y una dosis
diaria de esta cantidad mostraron que la absorción, los picos máximos en los niveles plasmáticos,
la vida media biológica, la diferencia entre los valores máximos y mínimos y la recuperación
urinaria eran equivalentes. La dosis de una vez al día es más conveniente para los pacientes
individuales o para el personal de cuidado de los pacientes hospitalizados y debe ser utilizado
solamente en los pacientes que necesiten esta dosis de fármaco diaria. El gran problema para
motivar a los pacientes incumplidores podría disminuirse si el paciente puede tomar el fármaco

14
una vez al día. Sin embargo, los pacientes deberán tener la precaución de no olvidar una dosis
involuntariamente.
Solamente se recomienda Dilantin (cápsula de fenitoína sódica de liberación prolongada) para la
dosificación una vez al día. Las diferencias inherentes en las características de disolución y tasas
de absorción resultantes de la fenitoína debido a los diferentes procesos de fabricación y/o forma
farmacéutica excluyen la recomendación de las otras formas de productos de fenitoína. Se debe
realizar un monitoreo cuidadoso de los niveles séricos de fenitoína cuando se recomiendan
cambios en la dosis o en la marca comercial.
Dosis de carga: Algunos especialistas han recomendado el uso de dosis de carga oral de
fenitoína en adultos que necesitan niveles plasmáticos de estado estacionario urgente y donde no
se recomienda la administración intravenosa. La administración de la dosis deberá reservarse
para pacientes en clínicas u hospitales donde puede monitorearse atentamente los niveles
plasmáticos de fenitoína. Los pacientes con antecedentes de enfermedades renales o hepáticas no
deberían recibir la dosis de carga oral.
Al comienzo se divide un gramo de Dilantin (cápsula de fenitoína sódica de liberación
prolongada) en tres dosis (400 mg, 300 mg, 300 mg) y se administra con intervalos de dos horas.
La dosis de mantenimiento normal se implementa 24 horas después de la dosis de carga, con
determinaciones de nivel sérico frecuentes.
Dosis en poblaciones especiales
Pacientes con enfermedad hepática o renal: La interpretación de la concentración total de
fenitoína en el plasma deberá realizarse cuidadosamente debido al aumento de fracciones no
ligadas de fenitoína en pacientes con enfermedades hepáticas o renales, o en los que sufren
hipoalbuminemia. Las concentraciones de fenitoína no ligadas pueden resultar más útiles en
estas poblaciones de pacientes. Pacientes de edad avanzada: La depuración disminuye
ligeramente en los pacientes de edad avanzada y pueden necesitar dosis menores o menos
frecuentes. Pediátrico: Al comienzo, deben administrarse 5 mg/kg/día en dos o tres dosis
divididas equitativamente, con una dosificación subsiguiente individual de hasta un máximo de
300 mg al día. La dosis diaria de mantenimiento recomendada es, generalmente, de 4 a 8 mg/kg.
Los niños mayores a 6 años y adolescentes pueden necesitar la dosis mínima recomendada en
adultos (300 mg/día).
Almacenar a temperatura menor de 30 oC. Proteger de la luz y la humedad.
Presentaciones comerciales
Frasco con 100 cápsulas
No todas las presentaciones comerciales se encuentran disponibles en cada país.
Para mayor información contactar al departamento de información médica de Pfizer a:

15
Fecha de Revisión: Diciembre de 2015
Documento de referencia: USPI LAB-0375-19.0
Para la región de Centroamérica y Caribe ver información referente a titular, fabricante y
empacador en el empaque secundario.

16
DILANTIN*
(phenytoin sodium)
Dilantin®
(extended phenytoin sodium capsules)
DESCRIPTION
Each 100 mg Dilantin (extended phenytoin sodium capsule) contains 100 mg phenytoin sodium.
Product in vivo performance is characterized by a slow and extended rate of absorption with peak
blood concentrations expected in 4 to 12 hours as contrasted to Prompt Phenytoin Sodium
Capsules, with a rapid rate of absorption with peak blood concentration expected in 1½ to 3
hours.
INDICATIONS AND USAGE
Phenytoin is indicated for the control of tonic-clonic (grand mal) and psychomotor (temporal
lobe) seizures and prevention and treatment of seizures occurring during or following
neurosurgery.
Phenytoin serum level determinations may be necessary for optimal dosage adjustments (see
DOSAGE AND ADMINISTRATION).
CONTRAINDICATIONS
Phenytoin is contraindicated in those patients with a history of hypersensitivity to phenytoin, its
inactive ingredients, or other hydantoins.
Coadministration of Dilantin is contraindicated with delavirdine due to potential for loss of
virologic response and possible resistance to delavirdine or to the class of non-nucleoside reverse
transcriptase inhibitors.
WARNINGS
Effects of Abrupt Withdrawal
Abrupt withdrawal of phenytoin in epileptic patients may precipitate status epilepticus. When, in
the judgment of the clinician, the need for dosage reduction, discontinuation, or substitution of
17
alternative anticonvulsant medication arises, this should be done gradually. In the event of an
allergic or hypersensitivity reaction, more rapid substitution of alternative therapy may be
necessary. In this case, alternative therapy should be an anticonvulsant drug not belonging to the
hydantoin chemical class.
Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including Dilantin, increase the risk of suicidal thoughts or behavior
in patients taking these drugs for any indication. Patients treated with any AED for any
indication should be monitored for the emergence or worsening of depression, suicidal thoughts
or behavior, and/or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11
different AEDs showed that patients randomized to one of the AEDs had approximately twice
the risk (adjusted Relative Risk 1.8, 95% CI:1.2, 2.7) of suicidal thinking or behavior compared
to patients randomized to placebo. In these trials, which had a median treatment duration of 12
weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED-treated
patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an
increase of approximately one case of suicidal thinking or behavior for every 530 patients
treated. There were four suicides in drug-treated patients in the trials and none in placebo-treated
patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as one
week after starting drug treatment with AEDs and persisted for the duration of treatment
assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk
of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data
analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a
range of indications suggests that the risk applies to all AEDs used for any indication. The risk
did not vary substantially by age (5 to 100 years) in the clinical trials analyzed.
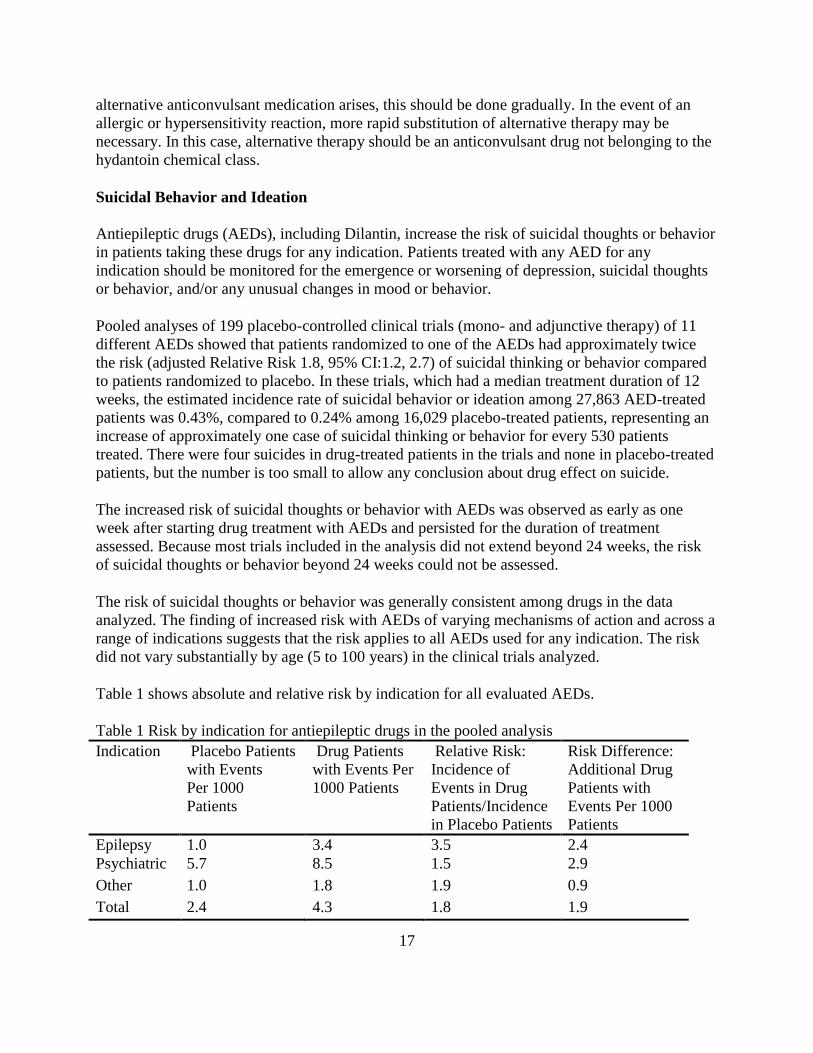
Table 1 shows absolute and relative risk by indication for all evaluated AEDs.
Table 1 Risk by indication for antiepileptic drugs in the pooled analysis
Indication Placebo Patients
with Events
Per 1000
Patients
Drug Patients
with Events Per
1000 Patients
Relative Risk:
Incidence of
Events in Drug
Patients/Incidence
in Placebo Patients
Risk Difference:
Additional Drug
Patients with
Events Per 1000
Patients
Epilepsy 1.0 3.4 3.5 2.4
Psychiatric 5.7 8.5 1.5 2.9
Other 1.0 1.8 1.9 0.9
Total 2.4 4.3 1.8 1.9

18
The relative risk for suicidal thoughts or behavior was higher in clinical trials for epilepsy than in
clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for
the epilepsy and psychiatric indications.
Anyone considering prescribing Dilantin or any other AED must balance the risk of suicidal
thoughts or behavior with the risk of untreated illness. Epilepsy and many other illnesses for
which AEDs are prescribed are themselves associated with morbidity and mortality and an
increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge
during treatment, the prescriber needs to consider whether the emergence of these symptoms in
any given patient may be related to the illness being treated.
Patients, their caregivers, and families should be informed that AEDs increase the risk of suicidal
thoughts and behavior and should be advised of the need to be alert for the emergence or
worsening of the signs and symptoms of depression, any unusual changes in mood or behavior,
or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Behaviors of
concern should be reported immediately to healthcare providers.
Serious Dermatologic Reactions
Serious and sometimes fatal dermatologic reactions, including toxic epidermal necrolysis (TEN)
and Stevens-Johnson syndrome (SJS), have been reported with phenytoin treatment. The onset of
symptoms is usually within 28 days, but can occur later. Dilantin should be discontinued at the
first sign of a rash, unless the rash is clearly not drug-related. If signs or symptoms suggest
SJS/TEN, use of this drug should not be resumed and alternative therapy should be considered. If
a rash occurs, the patient should be evaluated for signs and symptoms of Drug Reaction with
Eosinophilia and Systemic Symptoms (see DRESS/Multiorgan hypersensitivity below).
Studies in patients of Chinese ancestry have found a strong association between the risk of
developing SJS/TEN and the presence of HLA-B*1502, an inherited allelic variant of the HLA B
gene, in patients using carbamazepine. Limited evidence suggests that HLA-B*1502 may be a
risk factor for the development of SJS/TEN in patients of Asian ancestry taking other
antiepileptic drugs associated with SJS/TEN, including phenytoin. Consideration should be given
to avoiding phenytoin as an alternative for carbamazepine in patients positive for HLA-B*1502.
The use of HLA-B*1502 genotyping has important limitations and must never substitute for
appropriate clinical vigilance and patient management. The role of other possible factors in the
development of, and morbidity from, SJS/TEN, such as antiepileptic drug (AED) dose,
compliance, concomitant medications, comorbidities, and the level of dermatologic monitoring
have not been studied.
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan
hypersensitivity
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), also known as Multiorgan
hypersensitivity, has been reported in patients taking antiepileptic drugs, including Dilantin.

19
Some of these events have been fatal or life-threatening. DRESS typically, although not
exclusively, presents with fever, rash, and/or lymphadenopathy, in association with other organ
system involvement, such as hepatitis, nephritis, hematological abnormalities, myocarditis, or
myositis sometimes resembling an acute viral infection. Eosinophilia is often present. Because
this disorder is variable in its expression, other organ systems not noted here may be involved. It
is important to note that early manifestations of hypersensitivity, such as fever or
lymphadenopathy, may be present even though rash is not evident. If such signs or symptoms are
present, the patient should be evaluated immediately. Dilantin should be discontinued if an
alternative etiology for the signs or symptoms cannot be established.
Hypersensitivity
Dilantin and other hydantoins are contraindicated in patients who have experienced phenytoin
hypersensitivity (see CONTRAINDICATIONS). Additionally, consider alternatives to
structurally similar drugs such as carboxamides (e.g., carbamazepine), barbiturates,
succinimides, and oxazolidinediones (e.g., trimethadione) in these same patients. Similarly, if
there is a history of hypersensitivity reactions to these structurally similar drugs in the patient or
immediate family members, consider alternatives to Dilantin.
Hepatic Injury
Cases of acute hepatotoxicity, including infrequent cases of acute hepatic failure, have been
reported with Dilantin. These events may be part of the spectrum of DRESS or may occur in
isolation. Other common manifestations include jaundice, hepatomegaly, elevated serum
transaminase levels, leukocytosis, and eosinophilia. The clinical course of acute phenytoin
hepatotoxicity ranges from prompt recovery to fatal outcomes. In these patients with acute
hepatotoxicity, Dilantin should be immediately discontinued and not readministered.
Hematopoietic System
Hematopoietic complications, some fatal, have occasionally been reported in association with
administration of Dilantin. These have included thrombocytopenia, leukopenia,
granulocytopenia, agranulocytosis, and pancytopenia with or without bone marrow suppression.
There have been a number of reports suggesting a relationship between phenytoin and the
development of lymphadenopathy (local or generalized) including benign lymph node
hyperplasia, pseudolymphoma, lymphoma, and Hodgkin’s disease. Although a cause and effect
relationship has not been established, the occurrence of lymphadenopathy indicates the need to
differentiate such a condition from other types of lymph node pathology. Lymph node
involvement may occur with or without symptoms and signs of DRESS.
In all cases of lymphadenopathy, follow-up observation for an extended period is indicated and
every effort should be made to achieve seizure control using alternative antiepileptic drugs.

20
Effects on Vitamin D and Bone
The chronic use of phenytoin in patients with epilepsy has been associated with decreased bone
mineral density (osteopenia, osteoporosis, and osteomalacia) and bone fractures. Phenytoin
induces hepatic metabolizing enzymes. This may enhance the metabolism of vitamin D and
decrease vitamin D levels, which may lead to vitamin D deficiency, hypocalcemia, and
hypophosphatemia. Consideration should be given to screening with bone-related laboratory and
radiological tests as appropriate and initiating treatment plans according to established
guidelines.
Effects of Alcohol Use on Phenytoin Serum Levels
Acute alcoholic intake may increase phenytoin serum levels, while chronic alcohol use may
decrease serum levels.
Exacerbation of Porphyria
In view of isolated reports associating phenytoin with exacerbation of porphyria, caution should
be exercised in using this medication in patients suffering from this disease.
Usage In Pregnancy:
Clinical:
Risks to Mother. An increase in seizure frequency may occur during pregnancy because of
altered phenytoin pharmacokinetics. Periodic measurement of plasma phenytoin concentrations
may be valuable in the management of pregnant women as a guide to appropriate adjustment of
dosage (see PRECAUTIONS, Laboratory Tests). However, postpartum restoration of the
original dosage will probably be indicated.
Risks to the Fetus. If this drug is used during pregnancy, or if the patient becomes pregnant while
taking the drug, the patient should be apprised of the potential harm to the fetus.
Prenatal exposure to phenytoin may increase the risks for congenital malformations and other
adverse developmental outcomes. Increased frequencies of major malformations (such as
orofacial clefts and cardiac defects), minor anomalies (dysmorphic facial features, nail and digit
hypoplasia), growth abnormalities (including microcephaly), and mental deficiency have been
reported among children born to epileptic women who took phenytoin alone or in combination
with other antiepileptic drugs during pregnancy. There have also been several reported cases of
malignancies, including neuroblastoma, in children whose mothers received phenytoin during
pregnancy. The overall incidence of malformations for children of epileptic women treated with
antiepileptic drugs (phenytoin and/or others) during pregnancy is about 10%, or two- to three-
fold that in the general population. However, the relative contributions of antiepileptic drugs and
other factors associated with epilepsy to this increased risk are uncertain and in most cases it has
not been possible to attribute specific developmental abnormalities to particular antiepileptic
drugs.

21
Patients should consult with their physicians to weigh the risks and benefits of phenytoin during
pregnancy.
Postpartum Period. A potentially life-threatening bleeding disorder related to decreased levels of
vitamin K dependent clotting factors may occur in newborns exposed to phenytoin in utero. This
drug-induced condition can be prevented with vitamin K administration to the mother before
delivery and to the neonate after birth.
Nonclinical.
Administration of phenytoin to pregnant animals resulted in teratogenicity (increased incidences
of fetal malformations) and other developmental toxicity (including embryofetal death, growth
impairment, and behavioral abnormalities) in multiple animal species at clinically relevant doses.
PRECAUTIONS
General: The liver is the chief site of biotransformation of phenytoin; patients with impaired
liver function, elderly patients, or those who are gravely ill may show early signs of toxicity.
A small percentage of individuals who have been treated with phenytoin have been shown to
metabolize the drug slowly. Slow metabolism may be due to limited enzyme availability and
lack of induction; it appears to be genetically determined. If early signs of dose-related CNS
toxicity develop, plasma levels should be checked immediately.
Hyperglycemia, resulting from the drug’s inhibitory effects on insulin release, has been reported.
Phenytoin may also raise the serum glucose level in diabetic patients.
Phenytoin is not indicated for seizures due to hypoglycemic or other metabolic causes.
Appropriate diagnostic procedures should be performed as indicated.
Phenytoin is not effective for absence (petit mal) seizures. If tonic-clonic (grand mal) and
absence (petit mal) seizures are present, combined drug therapy is needed.
Serum levels of phenytoin sustained above the optimal range may produce confusional states
referred to as “delirium,” “psychosis,” or “encephalopathy,” or rarely irreversible cerebellar
dysfunction and/or cerebellar atrophy. Accordingly, at the first sign of acute toxicity, plasma
levels are recommended. Dose reduction of phenytoin therapy is indicated if plasma levels are
excessive; if symptoms persist, termination is recommended. (See WARNINGS section.)
Information for Patients:
Instruct patients to take Dilantin only as prescribed.
Patients taking phenytoin should be advised of the importance of adhering strictly to the
prescribed dosage regimen, and of informing the physician of any clinical condition in which it is
not possible to take the drug orally as prescribed, e.g., surgery, etc.

22
Patients should be made aware of the early toxic signs and symptoms of potential hematologic,
dermatologic, hypersensitivity, or hepatic reactions. These symptoms may include, but are not
limited to, fever, sore throat, rash, ulcers in the mouth, easy bruising, lymphadenopathy and
petechial or purpuric hemorrhage, and in the case of liver reactions, anorexia, nausea/vomiting,
or jaundice. The patient should be advised that, because these signs and symptoms may signal a
serious reaction, that they must report any occurrence immediately to a physician. In addition,
the patient should be advised that these signs and symptoms should be reported even if mild or
when occurring after extended use.
Patients should also be cautioned on the use of other drugs or alcoholic beverages without first
seeking the physician’s advice.
The importance of good dental hygiene should be stressed in order to minimize the development
of gingival hyperplasia and its complications.
Patients, their caregivers, and families should be counseled that AEDs, including Dilantin, may
increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert
for the emergence or worsening of symptoms of depression, any unusual changes in mood or
behavior, or the emergence of suicidal thoughts, behavior, or thoughts about self-harm.
Behaviors of concern should be reported immediately to healthcare providers.
Do not use capsules which are discolored.
Laboratory Tests: Phenytoin serum level determinations may be necessary to achieve optimal
dosage adjustments. Phenytoin doses are usually selected to attain therapeutic plasma total
phenytoin concentrations of 10 to 20 µg/mL (unbound phenytoin concentrations of 1 to 2
µg/mL).
Drug Interactions: Phenytoin is extensively bound to serum plasma proteins and is prone to
competitive displacement. Phenytoin is metabolized by hepatic cytochrome P450 enzymes
CYP2C9 and CYP2C19, and is particularly susceptible to inhibitory drug interactions because it
is subject to saturable metabolism. Inhibition of metabolism may produce significant increases in
circulating phenytoin concentrations and enhance the risk of drug toxicity. Phenytoin is a potent
inducer of hepatic drug-metabolizing enzymes. Serum level determinations for phenytoin are
especially helpful when possible drug interactions are suspected.
The most commonly occurring drug interactions are listed below:
Note: The list is not intended to be inclusive or comprehensive. Individual drug package inserts
should be consulted.
Drugs that affect phenytoin concentrations:
Drugs that may increase phenytoin serum levels include: acute alcohol intake,
amiodarone, anti-epileptic agents (ethosuximide,felbamate, oxcarbazepine,

23
methsuximide, topiramate), azoles (fluconazole, ketoconazole, itraconazole, miconazole,
voriconazole), capecitabine, chloramphenicol, chlordiazepoxide, disulfiram, estrogens,
fluorouracil, fluoxetine, fluvastatin, fluvoxamine, H2-antagonists (e.g., cimetidine),
halothane, isoniazid, methylphenidate, omeprazole, phenothiazines, salicylates, sertraline,
succinimides, sulfonamides (e.g., sulfamethizole, sulfaphenazole, sulfadiazine,
sulfamethoxazole-trimethoprim), ticlopidine, tolbutamide, trazodone, and warfarin.
Drugs that may decrease phenytoin levels include: anticancer drugs usually in
combination (e.g., bleomycin, carboplatin, cisplatin, doxorubicin, methotrexate),
carbamazepine, chronic alcohol abuse, diazepam, diazoxide, folic acid, fosamprenavir,
nelfinavir, reserpine, rifampin, ritonavir, St. John’s Wort, sucralfate, theophylline and
vigabatrin.
Administration of phenytoin with preparations that increase gastric pH (e.g., supplements
or antacids containing calcium carbonate, aluminum hydroxide, and magnesium
hydroxide) may affect the absorption of phenytoin. In most cases where interactions were
seen, the effect is a decrease in phenytoin levels when the drugs are taken at the same
time. When possible, phenytoin and these products should not be taken at the same time
of day.
Drugs that may either increase or decrease phenytoin serum levels include: phenobarbital,
sodium valproate, and valproic acid. Similarly, the effect of phenytoin on phenobarbital,
valproic acid, and sodium valproate serum levels is unpredictable.
The addition or withdrawal of these agents in patients on phenytoin therapy may require
an adjustment of the phenytoin dose to achieve optimal clinical outcome.
Drugs affected by phenytoin:
Drugs that should not be coadministered with phenytoin: Delavirdine (see
CONTRAINDICATIONS).
Drugs whose efficacy is impaired by phenytoin include: azoles (fluconazole,
ketoconazole, itraconazole, voriconazole), posaconazole, corticosteroids, doxycycline,
estrogens, furosemide, irinotecan, oral contraceptives, paclitaxel, paroxetine, quinidine,
rifampin, sertraline, teniposide, theophylline, and vitamin D.
Increased and decreased PT/INR responses have been reported when phenytoin is
coadministered with warfarin.
Phenytoin decreases plasma concentrations of active metabolites of albendazole, certain
HIV antivirals (efavirenz, lopinavir/ritonavir, indinavir, nelfinavir, ritonavir, saquinavir),
anti-epileptic agents (carbamazepine, felbamate, lamotrigine, topiramate, oxcarbazepine,
quetiapine), atorvastatin, chlorpropamide, clozapine, cyclosporine, digoxin, fluvastatin,
folic acid, methadone, mexiletine, nifedipine, nimodipine, nisoldipine, praziquantel,
simvastatin, and verapamil.
Phenytoin when given with fosamprenavir alone may decrease the concentration of
amprenavir, the active metabolite. Phenytoin when given with the combination of
fosamprenavir and ritonavir may increase the concentration of amprenavir.
Resistance to the neuromuscular blocking action of the non-depolarizing neuromuscular
blocking agents pancuronium, vecuronium, rocuronium, and cisatracurium has occurred
in patients chronically administered phenytoin. Whether or not phenytoin has the same

24
effect on other non-depolarizing agents is unknown. Patients should be monitored closely
for more rapid recovery from neuromuscular blockade than expected, and infusion rate
requirements may be higher.
The addition or withdrawal of phenytoin during concomitant therapy with these agents
may require adjustment of the dose of these agents to achieve optimal clinical outcome.
Drug Enteral Feeding/Nutritional Preparations Interaction: Literature reports suggest that
patients who have received enteral feeding preparations and/or related nutritional supplements
have lower than expected phenytoin plasma levels. It is therefore suggested that phenytoin not be
administered concomitantly with an enteral feeding preparation. More frequent serum phenytoin
level monitoring may be necessary in these patients.
Drug/Laboratory Test Interactions: Phenytoin may decrease serum concentrations of T4. It
may also produce lower than normal values for dexamethasone or metyrapone tests. Phenytoin
may cause increased serum levels of glucose, alkaline phosphatase, and gamma glutamyl
transpeptidase (GGT).
Care should be taken when using immunoanalytical methods to measure plasma phenytoin
concentrations.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis: See WARNINGS (Hematopoietic System) section
In carcinogenicity studies, phenytoin was administered in the diet to mice (10, 25, or
45 mg/kg/day) and rats (25, 50, or 100 mg/kg/day) for 2 years. The incidence of hepatocellular
tumors were increased in male and female mice at the highest dose. No increases in tumor
incidence were observed in rats. The highest doses tested in these studies were associated with
peak plasma phenytoin levels below human therapeutic concentrations.
In carcinogenicity studies reported in the literature, phenytoin was administered in the diet for 2
years at doses up to 600 ppm (approximately 90 mg/kg/day) to mice and up to 2400 ppm
(approximately 120 mg/kg/day) to rats. The incidences of hepatocellular tumors were increased
in female mice at all but the lowest dose tested. No increases in tumor incidence were observed
in rats.
Mutagenesis
Phenytoin was negative in the Ames test and in the in vitro clastogenicity assay in Chinese
hamster ovary (CHO) cells.
In studies reported in the literature, phenytoin was negative in the in vitro mouse lymphoma
assay and the in vivo micronucleus assay in mouse. Phenytoin was clastogenic in the in vitro
sister chromatid exchange assay in CHO cells.
Fertility
Phenytoin has not been adequately assessed for effects on male or female fertility.

25
Pregnancy:
See WARNINGS section.
Nursing Mothers: Infant breast-feeding is not recommended for women taking this drug
because phenytoin appears to be secreted in low concentrations in human milk.
Pediatric Use: See DOSAGE AND ADMINISTRATION section.
Geriatric Use: Phenytoin clearance tends to decrease with increasing age.
ADVERSE REACTIONS
Body as a Whole: Allergic reactions in the form of rash and rarely more serious forms (see Skin
and Appendages paragraph below) and DRESS (see WARNINGS) have been observed.
Anaphylaxis has also been reported.
There have also been reports of coarsening of facial features, systemic lupus erythematosus,
periarteritis nodosa, and immunoglobulin abnormalities.
Nervous System: The most common adverse reactions encountered with phenytoin therapy are
nervous system reactions and are usually dose-related. Reactions include nystagmus, ataxia,
slurred speech, decreased coordination, somnolence, and mental confusion. Dizziness, vertigo,
insomnia, transient nervousness, motor twitchings, paresthesias, and headaches have also been
observed. There have also been rare reports of phenytoin-induced dyskinesias, including chorea,
dystonia, tremor, and asterixis, similar to those induced by phenothiazine and other neuroleptic
drugs. Cerebellar atrophy has been reported, and appears more likely in settings of elevated
phenytoin levels and/or long-term phenytoin use.
A predominantly sensory peripheral polyneuropathy has been observed in patients receiving
long-term phenytoin therapy.
Digestive System: Acute hepatic failure, toxic hepatitis, liver damage, nausea, vomiting,
constipation, enlargement of the lips, and gingival hyperplasia.
Skin and Appendages: Dermatological manifestations sometimes accompanied by fever have
included scarlatiniform or morbilliform rashes. A morbilliform rash (measles-like) is the most
common; other types of dermatitis are seen more rarely. Other more serious forms which may be
fatal have included bullous, exfoliative or purpuric dermatitis, Stevens-Johnson syndrome, and
toxic epidermal necrolysis (see WARNINGS section). There have also been reports of
hypertrichosis.
Hematologic and Lymphatic System: Hematopoietic complications, some fatal, have
occasionally been reported in association with administration of phenytoin. These have included
thrombocytopenia, leukopenia, granulocytopenia, agranulocytosis, and pancytopenia with or

26
without bone marrow suppression. While macrocytosis and megaloblastic anemia have occurred,
these conditions usually respond to folic acid therapy. Lymphadenopathy including benign
lymph node hyperplasia, pseudolymphoma, lymphoma, and Hodgkin’s disease have been
reported (see WARNINGS section).
Special Senses: Altered taste sensation including metallic taste.
Urogenital: Peyronie’s disease
OVERDOSAGE
The lethal dose in pediatric patients is not known. The lethal dose in adults is estimated to be 2 to
5 grams. The initial symptoms are nystagmus, ataxia, and dysarthria. Other signs are tremor,
hyperreflexia, lethargy, slurred speech, blurred vision, nausea, and vomiting. The patient may
become comatose and hypotensive. Death is due to respiratory and circulatory depression.
There are marked variations among individuals with respect to phenytoin plasma levels where
toxicity may occur. Nystagmus, on lateral gaze, usually appears at 20 mcg/mL, ataxia at 30
mcg/mL; dysarthria and lethargy appear when the plasma concentration is over 40 mcg/mL, but
as high a concentration as 50 mcg/mL has been reported without evidence of toxicity. As much
as 25 times the therapeutic dose has been taken to result in a serum concentration over 100
mcg/mL with complete recovery. Irreversible cerebellar dysfunction and atrophy have been
reported.
Treatment: Treatment is nonspecific since there is no known antidote.
The adequacy of the respiratory and circulatory systems should be carefully observed and
appropriate supportive measures employed. Hemodialysis can be considered since phenytoin is
not completely bound to plasma proteins. Total exchange transfusion has been used in the
treatment of severe intoxication in pediatric patients.
In acute overdosage, the possibility of other CNS depressants, including alcohol, should be borne
in mind.
DOSAGE AND ADMINISTRATION
Serum concentrations should be monitored in changing from Dilantin (extended phenytoin
sodium capsules) to Prompt Phenytoin Sodium Capsules, and from the sodium salt to the free
acid form.
Dilantin (extended phenytoin sodium capsules,) are formulated with the sodium salt of
phenytoin. The free acid form of phenytoin is used in Dilantin-125 Suspension and Dilantin
Infatabs. Because there is approximately an 8% increase in drug content with the free acid form
over that of the sodium salt, dosage adjustments and serum level monitoring may be necessary

27
when switching from a product formulated with the free acid to a product formulated with the
sodium salt and vice versa.
General: Dosage should be individualized to provide maximum benefit. In some cases, serum
blood level determinations may be necessary for optimal dosage adjustments—the clinically
effective serum level is usually 10 to 20 mcg/mL. With recommended dosage, a period of seven
to ten days may be required to achieve steady-state blood levels with phenytoin and changes in
dosage (increase or decrease) should not be carried out at intervals shorter than seven to ten days.
Adult Dosage:
Divided daily dosage: Patients who have received no previous treatment may be started on one
100-mg Dilantin (extended phenytoin sodium capsule) three times daily and the dosage then
adjusted to suit individual requirements. For most adults, the satisfactory maintenance dosage
will be one capsule three to four times a day. An increase up to two capsules three times a day
may be made, if necessary.
Once-a-day dosage: In adults, if seizure control is established with divided doses of three 100-
mg Dilantin (extended phenytoin sodium capsules,) daily, once-a-day dosage with 300 mg of
Dilantin (extended phenytoin sodium capsules) may be considered. Studies comparing divided
doses of 300 mg with a single daily dose of this quantity indicated absorption, peak plasma
levels, biologic half-life, difference between peak and minimum values, and urinary recovery
were equivalent. Once-a-day dosage offers a convenience to the individual patient or to nursing
personnel for institutionalized patients and is intended to be used only for patients requiring this
amount of drug daily. A major problem in motivating noncompliant patients may also be
lessened when the patient can take this drug once a day. However, patients should be cautioned
not to miss a dose, inadvertently.
Only Dilantin (extended phenytoin sodium capsules) are recommended for once-a-day dosing.
Inherent differences in dissolution characteristics and resultant absorption rates of phenytoin due
to different manufacturing procedures and/or dosage forms preclude such recommendation for
other phenytoin products. When a change in the dosage form or brand is prescribed, careful
monitoring of phenytoin serum levels should be carried out.
Loading dose: Some authorities have advocated use of an oral loading dose of phenytoin in
adults who require rapid steady-state serum levels and where intravenous administration is not
desirable. This dosing regimen should be reserved for patients in a clinic or hospital setting
where phenytoin serum levels can be closely monitored. Patients with a history of renal or liver
disease should not receive the oral loading regimen.
Initially, one gram of Dilantin (extended phenytoin sodium capsules) is divided into three doses
(400 mg, 300 mg, 300 mg) and administered at two-hour intervals. Normal maintenance dosage
is then instituted 24 hours after the loading dose, with frequent serum level determinations.

28
Dosing in Special Populations
Patients with Renal or Hepatic Disease: Due to an increased fraction of unbound phenytoin in
patients with renal or hepatic disease, or in those with hypoalbuminemia, the interpretation of
total phenytoin plasma concentrations should be made with caution. Unbound phenytoin
concentrations may be more useful in these patient populations.
Elderly Patients: Phenytoin clearance is decreased slightly in elderly patients and lower or less
frequent dosing may be required.
Pediatric: Initially, 5 mg/kg/day in two or three equally divided doses, with subsequent dosage
individualized to a maximum of 300 mg daily. A recommended daily maintenance dosage is
usually 4 to 8 mg/kg. Children over 6 years old and adolescents may require the minimum adult
dose (300 mg/day).
Store below 30° C. Preserve in tight, light-resistant containers. Protect from moisture.
Commercial presentation
Bottle with 100 capsules.
Not all commercial presentations are available in each country.
For more information please contact Pfizer Medical Information at: [email protected]
Revision Date: December 2015
Reference Document: USPI LAB-0375-19.0
For Central America and the Caribbean region see information about the license holder,
manufacturer and packager in the secondary packaging.


















